Gut Microbiota in Hypertension and Atherosclerosis: A Review




Abstract
1. Introduction
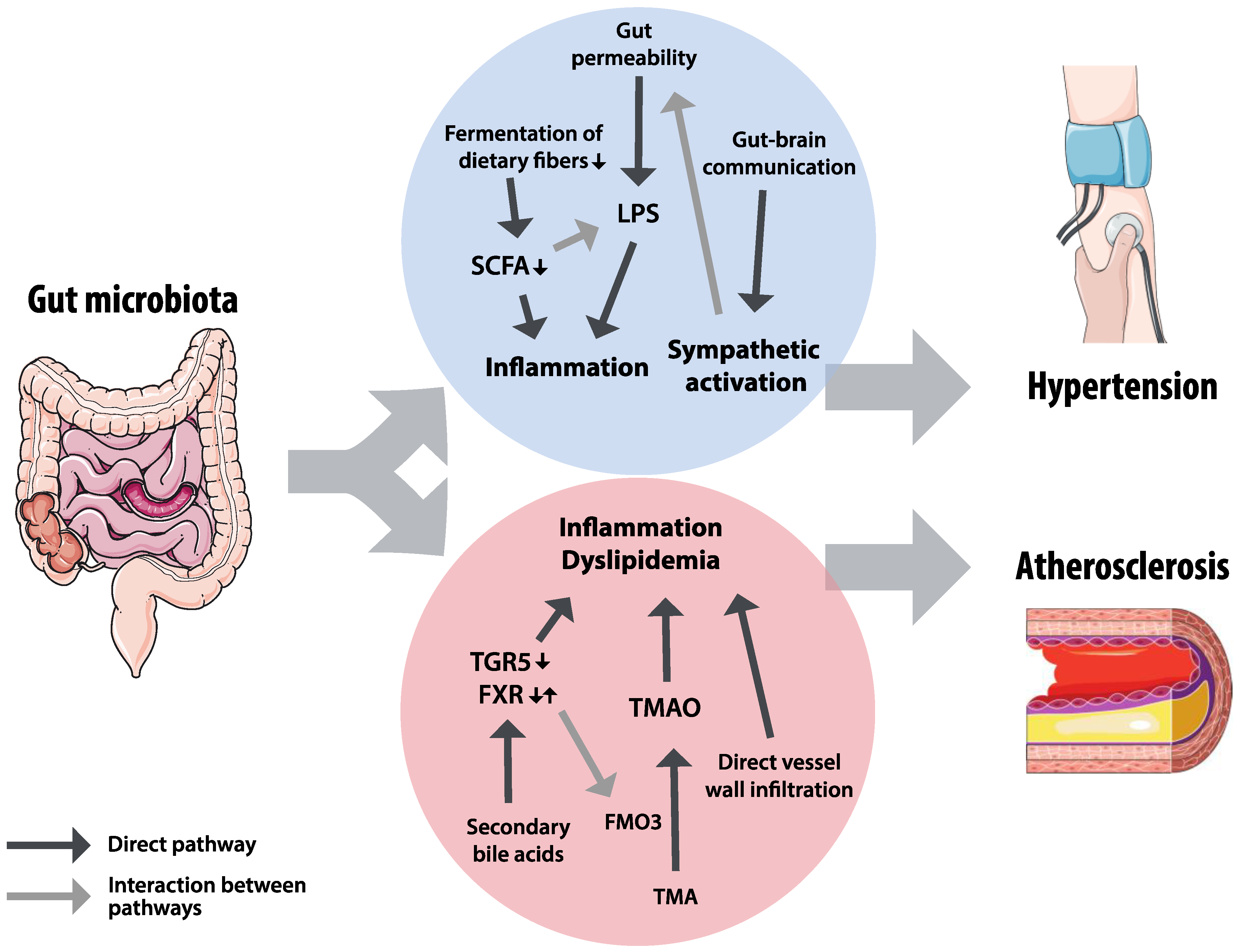
2. Gut Microbiota in Hypertension
2.1. Gut Microbiota Composition in Hypertension
2.2. Short Chain Fatty Acids
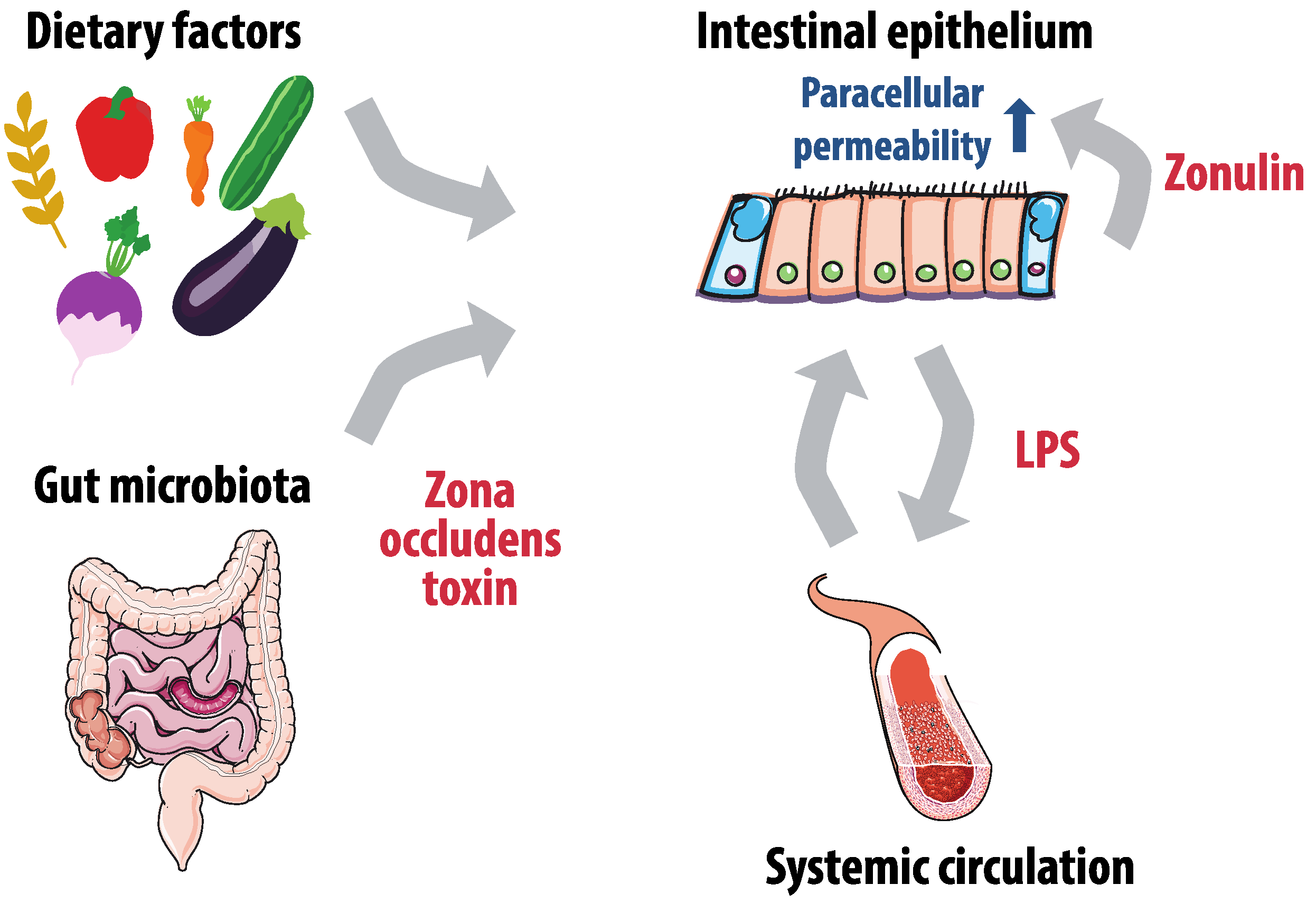
2.3. Gut Permeability and Lipopolysaccharides
2.4. Gut-Brain Interactions and Sympathetic Activation
3. Gut Microbiota in Atherosclerosis
3.1. Atherosclerosis and Gut Microbiota
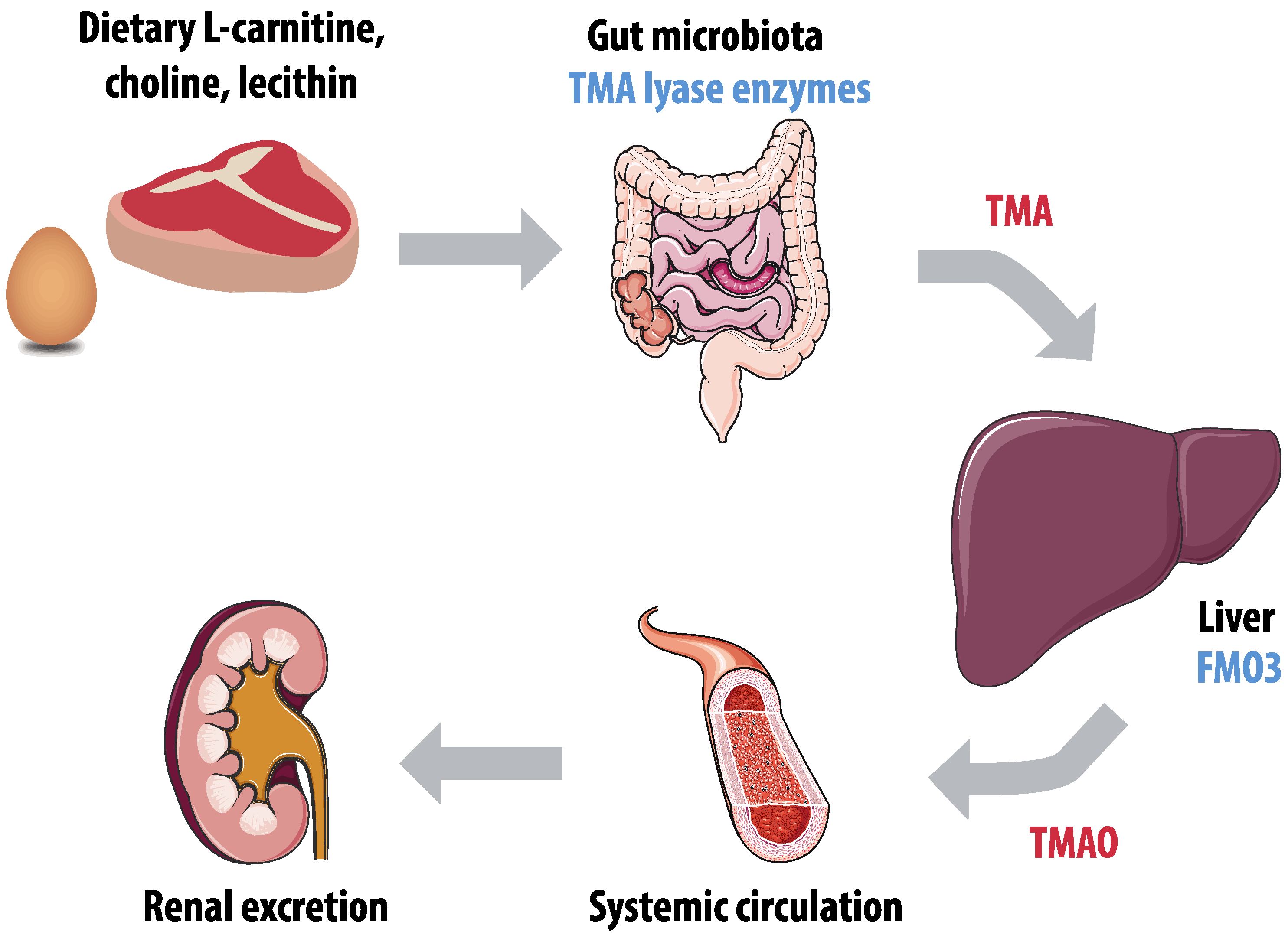
3.2. Trimethylamine-N-Oxide
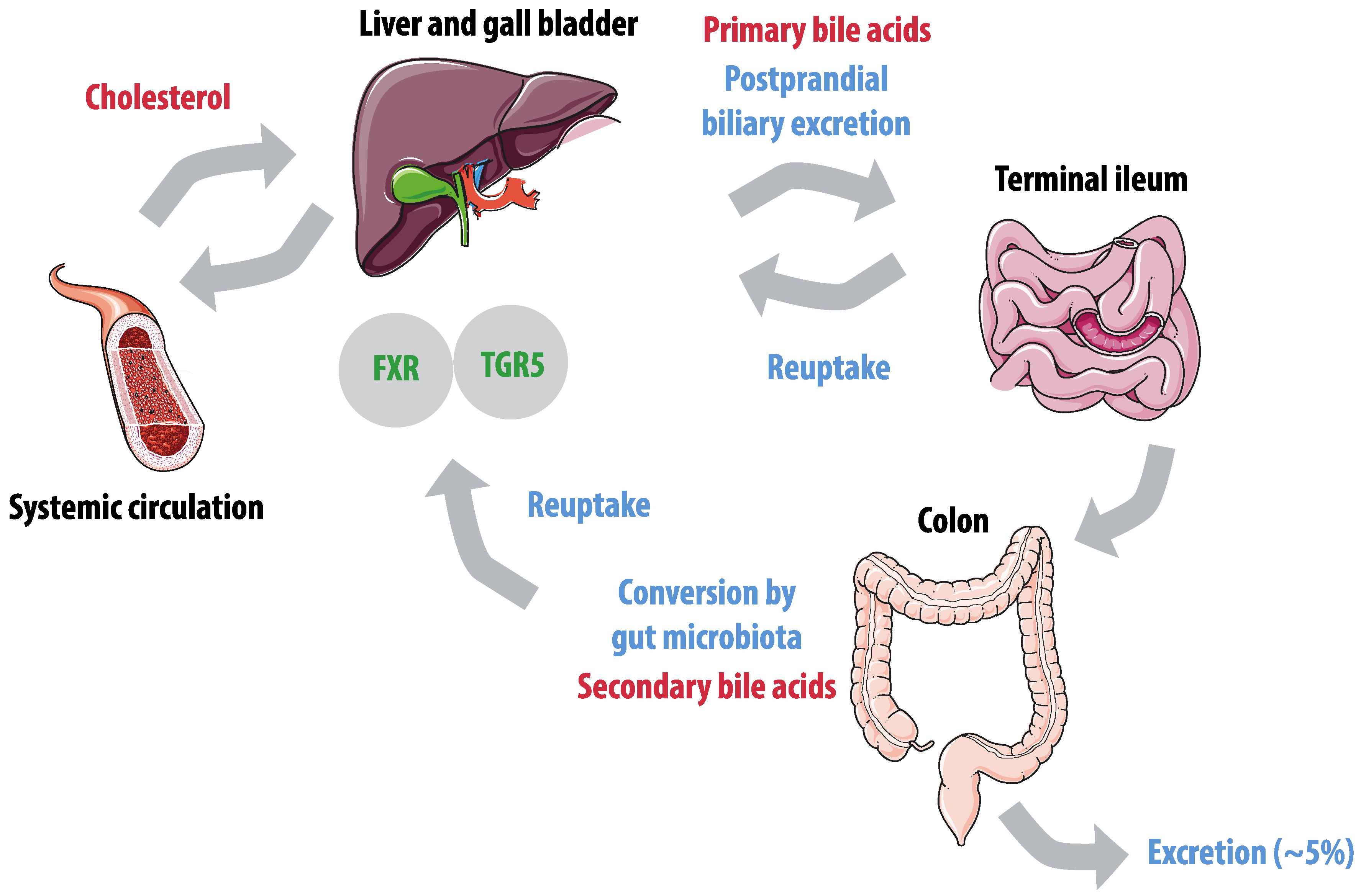
3.3. Bile Acids
4. Therapeutic Strategies
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Global action plan for the prevention and control of noncommunicable diseases 2013–2020. In Follow-up to the Political Declaration of the High-level Meeting of the General Assembly on the Prevention and Control of Non-communicable Diseases; World Health Organization: Geneva, Switzerland, 2013; pp. 68–69. ISBN 978-92-4-1506236. Available online: https://www.who.int/nmh/events/ncd_action_plan/en/ (accessed on 28 September 2020).
- World Health Organization Cardiovascular Diseases (CVDs) Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 28 September 2020).
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and Evolutionary Forces Shaping Microbial Diversity in the Human Intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.-C.; Khelaifia, S.; Alou, M.T.; Ndongo, S.; Dione, N.; Hugon, P.; Caputo, A.; Cadoret, F.; Traore, S.I.; Seck, E.H.; et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol. 2016, 1, 16203. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.P.; Gratz, S.; Sheridan, P.O.; Flint, H.J.; Duncan, S.H. The influence of diet on the gut microbiota. Pharmacol. Res. 2013, 69, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory Bowel Disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef]
- Wang, Q.; Li, F.; Liang, B.; Liang, Y.; Chen, S.; Mo, X.; Ju, Y.; Zhao, H.; Jia, H.; Spector, T.D.; et al. A metagenome-wide association study of gut microbiota in asthma in UK adults. BMC Microbiol. 2018, 18, 114. [Google Scholar] [CrossRef]
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C.; et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632. [Google Scholar] [CrossRef]
- Tang, W.W.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef]
- Li, Z.; Yi, C.-X.; Katiraei, S.; Kooijman, S.; Zhou, E.; Chung, C.K.; Gao, Y.; Heuvel, J.K.V.D.; Meijer, O.C.; Berbée, J.F.P.; et al. Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut 2017, 67, 1269–1279. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-Generated Metabolites Promote Metabolic Benefits via Gut-Brain Neural Circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. Inflammation, atherosclerosis, and cardiovascular risk: An epidemiologic view. Blood Coagul. Fibrinolysis 1999, 10 (Suppl. 1), S9–S12. [Google Scholar]
- Sesso, H.D.; Buring, J.; Rifai, N.; Blake, G.J.; Gaziano, J.M.; Ridker, P.M. C-Reactive Protein and the Risk of Developing Hypertension. J. Am. Med. Assoc. 2003, 290, 2945–2951. [Google Scholar] [CrossRef] [PubMed]
- Vlachopoulos, C.; Ioakeimidis, N.; Aznaouridis, K.; Bratsas, A.; Baou, K.; Xaplanteris, P.; Lazaros, G.; Stefanadis, C. Association of Interleukin-18 Levels With Global Arterial Function and Early Structural Changes in Men Without Cardiovascular Disease. Am. J. Hypertens. 2010, 23, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, A.; Feely, J. Arterial Stiffness Is Related to Systemic Inflammation in Essential Hypertension. Hypertension 2005, 46, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Osto, M.; Geurts, L.; Everard, A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 2012, 3, 279–288. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Afshin, A.; Gakidou, E.; Lim, S.S.; Abate, D.; Abate, K.H.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1923–1994. [Google Scholar] [CrossRef]
- Giri, A.; Hellwege, J.N.; Keaton, J.M.; Park, J.; Qiu, C.; Warren, H.R.; Torstenson, E.S.; Kovesdy, C.P.; Sun, Y.V.; Wilson, O.D.; et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat. Genet. 2018, 51, 51–62. [Google Scholar] [CrossRef]
- Pazoki, R.; Dehghan, A.; Evangelou, E.; Warren, H.; Gao, H.; Caulfield, M.; Elliott, P.; Tzoulaki, I. Genetic predisposition to high blood pressure and lifestyle factors: Associations with midlife blood pressure levels and cardiovascular events. Circulation 2018, 137, 653–661. [Google Scholar] [CrossRef]
- Appel, L.J.; Moore, T.J.; Obarzanek, E.; Vollmer, W.M.; Svetkey, L.P.; Sacks, F.M.; Bray, G.A.; Vogt, T.M.; Cutler, J.A.; Windhauser, M.M.; et al. A Clinical Trial of the Effects of Dietary Patterns on Blood Pressure. N. Engl. J. Med. 1997, 336, 1117–1124. [Google Scholar] [CrossRef]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gomez-Gracia, E.; Ruiz-Gutierrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet. N. Engl. J. Med. 2013, 368, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016, 65, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Mell, B.; Jala, V.R.; Mathew, A.V.; Byun, J.; Waghulde, H.; Zhang, Y.; Haribabu, B.; Vijay-Kumar, M.; Pennathur, S.; Joe, B. Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiol. Genom. 2015, 47, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Adnan, S.; Nelson, J.W.; Ajami, N.J.; Venna, V.R.; Petrosino, J.F.; Bryan, R.M.; Durgan, D.J. Alterations in the gut microbiota can elicit hypertension in rats. Physiol. Genom. 2017, 49, 96–104. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertensioin 2015, 65, 1331–1340. [Google Scholar] [CrossRef]
- Marques, F.Z.; Nelson, E.; Chu, P.-Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef]
- Dan, X.; Mushi, Z.; Baili, W.; Han, L.; Enqi, W.; Huanhu, Z.; Shuchun, L. Differential Analysis of Hypertension-Associated Intestinal Microbiota. Int. J. Med. Sci. 2019, 16, 872–881. [Google Scholar] [CrossRef]
- De La Cuesta-Zuluaga, J.; Mueller, N.T.; Alvarez, R.Q.; Velásquez-Mejía, E.P.; Sierra, J.A.; Corrales-Agudelo, V.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Higher Fecal Short-Chain Fatty Acid Levels Are Associated with Gut Microbiome Dysbiosis, Obesity, Hypertension and Cardiometabolic Disease Risk Factors. Nutrients 2018, 11, 51. [Google Scholar] [CrossRef]
- Huart, J.; Leenders, J.; Taminiau, B.; Descy, J.; Saint-Remy, A.; Daube, G.; Krzesinski, J.-M.; Melin, P.; De Tullio, P.; Jouret, F. Gut Microbiota and Fecal Levels of Short-Chain Fatty Acids Differ Upon 24-Hour Blood Pressure Levels in Men. Hypertension 2019, 74, 1005–1013. [Google Scholar] [CrossRef]
- Jackson, M.A.; Verdi, S.; Maxan, M.-E.; Shin, C.M.; Zierer, J.; Bowyer, R.C.E.; Martin, T.; Williams, F.M.; Menni, C.; Bell, J.T.; et al. Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nat. Commun. 2018, 9, 2655. [Google Scholar] [CrossRef]
- Kim, S.; Goel, R.; Kumar, A.; Qi, Y.; Lobaton, G.; Hosaka, K.; Mohammed, M.; Handberg, E.; Richards, E.M.; Pepine, C.J.; et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. 2018, 132, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Lulla, A.; Sioda, M.; Winglee, K.; Wu, M.C.; Jr, D.R.J.; Shikany, J.M.; Lloyd-Jones, D.M.; Launer, L.J.; Fodor, A.A.; et al. Gut microbiota composition and blood pressure: The CARDIA study. Hypertension 2019, 73, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Verhaar, B.J.H.; Collard, D.; Prodan, A.; Levels, J.H.M.; Zwinderman, A.H.; Bäckhed, F.; Vogt, L.; Peters, M.J.L.; Muller, M.; Nieuwdorp, M.; et al. Associations between gut microbiota, faecal short-chain fatty acids, and blood pressure across ethnic groups: The HELIUS study. Eur. Heart J. 2020, ehaa704. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Gu, Y.; Li, X.; Yang, W.; Jia, L.; Chen, C.; Han, X.; Huang, Y.; Zhao, L.; Li, P.; et al. Alterations of the Gut Microbiome in Hypertension. Front. Cell. Infect. Microbiol. 2017, 7, 381. [Google Scholar] [CrossRef]
- Bier, A.; Braun, T.; Khasbab, R.; Di Segni, A.; Grossman, E.; Haberman, Y.; Leibowitz, A. A High Salt Diet Modulates the Gut Microbiota and Short Chain Fatty Acids Production in a Salt-Sensitive Hypertension Rat Model. Nutrients 2018, 10, 1154. [Google Scholar] [CrossRef]
- Miranda, P.; Serkis, V.; De Palma, G.; Pigrau, M.; Lu, J.; Collins, S.; Bercík, P. High salt diet increases susceptibility to experimental colitis: A putative role of gut microbiota. Gastroenterology 2016, 150, S583. [Google Scholar] [CrossRef]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mähler, A.; Balogh, A.; Markó, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef]
- Tanida, M.; Yamano, T.; Maeda, K.; Okumura, N.; Fukushima, Y.; Nagai, K. Effects of intraduodenal injection of Lactobacillus johnsonii La1 on renal sympathetic nerve activity and blood pressure in urethane-anesthetized rats. Neurosci. Lett. 2005, 389, 109–114. [Google Scholar] [CrossRef]
- Gomez-Guzman, M.; Toral, M.; Romero, M.; Jiménez, R.; Galindo, P.; Sanchez, M.; Zarzuelo, M.J.; Olivares, M.; Gálvez, J.; Duarte, J. Antihypertensive effects of probioticsLactobacillusstrains in spontaneously hypertensive rats. Mol. Nutr. Food Res. 2015, 59, 2326–2336. [Google Scholar] [CrossRef]
- Khalesi, S.; Sun, J.; Buys, N.; Jayasinghe, R. Effect of probiotics on blood pressure: A systematic review and meta-analysis of randomized, controlled trials. Hypertension 2014, 64, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Hashimoto, H.; Hosoda, M.; Morita, H.; Hosono, A. Effect of Administration of Fermented Milk Containing Whey Protein Concentrate to Rats and Healthy Men on Serum Lipids and Blood Pressure. J. Dairy Sci. 2000, 83, 255–263. [Google Scholar] [CrossRef]
- Hata, Y.; Yamamoto, M.; Ohni, M.; Nakajima, K.; Nakamura, Y.; Takano, T. A placebo-controlled study of the effect of sour milk on blood pressure in hypertensive subjects. Am. J. Clin. Nutr. 1996, 64, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef]
- Calderón-Pérez, L.; Gosalbes, M.J.; Yuste, S.; Valls, R.M.; Pedret, A.; Llauradó, E.; Jimenez-Hernandez, N.; Artacho, A.; Pla-Pagà, L.; Companys, J.; et al. Gut metagenomic and short chain fatty acids signature in hypertension: A cross-sectional study. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Cuervo, A.; Salazar, N.; Ruas-Madiedo, P.; Gueimonde, M.; González, S. Fiber from a regular diet is directly associated with fecal short-chain fatty acid concentrations in the elderly. Nutr. Res. 2013, 33, 811–816. [Google Scholar] [CrossRef]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Boets, E.; Gomand, S.V.; Deroover, L.; Preston, T.; Vermeulen, K.; De Preter, V.; Hamer, H.M.; Mooter, G.V.D.; De Vuyst, L.; Courtin, C.M.; et al. Systemic availability and metabolism of colonic-derived short-chain fatty acids in healthy subjects: A stable isotope study. J. Physiol. 2017, 595, 541–555. [Google Scholar] [CrossRef]
- Roediger, W.E. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980, 21, 793–798. [Google Scholar] [CrossRef]
- Yang, T.; Magee, K.L.; Colon-Perez, L.M.; Larkin, R.; Liao, Y.-S.; Balazic, E.; Cowart, J.R.; Arocha, R.; Redler, T.; Febo, M.; et al. Impaired butyrate absorption in the proximal colon, low serum butyrate and diminished central effects of butyrate on blood pressure in spontaneously hypertensive rats. Acta Physiol. 2019, 226, e13256. [Google Scholar] [CrossRef]
- Bouter, K.E.; Bakker, G.J.; Levin, E.; Hartstra, A.V.; Kootte, R.S.; Udayappan, S.D.; Katiraei, S.; Bähler, L.; Gilijamse, P.W.; Tremaroli, V.; et al. Differential metabolic effects of oral butyrate treatment in lean versus metabolic syndrome subjects. Clin. Transl. Gastroenterol. 2018, 9, e155-10. [Google Scholar] [CrossRef] [PubMed]
- Roshanravan, N.; Jafarabadi, M.A.; Ghavami, A.; Alipour, S.; Alamdari, N.M.; Barati, M.; Namin, S.A.M.; Mahdavi, R.; Alizadeh, E.; Hedayati, M. Effect of Butyrate and Inulin Supplementation on Glycemic Status, Lipid Profile and Glucagon-Like Peptide 1 Level in Patients with Type 2 Diabetes: A Randomized Double-Blind, Placebo-Controlled Trial. Horm. Metab. Res. 2017, 49, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L. Gut microbiota in renal physiology: Focus on short-chain fatty acids and their receptors. Kidney Int. 2016, 90, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.-Y.; Lannoy, V.; Decobecq, M.-E.; Brézillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional Characterization of Human Receptors for Short Chain Fatty Acids and Their Role in Polymorphonuclear Cell Activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L.; Protzko, R.J.; Gevorgyan, H.; Peterlin, Z.; Sipos, A.; Han, J.; Brunet, I.; Wan, L.-X.; Rey, F.; Wang, T.; et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 4410–4415. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, Q.; Lu, A.; Liu, X.; Zhang, L.; Xu, C.; Liu, X.; Li, H.; Yang, T. Sodium butyrate suppresses angiotensin II-induced hypertension by inhibition of renal (pro)renin receptor and intrarenal renin–angiotensin system. J. Hypertens. 2017, 35, 1899–1908. [Google Scholar] [CrossRef]
- Pluznick, J.L. Microbial Short-Chain Fatty Acids and Blood Pressure Regulation. Curr. Hypertens. Rep. 2017, 19, 25. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Aguilar, E.; Leonel, A.; Teixeira, L.; Silva, A.; Silva, J.; Pelaez, J.; Capettini, L.; Lemos, V.S.; Santos, R.; Alvarez-Leite, J.I. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFκB activation. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 606–613. [Google Scholar] [CrossRef]
- Saemann, M.D.; Böhmig, G.A.; Österreicher, C.H.; Burtscher, H.; Parolini, O.; Diakos, C.; Stockl, J.; Hörl, W.H.; Zlabinger, G.J. Anti-inflammatory effects of sodium butyrate on human monocytes: Potent inhibition of IL-12 and up-regulation of IL-10 production. FASEB J. 2000, 14, 2380–2382. [Google Scholar] [CrossRef]
- Li, M.; Van Esch, B.C.A.M.; Henricks, P.A.J.; Folkerts, G.; Garssen, J. The Anti-inflammatory Effects of Short Chain Fatty Acids on Lipopolysaccharide- or Tumor Necrosis Factor α-Stimulated Endothelial Cells via Activation of GPR41/43 and Inhibition of HDACs. Front. Pharmacol. 2018, 9, 533. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, J.P.; Sriramula, S.; Pariaut, R.; Guggilam, A.; Mariappan, N.; Elks, C.M.; Francis, J. HDAC Inhibition Attenuates Inflammatory, Hypertrophic, and Hypertensive Responses in Spontaneously Hypertensive Rats. Hypertension 2010, 56, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Gogulamudi, V.R.; Periasamy, R.; Raghavaraju, G.; Subramanian, U.; Pandey, K.N. Inhibition of HDAC enhances STAT acetylation, blocks NF-κB, and suppresses the renal inflammation and fibrosis in Npr1 haplotype male mice. Am. J. Physiol. Renal Physiol. 2017, 313, F781–F795. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Kirkup, A.J.; Brunsden, A.M.; Thompson, D.G.; Grundy, D. Vagal afferent responses to fatty acids of different chain length in the rat. Am. J. Physiol. Liver Physiol. 2001, 281, G907–G915. [Google Scholar] [CrossRef]
- Onyszkiewicz, M.; Gawrys-Kopczynska, M.; Konopelski, P.; Aleksandrowicz, M.; Sawicka, A.; Koźniewska, E.; Samborowska, E.; Ufnal, M. Butyric acid, a gut bacteria metabolite, lowers arterial blood pressure via colon-vagus nerve signaling and GPR41/43 receptors. Pflugers Arch. Eur. J. Physiol. 2019, 471, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, M.C.; Bistritz, L.; Meddings, J. Alterations in intestinal permeability. Gut 2006, 55, 1512–1520. [Google Scholar] [CrossRef]
- Wang, W.; Uzzau, S.; Goldblum, S.E.; Fasano, A. Human zonulin, a potential modulator of intestinal tight junctions. J. Cell Sci. 2000, 113, 4435–4440. [Google Scholar]
- Fasano, A.; Not, T.; Wang, W.; Uzzau, S.; Berti, I.; Tommasini, A.; Goldblum, S.E. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet 2000, 355, 1518–1519. [Google Scholar] [CrossRef]
- Toral, M.; Robles-Vera, I.; De La Visitacion, N.; Romero, M.; Yang, T.; Sánchez, M.; Gomez-Guzman, M.; Jiménez, R.; Raizada, M.K.; Duarte, J. Critical Role of the Interaction Gut Microbiota—Sympathetic Nervous System in the Regulation of Blood Pressure. Front. Physiol. 2019, 10, 231. [Google Scholar] [CrossRef]
- Santisteban, M.M.; Qi, Y.F.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ. Res. 2017, 120, 312–323. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide Endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed]
- Aderem, A.; Underhill, D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Tobias, P.S.; Soldau, K.; Gegner, J.A.; Mintz, D.; Ulevitch, R.J. Lipopolysaccharide Binding Protein-mediated Complexation of Lipopolysaccharide with Soluble CD14. J. Biol. Chem. 1995, 270, 10482–10488. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.; Ramos, R.; Tobias, P.; Ulevitch, R.; Mathison, J.; Nishibe, S.; Wahl, M.; Hernandez-Sotomayor, S.; Tonks, N.; Rhee, S.; et al. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef]
- Maa, M.-C.; Chang, M.Y.; Hsieh, M.-Y.; Chen, Y.-J.; Yang, C.-J.; Chen, Z.-C.; Li, Y.K.; Yen, C.-K.; Wu, R.-R.; Leu, T.-H. Butyrate reduced lipopolysaccharide-mediated macrophage migration by suppression of Src enhancement and focal adhesion kinase activity. J. Nutr. Biochem. 2010, 21, 1186–1192. [Google Scholar] [CrossRef]
- Masson, G.S.; Nair, A.R.; Dange, R.B.; Silva-Soares, P.P.; Michelini, L.C.; Francis, J. Toll-Like Receptor 4 Promotes Autonomic Dysfunction, Inflammation and Microglia Activation in the Hypothalamic Paraventricular Nucleus: Role of Endoplasmic Reticulum Stress. PLoS ONE 2015, 10, e0122850. [Google Scholar] [CrossRef]
- Sandiego, C.M.; Gallezot, J.-D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.-F.; Matuskey, D.; Lee, J.-Y.; O’Connor, K.C.; Huang, Y.; et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc. Natl. Acad. Sci. USA 2015, 112, 12468–12473. [Google Scholar] [CrossRef]
- Mancia, G.; Grassi, G. The Autonomic Nervous System and Hypertension. Circ. Res. 2014, 114, 1804–1814. [Google Scholar] [CrossRef]
- Burnstock, G.; Loesch, A. Sympathetic innervation of the kidney in health and disease: Emphasis on the role of purinergic cotransmission. Auton. Neurosci. Basic Clin. 2017, 204, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.P.; Young, C.N.; Fadel, P.J. Central sympathetic overactivity: Maladies and mechanisms. Auton. Neurosci. Basic Clin. 2009, 148, 5–15. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, A.; Liu, M.; Rodríguez, V.; Krause, E.G.; Sumners, C. Role of neurons and glia in the CNS actions of the renin-angiotensin system in cardiovascular control. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R444–R458. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Grobe, J.L.; Desland, F.A.; Zhou, G.; Shen, X.Z.; Shan, Z.; Liu, M.; Raizada, M.K.; Sumners, C. Direct Pro-Inflammatory Effects of Prorenin on Microglia. PLoS ONE 2014, 9, e92937. [Google Scholar] [CrossRef]
- Berthoud, H.R.; Blackshaw, L.A.; Brookes, S.J.H.; Grundy, D. Neuroanatomy of extrinsic afferents supplying the gastrointestinal tract. Neurogastroenterol. Motil. 2004, 16, 28–33. [Google Scholar] [CrossRef]
- Furness, J.B.; Callaghan, B.P.; Rivera, L.R.; Cho, H.J. The enteric nervous system and gastrointestinal innervation: Integrated local and central control. Adv. Exp. Med. Biol. 2014, 817, 39–71. [Google Scholar] [CrossRef]
- Gershon, M.D. The enteric nervous system: A second brain. Hosp. Pract. 1999, 34, 31–52. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef]
- Schäper, J.; Wagner, A.; Enigk, F.; Brell, B.; Mousa, H.A.; Habazettl, H.; Schäfer, M. Regional Sympathetic Blockade Attenuates Activation of Intestinal Macrophages and Reduces Gut Barrier Failure. Anesthesiology 2013, 118, 134–142. [Google Scholar] [CrossRef]
- Santisteban, M.M.; Ahmari, N.; Carvajal, J.M.; Zingler, M.B.; Qi, Y.; Kim, S.; Joseph, J.; Garcia-Pereira, F.; Johnson, R.D.; Shenoy, V.; et al. Involvement of Bone Marrow Cells and Neuroinflammation in Hypertension. Circ. Res. 2015, 117, 178–191. [Google Scholar] [CrossRef]
- Zubcevic, J.; Jun, J.Y.; Kim, S.; Perez, P.D.; Afzal, A.; Shan, Z.; Li, W.; Santisteban, M.M.; Yuan, W.; Febo, M.; et al. Altered inflammatory response is associated with an impaired autonomic input to the bone marrow in the spontaneously hypertensive rat. Hypertension 2014, 63, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-L.; Kim, S.-H. Pulse Wave Velocity in Atherosclerosis. Front. Cardiovasc. Med. 2019, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Van Popele, N.M.; Grobbee, D.E.; Bots, M.L.; Asmar, R.; Topouchian, J.; Reneman, R.S.; Hoeks, A.P.G.; Van Der Kuip, D.A.M.; Hofman, A.; Witteman, J.C.M. Association Between Arterial Stiffness and Atherosclerosis. Stroke 2001, 32, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E. C-Reactive Protein Levels and Outcomes after Statin Therapy. N. Engl. J. Med. 2005, 352, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ott, S.J.; El-Mokhtari, N.E.; Musfeldt, M.; Hellmig, S.; Freitag, S.; Rehman, A.; Kühbacher, T.; Nikolaus, S.; Namsolleck, P.; Blaut, M.; et al. Detection of Diverse Bacterial Signatures in Atherosclerotic Lesions of Patients with Coronary Heart Disease. Circulation 2006, 113, 929–937. [Google Scholar] [CrossRef]
- Koren, O.; Spor, A.; Felin, J.; Fåk, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2010, 108, 4592–4598. [Google Scholar] [CrossRef]
- Lanter, B.B.; Sauer, K.; Davies, D.G. Bacteria Present in Carotid Arterial Plaques Are Found as Biofilm Deposits Which May Contribute to Enhanced Risk of Plaque Rupture. mBio 2014, 5, e01206-14. [Google Scholar] [CrossRef]
- Mitra, S.; Drautz-Moses, D.I.; Alhede, M.; Maw, M.T.; Liu, Y.; Purbojati, R.W.; Hwee, Y.Z.; Kushwaha, K.K.; Gheorghe, A.G.; Bjarnsholt, T.; et al. In silico analyses of metagenomes from human atherosclerotic plaque samples. Microbiome 2015, 3, 38. [Google Scholar] [CrossRef]
- Jonsson, A.L.; Fåk, F.; Akrami, R.; Johansson, E.; Wester, P.; Arnerlöv, C.; Bäckhed, F.; Bergström, G. Bacterial profile in human atherosclerotic plaques. Atherosclerosis 2017, 263, 177–183. [Google Scholar] [CrossRef]
- Epstein, S.E.; Zhu, J.; Burnett, M.S.; Zhou, Y.F.; Vercellotti, G.; Hajjar, D. Infection and atherosclerosis: Potential roles of pathogen burden and molecular mimicry. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1417–1420. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.A.; Rosenfeld, M. Pathogens and atherosclerosis: Update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb. Haemost. 2011, 106, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Grayston, J.T. Antibiotic Treatment of Atherosclerotic Cardiovascular Disease. Circulation 2003, 107, 1228–1230. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Brassard, P.; Brophy, J.M. A meta-analysis of antibiotic use for the secondary prevention of cardiovascular diseases. Can. J. Cardiol. 2008, 24, 391–395. [Google Scholar] [CrossRef]
- Jonsson, A.L.; Bäckhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 79–87. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef]
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef]
- Liu, H.; Chen, X.; Hu, X.; Niu, H.; Tian, R.; Wang, H.; Pang, H.; Jiang, L.; Qiu, B.; Chen, X.; et al. Alterations in the gut microbiome and metabolism with coronary artery disease severity. Microbiome 2019, 7, 68. [Google Scholar] [CrossRef]
- Menni, C.; Lin, C.; Cecelja, M.; Mangino, M.; Hernandez, M.M.; Keehn, L.; Mohney, R.P.; Steves, C.J.; Spector, T.D.; Kuo, C.-F.; et al. Gut microbial diversity is associated with lower arterial stiffness in women. Eur. Hear. J. 2018, 39, 2390–2397. [Google Scholar] [CrossRef]
- Brandsma, E.; Kloosterhuis, N.J.; Koster, M.; Dekker, D.C.; Gijbels, M.J.; Van Der Velden, S.; Ríos-Morales, M.; Van Faassen, M.J.; Loreti, M.G.; De Bruin, A.; et al. A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ. Res. 2019, 124, 94–100. [Google Scholar] [CrossRef]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.; Vallim, T.Q.D.A.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, C.; Moré, M.; Bellamine, A. Trimethylamine N-Oxide in Relation to Cardiometabolic Health—Cause or Effect? Nutrients 2020, 12, 1330. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2014, 116, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zheng, X.; Feng, M.; Li, D.; Zhang, H. Gut Microbiota-Dependent Metabolite Trimethylamine N-Oxide Contributes to Cardiac Dysfunction in Western Diet-Induced Obese Mice. Front. Physiol. 2017, 8, 139. [Google Scholar] [CrossRef]
- Warrier, M.; Shih, D.M.; Burrows, A.C.; Ferguson, D.; Gromovsky, A.D.; Brown, A.L.; Marshall, S.; McDaniel, A.; Schugar, R.C.; Wang, Z.; et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015, 10, 326–338. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Heianza, Y.; Ma, W.; Manson, J.E.; Rexrode, K.M.; Qi, L. Gut Microbiota Metabolites and Risk of Major Adverse Cardiovascular Disease Events and Death: A Systematic Review and Meta-Analysis of Prospective Studies. J. Am. Hear. Assoc. 2017, 6, e004947. [Google Scholar] [CrossRef]
- Kaysen, G.A.; Johansen, K.L.; Chertow, G.M.; Dalrymple, L.S.; Kornak, J.; Grimes, B.; Dwyer, T.; Chassy, A.W.; Fiehn, O. Associations of Trimethylamine N-Oxide With Nutritional and Inflammatory Biomarkers and Cardiovascular Outcomes in Patients New to Dialysis. J. Ren. Nutr. 2015, 25, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Aldana-Hernández, P.; Leonard, K.-A.; Zhao, Y.-Y.; Curtis, J.M.; Field, C.J.; Jacobs, R.L. Dietary Choline or Trimethylamine N-oxide Supplementation Does Not Influence Atherosclerosis Development in Ldlr-/- and Apoe-/- Male Mice. J. Nutr. 2019, 150, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.M.; Allenspach, M.; Othman, A.; Saely, C.H.; Muendlein, A.; Vonbank, A.; Drexel, H.; Von Eckardstein, A. Plasma levels of trimethylamine-N-oxide are confounded by impaired kidney function and poor metabolic control. Atherosclerosis 2015, 243, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.; Wang, Z.; Levison, B.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Lever, M.; George, P.M.; Slow, S.; Bellamy, D.; Young, J.M.; Ho, M.; McEntyre, C.J.; Elmslie, J.L.; Atkinson, W.; Molyneux, S.L.; et al. Betaine and Trimethylamine-N-Oxide as Predictors of Cardiovascular Outcomes Show Different Patterns in Diabetes Mellitus: An Observational Study. PLoS ONE 2014, 9, e114969. [Google Scholar] [CrossRef]
- Qi, J.; You, T.; Li, J.; Pan, T.; Xiang, L.; Han, Y.; Zhu, L. Circulating trimethylamine N-oxide and the risk of cardiovascular diseases: A systematic review and meta-analysis of 11 prospective cohort studies. J. Cell. Mol. Med. 2017, 22, 185–194. [Google Scholar] [CrossRef]
- Smith, G.D.; Ebrahim, S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef]
- Jia, J.; Dou, P.; Gao, M.; Kong, X.; Li, C.; Liu, Z.; Huang, T. Assessment of Causal Direction Between Gut Microbiota–Dependent Metabolites and Cardiometabolic Health: A Bidirectional Mendelian Randomization Analysis. Diabetes 2019, 68, 1747–1755. [Google Scholar] [CrossRef]
- Wang, Z.; Roberts, A.; Buffa, J.A.; Levison, B.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef]
- Vrieze, A.; Out, C.; Fuentes, S.; Jonker, L.; Reuling, I.J.; Kootte, R.S.; Van Nood, E.; Holleman, F.; Knaapen, M.; Romijn, J.A.; et al. Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J. Hepatol. 2014, 60, 824–831. [Google Scholar] [CrossRef]
- Baars, A.; Oosting, A.; Knol, J.; Garssen, J.; Van Bergenhenegouwen, J. The Gut Microbiota as a Therapeutic Target in IBD and Metabolic Disease: A Role for the Bile Acid Receptors FXR and TGR5. Microorganisms 2015, 3, 641–666. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap-bile acids in metabolic control. Nat. Publ. Gr. 2014, 10, 488–498. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Dawson, P. The sodium bile salt cotransport family SLC10. Pflugers Arch. Eur. J. Physiol. 2004, 447, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Eckmann, L. How bile acids confer gut mucosal protection against bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 4333–4334. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Gahan, C.G.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [PubMed]
- Brufau, G.; Groen, A.K.; Kuipers, F. Reverse cholesterol transport revisited: Contribution of biliary versus intestinal cholesterol excretion. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1726–1733. [Google Scholar] [CrossRef]
- Selwyn, F.P.; Csanaky, I.L.; Zhang, Y.; Klaassen, C.D. Importance of Large Intestine in Regulating Bile Acids and Glucagon-Like Peptide-1 in Germ-Free Mice. Drug Metab. Dispos. 2015, 43, 1544–1556. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut Microbiota Regulates Bile Acid Metabolism by Reducing the Levels of Tauro-beta-muricholic Acid, a Naturally Occurring FXR Antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Hodge, R.J.; Nunez, D.J. Therapeutic potential of Takeda-G-protein-receptor-5 (TGR5) agonists. Hope or hype? Diabetes Obes. Metab. 2016, 18, 439–443. [Google Scholar] [CrossRef]
- Yoneno, K.; Hisamatsu, T.; Shimamura, K.; Kamada, N.; Ichikawa, R.; Kitazume, M.T.; Mori, M.; Uo, M.; Namikawa, Y.; Matsuoka, K.; et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology 2013, 139, 19–29. [Google Scholar] [CrossRef]
- Pols, T.W.; Nomura, M.; Harach, T.; Sasso, G.L.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 Activation Inhibits Atherosclerosis by Reducing Macrophage Inflammation and Lipid Loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.J.; Lin, J.; Johnson, L.S.V.; Gould, E.P.; Bowers, G.D.; Nunez, D.J.; on behalf of the SB-756050 Project Team. Safety, Pharmacokinetics, and Pharmacodynamic Effects of a Selective TGR5 Agonist, SB-756050, in Type 2 Diabetes. Clin. Pharmacol. Drug Dev. 2013, 2, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Pols, T.; Eggink, H.; Soeters, M. TGR5 ligands as potential therapeutics in inflammatory diseases. Int. J. Interf. Cytokine Mediat. Res. 2014, 6, 27–38. [Google Scholar] [CrossRef]
- Hanniman, E.A.; Lambert, G.; McCarthy, T.C.; Sinal, C.J. Loss of functional farnesoid X receptor increases atherosclerotic lesions in apolipoprotein E-deficient mice. J. Lipid Res. 2005, 46, 2595–2604. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Vales, C.; Lee, F.Y.; Lee, H.; Lusis, A.J.; Edwards, P.A. FXR Deficiency Causes Reduced Atherosclerosis in Ldlr −/− Mice. Thromb. Vasc. Biol. 2006, 26, 2316–2321. [Google Scholar] [CrossRef]
- Guo, G.L.; Santamarina-Fojo, S.; Akiyama, T.E.; Amar, M.J.; Paigen, B.J.; Brewer, B.; Gonzalez, F.J. Effects of FXR in foam-cell formation and atherosclerosis development. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2006, 1761, 1401–1409. [Google Scholar] [CrossRef]
- Hartman, H.B.; Gardell, S.J.; Petucci, C.J.; Wang, S.; Krueger, J.A.; Evans, M.J. Activation of farnesoid X receptor prevents atherosclerotic lesion formation in LDLR−/− and apoE−/− mice. J. Lipid Res. 2009, 50, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. Antiatherosclerotic effect of farnesoid X receptor. Am. J. Physiol. Hear. Circ. Physiol. 2009, 296, H272–H281. [Google Scholar] [CrossRef]
- Hambruch, E.; Miyazaki-Anzai, S.; Hahn, U.; Matysik, S.; Boettcher, A.; Perović-Ottstadt, S.; Schlüter, T.; Kinzel, O.; Krol, H.D.; Deuschle, U.; et al. Synthetic Farnesoid X Receptor Agonists Induce High-Density Lipoprotein-Mediated Transhepatic Cholesterol Efflux in Mice and Monkeys and Prevent Atherosclerosis in Cholesteryl Ester Transfer Protein Transgenic Low-Density Lipoprotein Receptor (−/−) Mice. J. Pharmacol. Exp. Ther. 2012, 343, 556–567. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Van Natta, M.L.; Connelly, M.A.; Vuppalanchi, R.; Neuschwander-Tetri, B.A.; Tonascia, J.; Guy, C.; Loomba, R.; Dasarathy, S.; Wattacheril, J.; et al. Impact of obeticholic acid on the lipoprotein profile in patients with non-alcoholic steatohepatitis. J. Hepatol. 2019, 72, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki-Anzai, S.; Masuda, M.; Levi, M.; Keenan, A.L.; Miyazaki, M. Dual Activation of the Bile Acid Nuclear Receptor FXR and G-Protein-Coupled Receptor TGR5 Protects Mice against Atherosclerosis. PLoS ONE 2014, 9, e108270. [Google Scholar] [CrossRef] [PubMed]
- Baghdasaryan, A.; Claudel, T.; Gumhold, J.; Silbert, D.; Adorini, L.; Roda, A.; Vecchiotti, S.; Gonzalez, F.J.; Schoonjans, K.; Strazzabosco, M.; et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2 −/− (Abcb4 −/−) mouse cholangiopathy model by promoting biliary HCO 3− output. Hepatology 2011, 54, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, K.; Xu, Y.; Xu, Y.; Li, Y.; Xu, J.; Zhu, Y.; Adorini, L.; Lee, Y.K.; Kasumov, T.; Yin, L.; et al. Reversal of metabolic disorders by pharmacological activation of bile acid receptors TGR5 and FXR. Mol. Metab. 2018, 9, 131–140. [Google Scholar] [CrossRef]
- Iracheta-Vellve, A.; Calenda, C.D.; Petrasek, J.; Ambade, A.; Kodys, K.; Adorini, L.; Szabo, G. FXR and TGR5 Agonists Ameliorate Liver Injury, Steatosis, and Inflammation After Binge or Prolonged Alcohol Feeding in Mice. Hepatol. Commun. 2018, 2, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Wang, N.; Luo, Y.; Myakala, K.; Dobrinskikh, E.; Rosenberg, A.Z.; Levi, J.; Kopp, J.B.; Field, A.; Hill, A.; et al. FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J. Am. Soc. Nephrol. 2017, 29, 118–137. [Google Scholar] [CrossRef]
- Hiippala, K.; Jouhten, H.; Ronkainen, A.; Hartikainen, A.; Kainulainen, V.; Jalanka, J.; Satokari, R. The Potential of Gut Commensals in Reinforcing Intestinal Barrier Function and Alleviating Inflammation. Nutrients 2018, 10, 988. [Google Scholar] [CrossRef]
- Gilijamse, P.W.; Hartstra, A.V.; Levin, E.; Wortelboer, K.; Serlie, M.J.; Ackermans, M.T.; Herrema, H.; Nederveen, A.J.; Imangaliyev, S.; Aalvink, S.; et al. Treatment with Anaerobutyricum soehngenii: A pilot study of safety and dose-response effects on glucose metabolism in human subjects with metabolic syndrome. npj Biofilms Microbiomes 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Quigley, E.M. Prebiotics and Probiotics in Digestive Health. Clin. Gastroenterol. Hepatol. 2019, 17, 333–344. [Google Scholar] [CrossRef]
- McFarland, L.V.; Evans, C.T.; Goldstein, E.J.C. Strain-Specificity and Disease-Specificity of Probiotic Efficacy: A Systematic Review and Meta-Analysis. Front. Med. 2018, 5, 124. [Google Scholar] [CrossRef]
- Dailey, F.E.; Turse, E.P.; Daglilar, E.; Tahan, V. The dirty aspects of fecal microbiota transplantation: A review of its adverse effects and complications. Curr. Opin. Pharmacol. 2019, 49, 29–33. [Google Scholar] [CrossRef]
- Hecht, G.A.; Blaser, M.J.; Gordon, J.; Kaplan, L.M.; Knight, R.; Laine, L.; Peek, R.; Sanders, M.E.; Sartor, B.; Wu, G.D.; et al. What is the value of a food and drug administration investigational new drug application for fecal microbiota transplantation to treat clostridium difficile infection? Clin. Gastroenterol. Hepatol. 2014, 12, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.Y.; Sung, J.; Cheng, F.; Tang, W.; Wong, S.H.; Chan, P.K.; Kamm, M.A.; Sung, J.J.; Kaplan, G.G.; Chan, F.K.; et al. Systematic review with meta-analysis: Review of donor features, procedures and outcomes in 168 clinical studies of faecal microbiota transplantation. Aliment. Pharmacol. Ther. 2019, 49, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Draper, L.A.; Ryan, F.J.; Smith, M.K.; Jalanka, J.; Mattila, E.; Arkkila, P.A.; Ross, R.P.; Satokari, R.; Hill, C. Long-term colonisation with donor bacteriophages following successful faecal microbial transplantation. Microbiome 2018, 6, 220. [Google Scholar] [CrossRef] [PubMed]
- Smits, L.P.; Kootte, R.S.; Levin, E.; Prodan, A.; Fuentes, S.; Zoetendal, E.G.; Wang, Z.; Levison, B.S.; Cleophas, M.C.P.; Kemper, E.M.; et al. Effect of Vegan Fecal Microbiota Transplantation on Carnitine- and Choline-Derived Trimethylamine-N-Oxide Production and Vascular Inflammation in Patients With Metabolic Syndrome. J. Am. Hear. Assoc. 2018, 7, e008342. [Google Scholar] [CrossRef]
- Kootte, R.S.; Levin, E.; Salojärvi, J.; Smits, L.P.; Hartstra, A.V.; Udayappan, S.D.; Hermes, G.; Bouter, K.E.; Koopen, A.M.; Holst, J.J.; et al. Improvement of Insulin Sensitivity after Lean Donor Feces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition. Cell Metab. 2017, 26, 611–619.e6. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga–Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of Intestinal Microbiota From Lean Donors Increases Insulin Sensitivity in Individuals With Metabolic Syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef]
- Holscher, H.D. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes 2017, 8, 172–184. [Google Scholar] [CrossRef]
- Martínez-González, M.A.; Gea, A.; Ruiz-Canela, M. The Mediterranean Diet and Cardiovascular Health: A Critical Review. Circ. Res. 2019, 124, 779–798. [Google Scholar] [CrossRef]
- Siervo, M.; Lara, J.; Chowdhury, S.; Ashor, A.W.; Oggioni, C.; Mathers, J.C. Effects of the Dietary Approach to Stop Hypertension (DASH) diet on cardiovascular risk factors: A systematic review and meta-analysis. Br. J. Nutr. 2014, 113, 1–15. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Population | Hypertension Definition | Sequencing Method | Higher Abundance in HT or Higher BP | Lower Abundance in HT or Higher BP | Alpha Diversity in HT or Higher BP | Covariates in Analyses | Ref. |
|---|---|---|---|---|---|---|---|---|
| Dan et al. 2019 | 67 HT, 62 controls | SBP ≥ 140 or DBP ≥ 90 mmHg | 16S | Acetobacteroides, Alistipes, Bacteroides, Christensenella, Clostridium sensu stricto, Desulfovibrio, Parabacteroides * | Acetobacteroides, Clostridium, Coprobacter, Enterococcus, Enterorhabdus, Lachnospiracea, Lactobacillus, Paraprevotella, Prevotella, Romboutsia, Ruminococcus, Veillonella * | No difference | Unadjusted | [29] |
| De la Cuesta-Zuluaga et al. 2019 | 441 subjects | No hypertension groups | 16S | NR | NR | Lower | Unadjusted | [30] |
| Huart et al. 2019 | 38 HT, 7 pre-HT, 9 controls | Antihypertensive medication use, mean 24 h BP SBP ≥ 130 or DBP ≥ 80 mmHg | 16S | Clostridum sensu stricto | Ruminococcaceae, Clostridiales | NR | Unadjusted | [31] |
| Jackson et al. 2019 | 756 HT, 1790 controls | Self-report or antihypertensive medication use | 16S | Lactobacillaceae, Streptococcaceae | Dehalobacteriaceae, Christensenellaceae, Oxalobacteraceae, Mollicutes, Rikenellaceae, Clostridia, Anaeroplasmataceae, Peptococcaceae | Lower | Age | [32] |
| Kim et al. 2018 | 22 HT, 18 controls | SBP ≥ 140 mmHg | Shotgun | Parabacteroides johnsonii, Eubacterium siraeum, Alistipes finegoldii | Bacteroides thetaiotaomicron | NR | Unadjusted | [33] |
| Li et al. 2017 | 99 HT, 56 pre-HT, 41 controls | SBP ≥ 140 or DBP ≥ 90 mmHg | Shotgun | Prevotella, Klebsiella, Desulfovibrio | Faecalibacterium, Oscillibacter, Roseburia, Bifidobacterium, Coprococcus, Butyrivibrio | Lower | Unadjusted | [34] |
| Sun et al. 2019 | 529 subjects (183 HT) | Antihypertensive medication use or elevated office BP: SBP ≥ 140 or DBP ≥ 90 mmHg | 16S | Anaerovorax, Butyricicoccus, Cellulosibacter, Clostridium IV, Methanobrevibacter, Mogibacterium, Oscillibacter, Oxalobacter, Papillobacter, Sporobacter, Vampirovibrio | Anaeroglobus, Atopobium, Lactobacillus, Megaspheara, Pseudocitrobacter, Rothia, | Lower | Age, ethnicity, sex, study center, sequencing run, education, smoking, physical activity, diet quality score | [35] |
| Verhaar et al. 2020 | 4672 subjects | No hypertension groups | Streptococcus | Roseburia, Clostridium sensu stricto, Roseburia hominis, Romboutsia, Ruminococcaceae, Enterorhabdus | Lower | Age, sex, BMI, smoking status, antihypertensive medication, diabetes | [36] | |
| Yan et al. 2017 | 60 HT, 60 controls | SBP ≥ 140 or DBP ≥ 90 mmHg | Shotgun | Klebsiella, Streptococcus, Parabacteroides | Roseburia, Faecalibacterium prausnitzii | Lower | Not adjusted, but age, sex−, and BMI-matched | [37] |
| Yang et al. 2015 | 7 HT, 10 controls | SBP ≥ 125 mmHg | 16S | NR | NR | Lower | Unadjusted | [27] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verhaar, B.J.H.; Prodan, A.; Nieuwdorp, M.; Muller, M. Gut Microbiota in Hypertension and Atherosclerosis: A Review. Nutrients 2020, 12, 2982. https://doi.org/10.3390/nu12102982
Verhaar BJH, Prodan A, Nieuwdorp M, Muller M. Gut Microbiota in Hypertension and Atherosclerosis: A Review. Nutrients. 2020; 12(10):2982. https://doi.org/10.3390/nu12102982
Chicago/Turabian StyleVerhaar, Barbara J. H., Andrei Prodan, Max Nieuwdorp, and Majon Muller. 2020. "Gut Microbiota in Hypertension and Atherosclerosis: A Review" Nutrients 12, no. 10: 2982. https://doi.org/10.3390/nu12102982
APA StyleVerhaar, B. J. H., Prodan, A., Nieuwdorp, M., & Muller, M. (2020). Gut Microbiota in Hypertension and Atherosclerosis: A Review. Nutrients, 12(10), 2982. https://doi.org/10.3390/nu12102982