Maternal High Fat Diet Programs Male Mice Offspring Hyperphagia and Obesity: Mechanism of Increased Appetite Neurons via Altered Neurogenic Factors and Nutrient Sensor AMPK




Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Maternal Diet
2.3. Offspring Studies
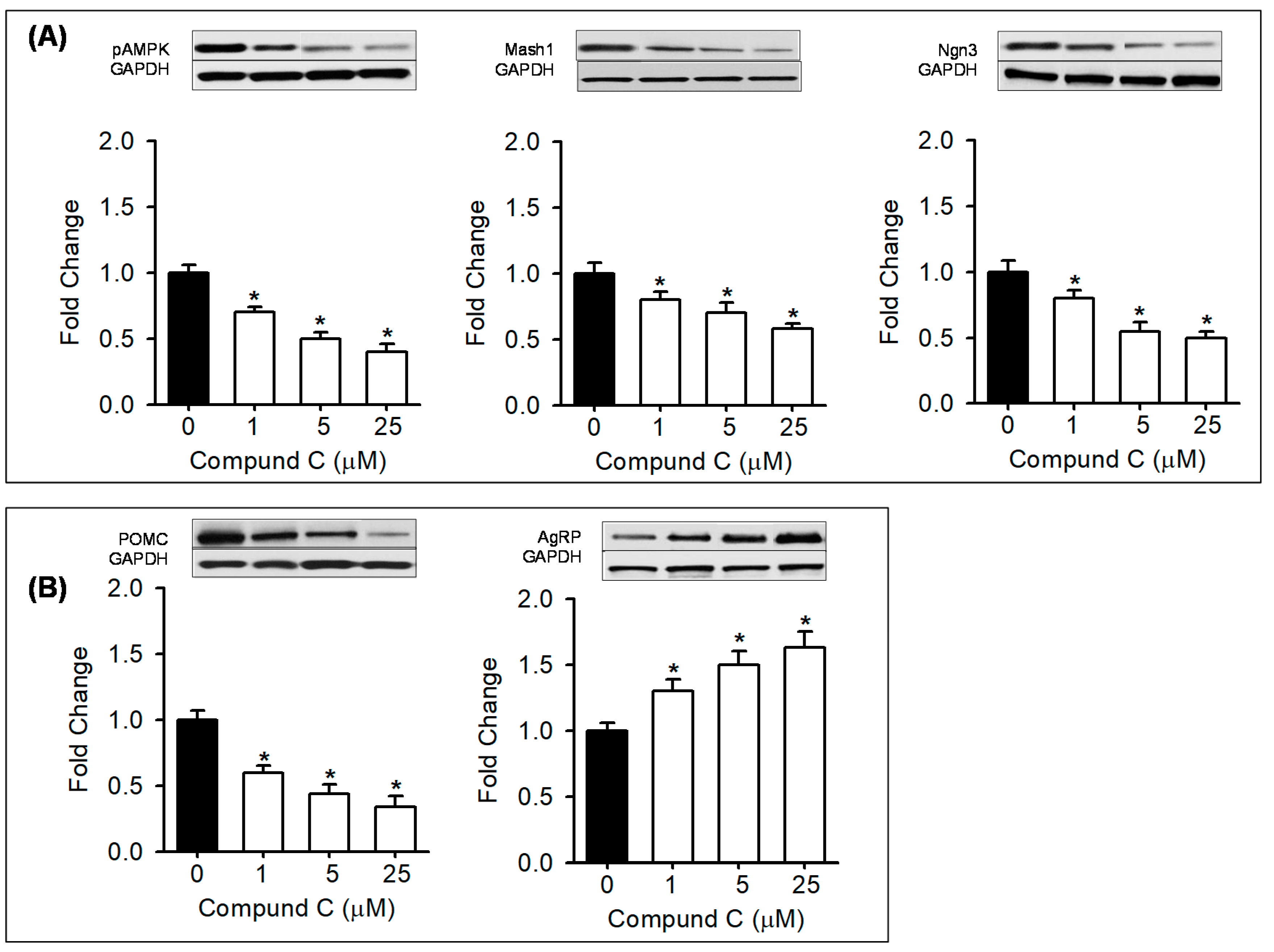
2.4. In Vitro Treatment with AMPK Modulators
2.5. Hypothalamus/ARC Dissection
2.6. Tissue Neuronal and NPY Counts
2.7. Neurosphere Cultures
2.8. Western Blot
2.9. Statistics
3. Results
3.1. Maternal Body Weight
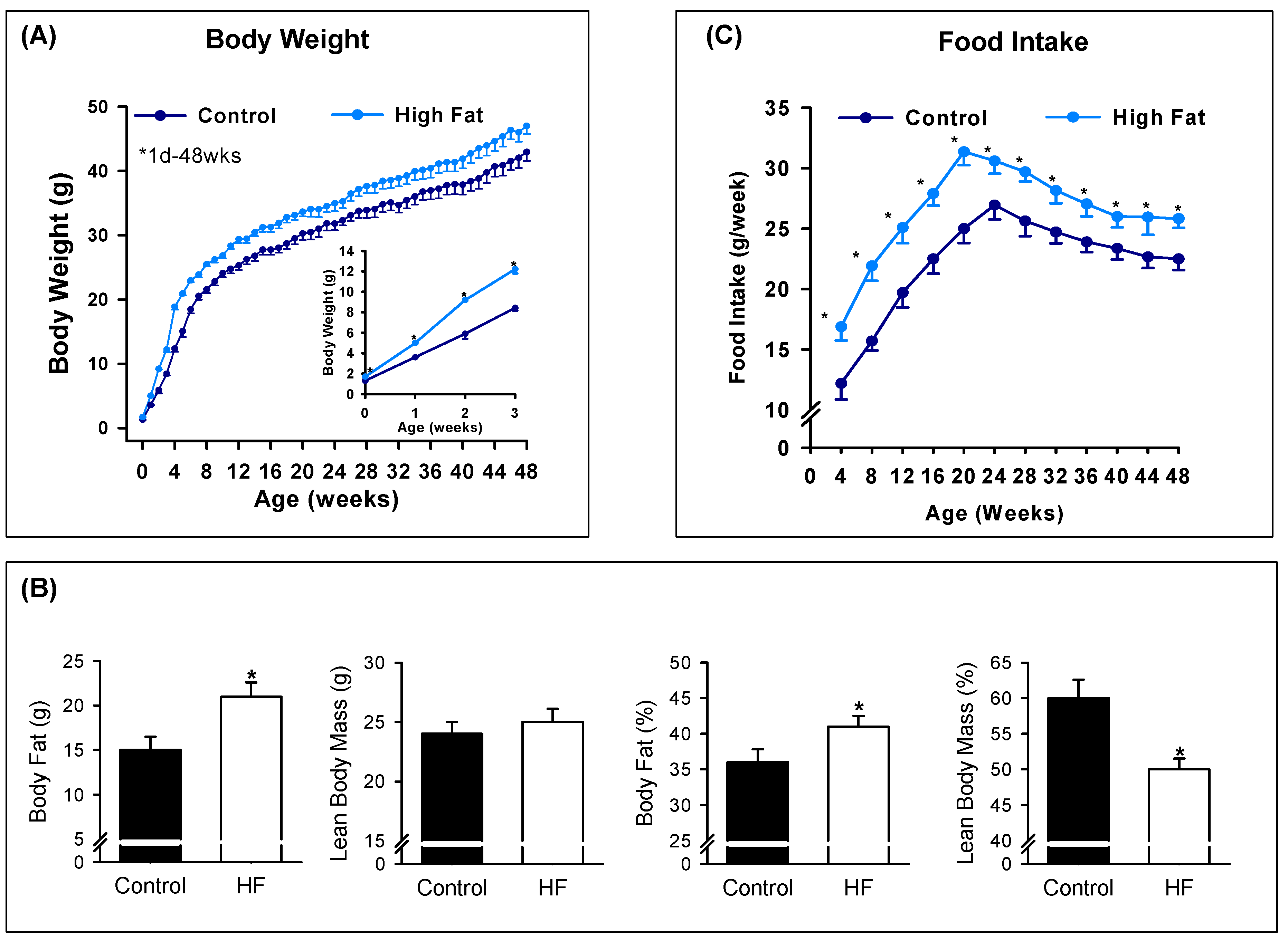
3.2. Offspring Phenotype
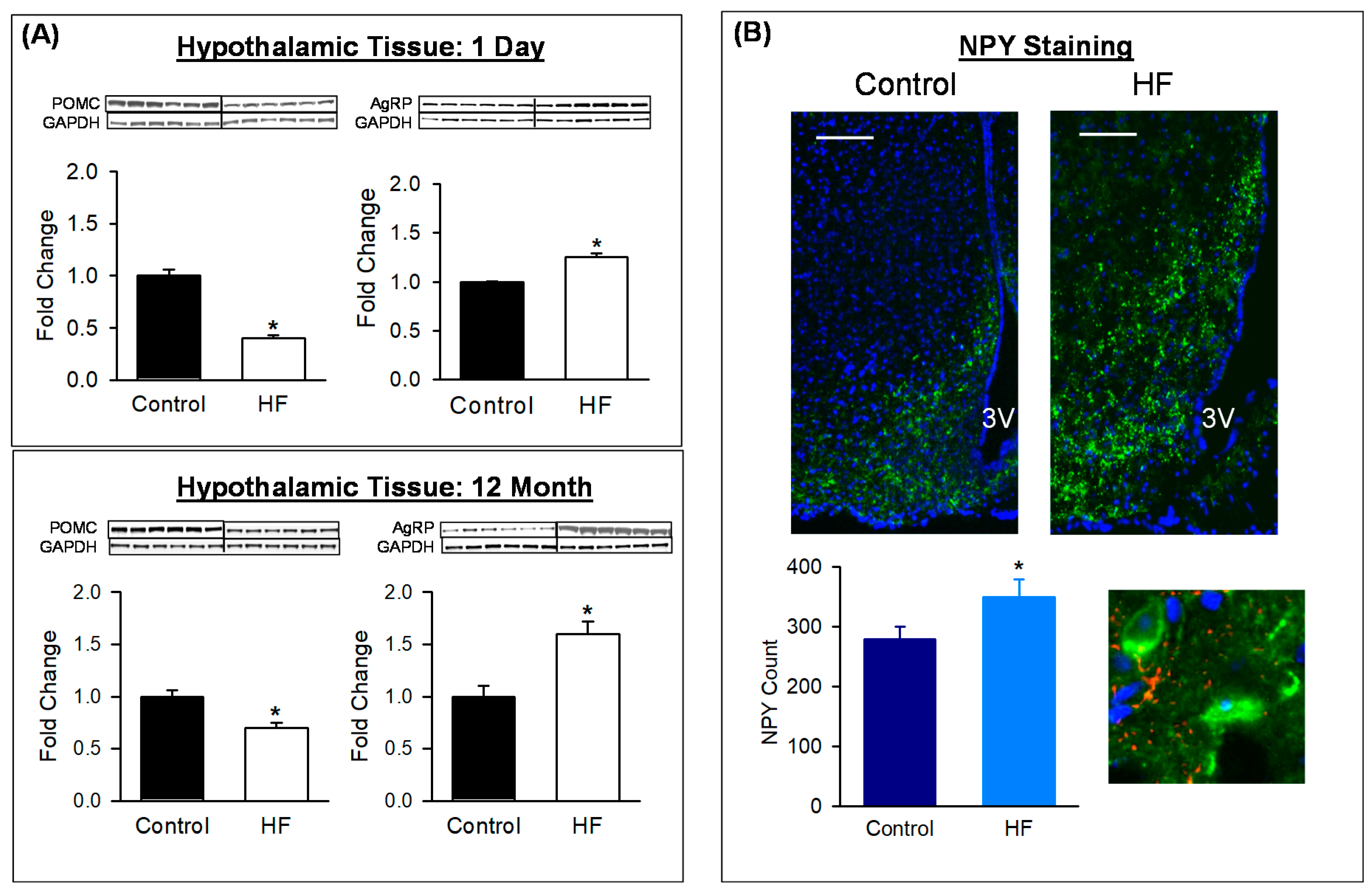
3.3. Hypothalamic/ARC Protein Expression and Neuronal Counts
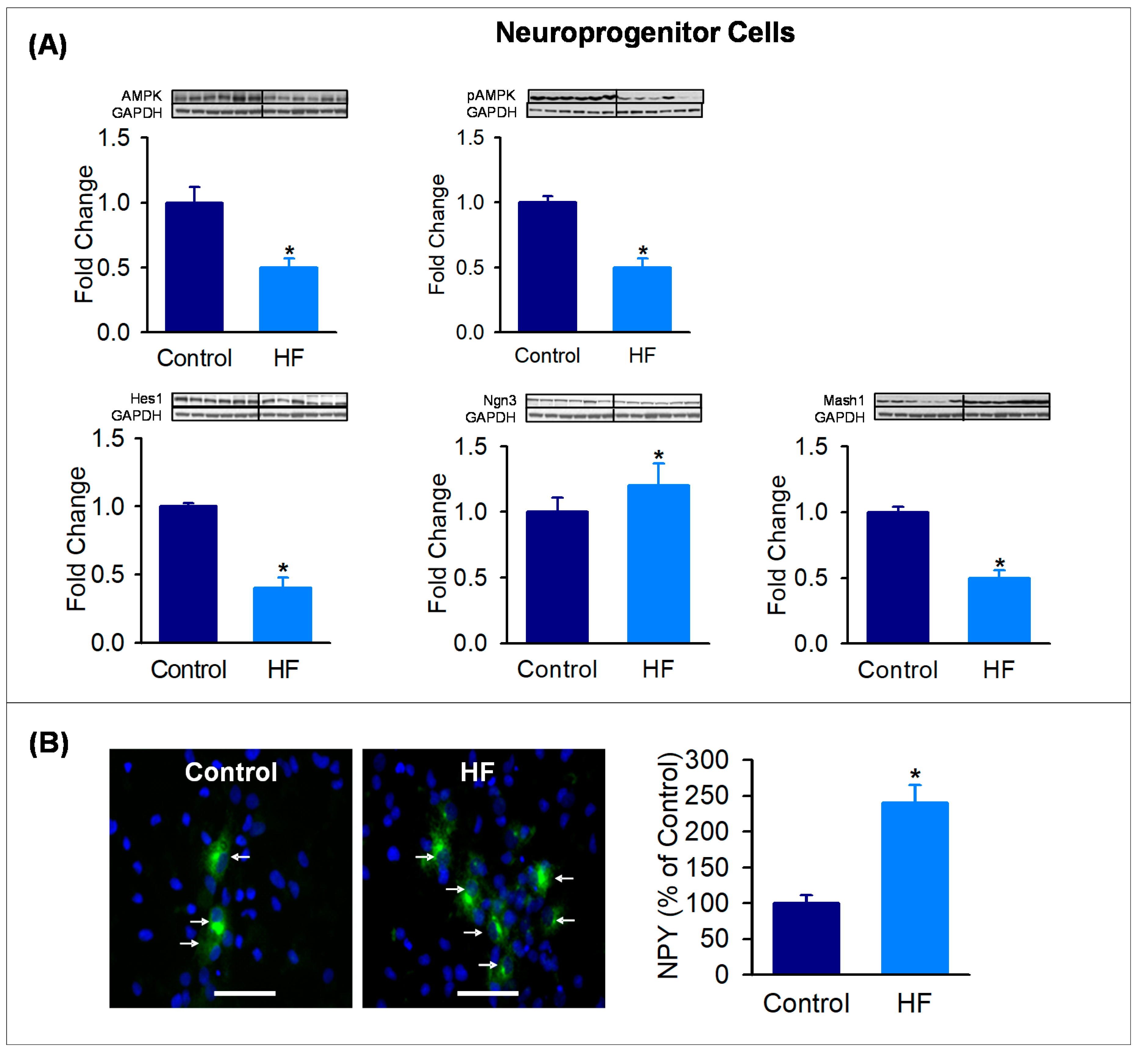
3.4. Hypothalamic Neuroprogenitor Cells
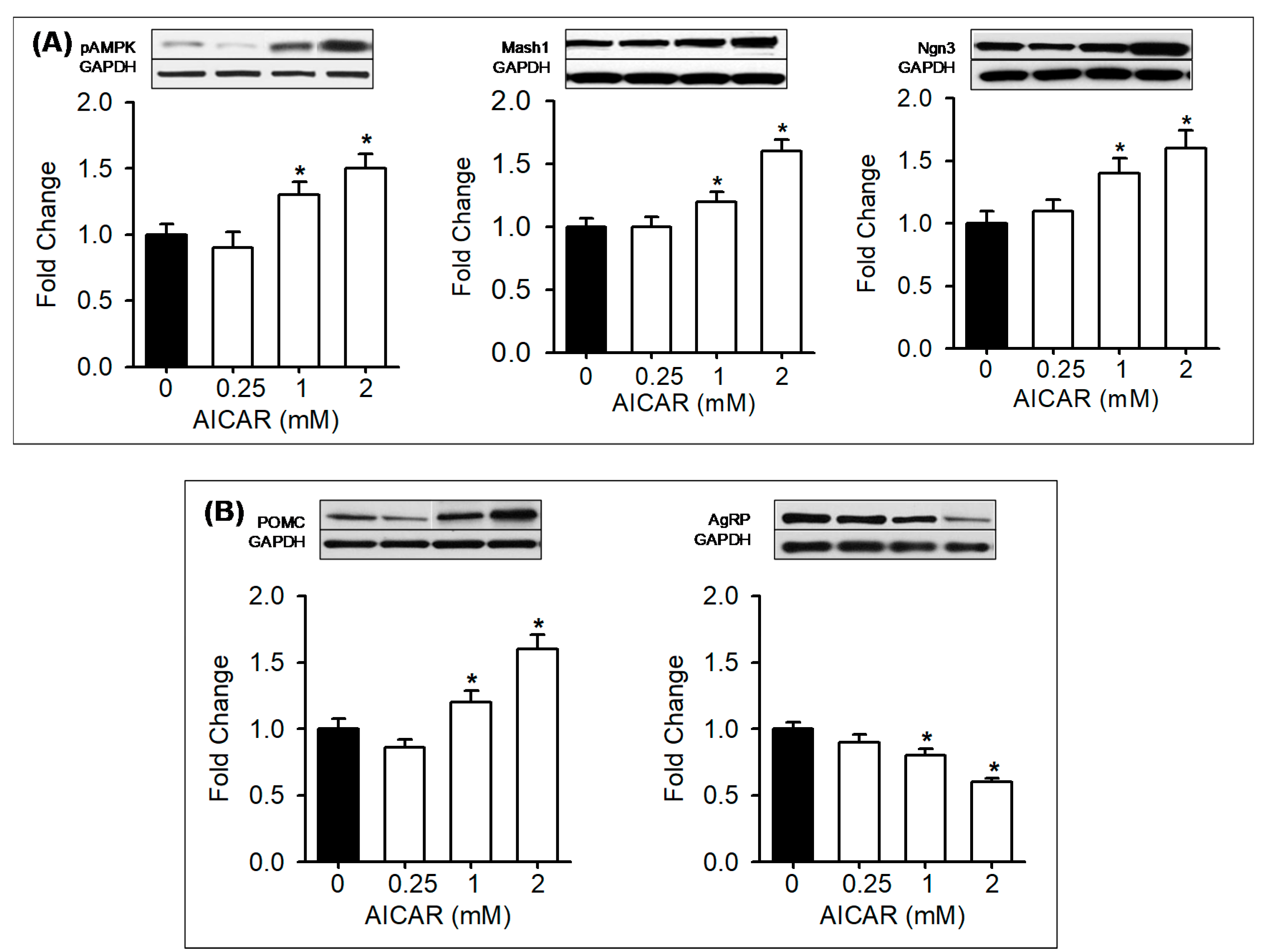
3.5. In Vitro Treatment with AMPK Modulators
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ogden, C.L.; Carroll, M.D.; Kit, B.K.; Flegal, K.M. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014, 311, 806–814. [Google Scholar] [CrossRef]
- Swinburn, B.; Sacks, G.; Ravussin, E. Increased food energy supply is more than sufficient to explain the US epidemic of obesity. Am. J. Clin. Nutr. 2009, 90, 1453–1456. [Google Scholar] [CrossRef]
- Swinburn, B.A.; Sacks, G.; Lo, S.K.; Westerterp, K.R.; Rush, E.C.; Rosenbaum, M.; Luke, A.; Schoeller, D.A.; Delany, J.P.; Butte, N.F.; et al. Estimating the changes in energy flux that characterize the rise in obesity prevalence. Am. J. Clin. Nutr. 2009, 89, 1723–1728. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Eriksson, J.G.; Forsén, T.; Osmond, C. Fetal origins of adult disease: Strength of effects and biological basis. Int. J. Epidemiol. 2002, 31, 1235–1239. [Google Scholar] [CrossRef]
- Ylihärsilä, H.; Kajantie, E.; Osmond, C.; Forsén, T.; Barker, D.J.P.; Eriksson, J.G. Birth size, adult body composition and muscle strength in later life. Int. J. Obes. 2007, 31, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.L.; Olsen, L.W.; Sorensen, T.I. Weight at birth and all-cause mortality in adulthood. Epidemiology 2008, 19, 197–203. [Google Scholar] [CrossRef]
- Santangeli, L.; Sattar, N.; Huda, S.S. Impact of maternal obesity on perinatal and childhood outcomes. Best Pract. Res. Clin. Obstet. Gynaecol. 2015, 29, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Boney, C.M.; Verma, A.; Tucker, R.; Vohr, B.R. Metabolic syndrome in childhood: Association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 2005, 115, e290–e296. [Google Scholar] [CrossRef]
- Hermann, G.M.; Dallas, L.M.; Haskell, S.E.; Roghair, R.D. Neonatal macrosomia is an independent risk factor for adult metabolic syndrome. Neonatology 2010, 98, 238–244. [Google Scholar] [CrossRef]
- Shankar, K.; Harrell, A.; Gilchrist, J.; Ronis, M.J.J.; Badger, T. Maternal Obesity at Conception Programs Obesity in the Offspring. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2008, 294, R528–R538. [Google Scholar] [CrossRef]
- Sousa-Ferreira, L.; Garrido, M.; Nascimento-Ferreira, I.; Nóbrega, C.; Santos-Carvalho, A.; Álvaro, A.R.; Rosmaninho-Salgado, J.; Kaster, M.P.; Kügler, S.; De Almeida, L.P.; et al. Moderate long-term modulation of neuropeptide Y in hypothalamic arcuate nucleus induces energy balance alterations in adult rats. PLoS ONE 2011, 6, e22333. [Google Scholar] [CrossRef]
- Pich, E.M.; Messori, B.; Zoli, M.; Ferraguti, F.; Marrama, P.; Biagini, G.; Fuxe, K.; Agnati, L.F. Feeding and drinking responses to neuropeptide Y injections in the paraventricular hypothalamic nucleus of aged rats. Brain Res. 1992, 575, 265–271. [Google Scholar] [CrossRef]
- Koutcherov, Y.; Ashwell, K.W.; Paxinos, G. Organization of human hypothalamus in fetal development. J. Comp. Neurol. 2002, 446, 301–324. [Google Scholar] [CrossRef]
- Miller, F.D.; Gauthier, A.S. Timing is everything: Making neurons versus glia in the developing cortex. Neuron 2007, 54, 357–369. [Google Scholar] [CrossRef]
- Padilla, S.L.; Carmody, J.S.; Zeltser, L.M. Pomc-expressing progenitors give rise to antagonistic neuronal populations in hypothalamic feeding circuits. Nat. Med. 2010, 16, 403–405. [Google Scholar] [CrossRef]
- Nilsson, I.A.K.; Johansen, J.E.; Schalling, M.; Hökfelt, T.; Fetissov, S.O. Maturation of the hypothalamic arcuate agouti-related protein system during postnatal development in the mouse. Brain Res. Dev. 2005, 155, 147–154. [Google Scholar] [CrossRef]
- Kageyama, R.; Ohtsuka, T.; Kobayashi, T. Roles of Hes genes in neural development. Dev. Growth Differ. 2008, 50 (Suppl. S1), S97-S103. [Google Scholar] [CrossRef]
- Hatakeyama, J.; Kageyama, R. Notch1 expression is spatiotemporally correlated with neurogenesis and negatively regulated by Notch1-independent Hes genes in the developing nervous system. Cereb. Cortex 2006, 16 (Suppl. S1), i132–i137. [Google Scholar] [CrossRef]
- McNay, D.E.G.; Pelling, M.; Claxton, S.; Guillemot, F.; Ang, S.-L. Mash1 is required for generic and subtype differentiation of hypothalamic neuroendocrine cells. Mol. Endocrinol. 2006, 20, 1623–1632. [Google Scholar] [CrossRef]
- MacKay, H.; Abizaid, A. Embryonic development of the hypothalamic feeding circuitry: Transcriptional, nutritional, and hormonal influences. Mol. Metab. 2014, 3, 813–822. [Google Scholar] [CrossRef]
- Pelling, M.; Anthwal, N.; McNay, D.; Gradwohl, G.; Leiter, A.B.; Guillemot, F.; Ang, S.-L. Differential requirements for neurogenin 3 in the development of POMC and NPY neurons in the hypothalamus. Dev. Biol. 2011, 349, 406–416. [Google Scholar] [CrossRef]
- Anthwal, N.; Pelling, M.; Claxton, S.; Mellitzer, G.; Collin, C.; Kessaris, N.; Richardson, W.D.; Gradwohl, G.; Ang, S.-L. Conditional deletion of neurogenin-3 using Nkx2.1iCre results in a mouse model for the central control of feeding, activity and obesity. Dis. Model. Mech. 2013, 6, 1133–1145. [Google Scholar] [CrossRef]
- Grundy, D. Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. Exp. Physiol. 2015, 100, 755–758. [Google Scholar] [CrossRef]
- Maske, C.B.; Jackson, C.M.; Terrill, S.J.; Eckel, L.A.; Williams, D.L. Estradiol modulates the anorexic response to central glucagon-like peptide 1. Horm. Behav. 2017, 93, 109–117. [Google Scholar] [CrossRef]
- Eckel, L.A.; Geary, N. Estradiol treatment increases feeding-induced c-Fos expression in the brains of ovariectomized rats. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2001, 281, R738–R746. [Google Scholar] [CrossRef]
- Gage, G.J.; Kipke, D.R.; Shain, W. Whole animal perfusion fixation for rodents. J. Vis. Exp. 2012, 2012, e3564. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kim, M.Y.; Kim, H.J.; Roh, G.S.; Ko, G.H.; Seo, H.G.; Lee, J.H.; Chang, K.C. Compound C independent of AMPK inhibits ICAM-1 and VCAM-1 expression in inflammatory stimulants-activated endothelial cells in vitro and in vivo. Atherosclerosis 2011, 219, 57–64. [Google Scholar] [CrossRef]
- Shimogori, T.; Lee, D.A.; Mirandaangulo, A.L.; Yang, Y.; Wang, H.; Jiang, L.; Yoshida, A.C.; Kataoka, A.; Mashiko, H.; Avetisyan, M.; et al. A genomic atlas of mouse hypothalamic development. Nat. Neurosci. 2010, 13, 767–775. [Google Scholar] [CrossRef]
- Burke, M.W.; Zangenehpour, S.; Mouton, P.R.; Ptito, M. Knowing what counts: Unbiased stereology in the non-human primate brain. J. Vis. Exp. 2009, 14, e1262. [Google Scholar] [CrossRef]
- Golub, V.M.; Ebrewer, J.; Ewu, X.; Ekuruba, R.; Eshort, J.; Emanchi, M.; Eswonke, M.; Eyounus, I.; Reddy, D.S. Neurostereology protocol for unbiased quantification of neuronal injury and neurodegeneration. Front. Aging Neurosci. 2015, 7, 196. [Google Scholar] [CrossRef]
- Desai, M.; Li, T.; Ross, M.G. Fetal Hypothalamic Neuroprogenitor Cell Culture: Preferential Differentiation Paths Induced by Leptin and Insulin. Endocrinology 2011, 152, 3192–3201. [Google Scholar] [CrossRef]
- Desai, M.; Li, T.; Ross, M.G. Hypothalamic neurosphere progenitor cells in low birth-weight rat newborns: Neurotrophic effects of leptin and insulin. Brain Res. 2011, 1378, 29–42. [Google Scholar] [CrossRef]
- Fukami, T.; Sun, X.; Li, T.; Yamada, M.; Desai, M.; Ross, M.G. Mechanism of programmed obesity: Altered central insulin sensitivity in growth-restricted juvenile female rats. J. Dev. Orig. Health Dis. 2013, 4, 239–248. [Google Scholar] [CrossRef]
- Desai, M.; Jellyman, J.K.; Han, G.; Beall, M.; Lane, R.H.; Ross, M.G. Maternal obesity and high-fat diet program offspring metabolic syndrome. Am. J. Obstet. Gynecol. 2014, 211, 237.e1–237.e13. [Google Scholar] [CrossRef]
- Lomas-Soria, C.; Reyes-Castro, L.A.; Rodríguez-González, G.L.; Ibáñez, C.A.; Bautista, C.J.; Cox, L.A.; Nathanielsz, P.W.; Zambrano, E. Maternal obesity has sex-dependent effects on insulin, glucose and lipid metabolism and the liver transcriptome in young adult rat offspring. J. Physiol. 2018, 596, 4611–4628. [Google Scholar] [CrossRef]
- Vasudevan, C.; Renfrew, M.; McGuire, W. Fetal and perinatal consequences of maternal obesity. Arch. Dis. Child.-Fetal Neonatal Ed. 2011, 96, F378–F382. [Google Scholar] [CrossRef]
- Catalano, P.M.; Shankar, K. Obesity and pregnancy: Mechanisms of short term and long term adverse consequences for mother and child. BMJ 2017, 356, j1. [Google Scholar] [CrossRef]
- Kirk, S.L.; Samuelsson, A.-M.; Argenton, M.; Dhonye, H.; Kalamatianos, T.; Poston, L.; Taylor, P.D.; Coen, C.W. Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS ONE 2009, 4, e5870. [Google Scholar] [CrossRef]
- Bautista, C.J.; Montaño, S.; Ramírez, V.; Morales, A.; Nathanielsz, P.W.; Bobadilla, N.A.; Zambrano, E. Changes in milk composition in obese rats consuming a high-fat diet. Br. J. Nutr. 2016, 115, 538–546. [Google Scholar] [CrossRef]
- Guidotti, S.; Jónás, I.; Schubert, K.A.; Garland, T.; Meijer, H.A.J.; Scheurink, A.J.W.; Van Dijk, G. High-saturated fat-sucrose feeding affects lactation energetics in control mice and mice selectively bred for high wheel-running behavior. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2013, 305, R1433–R1440. [Google Scholar] [CrossRef][Green Version]
- Del Prado, M.; Villalpando, S.; Gordillo, J.; Hernandez-Montes, H. A high dietary lipid intake during pregnancy and lactation enhances mammary gland lipid uptake and lipoprotein lipase activity in rats. J. Nutr. 1999, 129, 1574–1578. [Google Scholar] [CrossRef][Green Version]
- Del Prado, M.; Delgado, G.; Villalpando, S. Maternal lipid intake during pregnancy and lactation alters milk composition and production and litter growth in rats. J. Nutr. 1997, 127, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Blundell, J.E.; Stubbs, R.J.; Golding, C.; Croden, F.; Alam, R.; Whybrow, S.; Le Noury, J.; Lawton, C.L. Resistance and susceptibility to weight gain: Individual variability in response to a high-fat diet. Physiol. Behav. 2005, 86, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Mackenzie, K.C.; Gahagan, S. The effect of maternal obesity on the offspring. Clin. Obstet. Gynecol. 2014, 57, 508–515. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.; Eriksson, J.G.; Broekman, B.F.P. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef]
- Rising, R.; Lifshitz, F. Relationship between maternal obesity and infant feeding-interactions. Nutr. J. 2005, 4, 17. [Google Scholar] [CrossRef]
- Thompson, A.L. Intergenerational impact of maternal obesity and postnatal feeding practices on pediatric obesity. Nutr. Rev. 2013, 71 (Suppl. S1), S55–S61. [Google Scholar] [CrossRef]
- Morris, M.J.; Chen, H. Established maternal obesity in the rat reprograms hypothalamic appetite regulators and leptin signaling at birth. Int. J. Obes. 2009, 33, 115–122. [Google Scholar] [CrossRef]
- Desai, M.; Han, G.; Ross, M.G. Programmed hyperphagia in offspring of obese dams: Altered expression of hypothalamic nutrient sensors, neurogenic factors and epigenetic modulators. Appetite 2016, 99, 193–199. [Google Scholar] [CrossRef]
- Page, K.C.; Malik, R.E.; Ripple, J.A.; Anday, E.K. Maternal and postweaning diet interaction alters hypothalamic gene expression and modulates response to a high-fat diet in male offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1049–R1057. [Google Scholar] [CrossRef]
- Beck, B.; Richy, S.; Archer, Z.A.; Mercer, J.G. Early and persistent up-regulation of hypothalamic orexigenic peptides in rat offspring born to dams fed a high-carbohydrate supplement during gestation. Brain Res. 2012, 1477, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, R.; Ohtsuka, T.; Tomita, K. The bHLH gene Hes1 regulates differentiation of multiple cell types. Mol. Cells 2000, 10, 1–7. [Google Scholar] [CrossRef]
- Qu, X.; Afelik, S.; Jensen, J.N.; Bukys, M.A.; Kobberup, S.; Schmerr, M.; Xiao, F.; Nyeng, P.; Albertoni, M.V.; Grapin-Botton, A.; et al. Notch-mediated post-translational control of Ngn3 protein stability regulates pancreatic patterning and cell fate commitment. Dev. Biol. 2013, 376, 1–12. [Google Scholar] [CrossRef]
- Cavaliere, G.; Viggiano, E.; Trinchese, G.; De Filippo, C.; Messina, A.; Monda, V.; Valenzano, A.; Cincione, R.I.; Zammit, C.; Cimmino, F.; et al. Long Feeding High-Fat Diet Induces Hypothalamic Oxidative Stress and Inflammation, and Prolonged Hypothalamic AMPK Activation in Rat Animal Model. Front. Physiol. 2018, 9, 818. [Google Scholar] [CrossRef] [PubMed]
- López, M.; Nogueiras, R.; Tena-Sempere, M.; Diéguez, C. Hypothalamic AMPK: A canonical regulator of whole-body energy balance. Nat. Rev. Endocrinol. 2016, 12, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Dagon, Y.; Avraham, Y.; Magen, I.; Gertler, A.; Ben-Hur, T.; Berry, E.M. Nutritional status, cognition, and survival: A new role for leptin and AMP kinase. J. Biol. Chem. 2005, 280, 42142–42148. [Google Scholar] [CrossRef]
- Dasgupta, B.; Milbrandt, J. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev. Cell 2009, 16, 256–270. [Google Scholar] [CrossRef]
- Spasic, M.R.; Callaerts, P.; Norga, K.K. Drosophila alicorn is a neuronal maintenance factor protecting against activity-induced retinal degeneration. J. Neurosci. 2008, 28, 6419–6429. [Google Scholar] [CrossRef]
- Viollet, B.; Athea, Y.; Mounier, R.; Guigas, B.; Zarrinpashneh, E.; Horman, S.; Lantier, L.; Hebrard, S.; Devin-Leclerc, J.; Beauloye, C.; et al. AMPK: Lessons from transgenic and knockout animals. Front. Biosci. 2009, 14, 19–44. [Google Scholar] [CrossRef]
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.-B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferré, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef]
- Huynh, M.K.Q.; Kinyua, A.W.; Yang, D.J.; Kim, K.W. Hypothalamic AMPK as a Regulator of Energy Homeostasis. Neural Plast. 2016, 2016, 2754078. [Google Scholar] [CrossRef] [PubMed]
- Tamashiro, K.L.; Moran, T.H. Perinatal environment and its influences on metabolic programming of offspring. Physiol. Behav. 2010, 100, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Fukami, T.; Sun, X.; Li, T.; Desai, M.; Ross, M.G. Mechanism of programmed obesity in intrauterine fetal growth restricted offspring: Paradoxically enhanced appetite stimulation in fed and fasting states. Reprod. Sci. 2012, 19, 423–430. [Google Scholar] [CrossRef]
- Franco, J.; Lisboa, P.; Lima, N.S.; Peixoto-Silva, N.; Maia, L.; Oliveira, E.; Passos, M.; De Moura, E.G. Resveratrol prevents hyperleptinemia and central leptin resistance in adult rats programmed by early weaning. Horm. Metab. Res. 2014, 46, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; McCubbin, J.A.; Feinglos, M.N. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Seldin, M.F.; Kuhn, C.M.; Cochrane, C.; Feinglos, M.N. Control of expression of insulin resistance and hyperglycemia by different genetic factors in diabetic C57BL/6J mice. Diabetes 1991, 40, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.-M.; Matthews, P.A.; Argenton, M.; Christie, M.R.; McConnell, J.M.; Jansen, E.H.J.M.; Piersma, A.H.; Ozanne, S.E.; Twinn, D.F.; Remacle, C.; et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: A novel murine model of developmental programming. Hypertension 2008, 51, 383–392. [Google Scholar] [CrossRef]
- Kulhanek, D.; Weigel, R.; Paulsen, M.E. Maternal High-Fat-High-Carbohydrate Diet-Induced Obesity Is Associated with Increased Appetite in Peripubertal Male but Not Female C57Bl/6J Mice. Nutrients 2020, 12, 2919. [Google Scholar] [CrossRef]
- Collins, S.; Martin, T.L.; Surwit, R.S.; Robidoux, J. Genetic vulnerability to diet-induced obesity in the C57BL/6J mouse: Physiological and molecular characteristics. Physiol. Behav. 2004, 81, 243–248. [Google Scholar] [CrossRef]
- Gallou-Kabani, C.; Vigé, A.; Gross, M.-S.; Rabès, J.-P.; Boileau, C.; Larue-Achagiotis, C.; Tomé, D.; Jais, J.-P.; Junien, C. C57BL/6J and A/J mice fed a high-fat diet delineate components of metabolic syndrome. Obesity 2007, 15, 1996–2005. [Google Scholar] [CrossRef]
- Li, J.; Wu, H.; Liu, Y.; Yang, L. High fat diet induced obesity model using four strainsof mice: Kunming, C57BL/6, BALB/c and ICR. Exp. Anim. 2020, 69, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Plagemann, A.; Harder, T.; Brunn, M.; Harder, A.; Roepke, K.; Wittrock-Staar, M.; Ziska, T.; Schellong, K.; Rodekamp, E.; Melchior, K.; et al. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: An epigenetic model of obesity and the metabolic syndrome. J. Physiol. 2009, 587 Pt 20, 4963–4976. [Google Scholar] [CrossRef]
- Kuehnen, P.; Mischke, M.; Wiegand, S.; Sers, C.; Horsthemke, B.; Lau, S.; Keil, T.; Lee, Y.-A.; Grueters, A.; Krude, H. An Alu element-associated hypermethylation variant of the POMC gene is associated with childhood obesity. PLoS Genet. 2012, 8, e1002543. [Google Scholar] [CrossRef] [PubMed]
- Gugusheff, J.R.; Ong, Z.Y.; Muhlhausler, B.S. The early origins of food preferences: Targeting the critical windows of development. FASEB J. 2015, 29, 365–373. [Google Scholar] [CrossRef]
- Moraes, J.C.; Coope, A.; Morari, J.; Cintra, D.E.; Roman, E.A.; Pauli, J.R.; Romanatto, T.; Carvalheira, J.B.; Oliveira, A.L.R.; Saad, M.J.; et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS ONE 2009, 4, e5045. [Google Scholar] [CrossRef]
- McNay, D.E.; Briançon, N.; Kokoeva, M.V.; Maratos-Flier, E.; Flier, J.S. Remodeling of the arcuate nucleus energy-balance circuit is inhibited in obese mice. J. Clin. Investig. 2012, 122, 142–152. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | HF | |
|---|---|---|
| Body weight (g) at 3 weeks of age | 10.2 ± 0.3 | 10.3 ± 0.2 |
| Body weight (g) at 11 weeks | 19.1 ± 1.1 | 25.0 ± 1.5 * |
| Body weight (g) at term | 34.8 ± 0.5 | 36.8 ± 0.4 * |
| Body weight (g) at end of lactation | 25.5 ± 0.3 | 27.9 ± 0.4 * |
| Litter Size | 7.5 ± 0.5 | 6.9 ± 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desai, M.; Ferrini, M.G.; Han, G.; Narwani, K.; Ross, M.G. Maternal High Fat Diet Programs Male Mice Offspring Hyperphagia and Obesity: Mechanism of Increased Appetite Neurons via Altered Neurogenic Factors and Nutrient Sensor AMPK. Nutrients 2020, 12, 3326. https://doi.org/10.3390/nu12113326
Desai M, Ferrini MG, Han G, Narwani K, Ross MG. Maternal High Fat Diet Programs Male Mice Offspring Hyperphagia and Obesity: Mechanism of Increased Appetite Neurons via Altered Neurogenic Factors and Nutrient Sensor AMPK. Nutrients. 2020; 12(11):3326. https://doi.org/10.3390/nu12113326
Chicago/Turabian StyleDesai, Mina, Monica G. Ferrini, Guang Han, Kavita Narwani, and Michael G. Ross. 2020. "Maternal High Fat Diet Programs Male Mice Offspring Hyperphagia and Obesity: Mechanism of Increased Appetite Neurons via Altered Neurogenic Factors and Nutrient Sensor AMPK" Nutrients 12, no. 11: 3326. https://doi.org/10.3390/nu12113326
APA StyleDesai, M., Ferrini, M. G., Han, G., Narwani, K., & Ross, M. G. (2020). Maternal High Fat Diet Programs Male Mice Offspring Hyperphagia and Obesity: Mechanism of Increased Appetite Neurons via Altered Neurogenic Factors and Nutrient Sensor AMPK. Nutrients, 12(11), 3326. https://doi.org/10.3390/nu12113326