Maternal High-Fat–High-Carbohydrate Diet-Induced Obesity Is Associated with Increased Appetite in Peripubertal Male but Not Female C57Bl/6J Mice


Abstract
1. Introduction
2. Materials and Methods
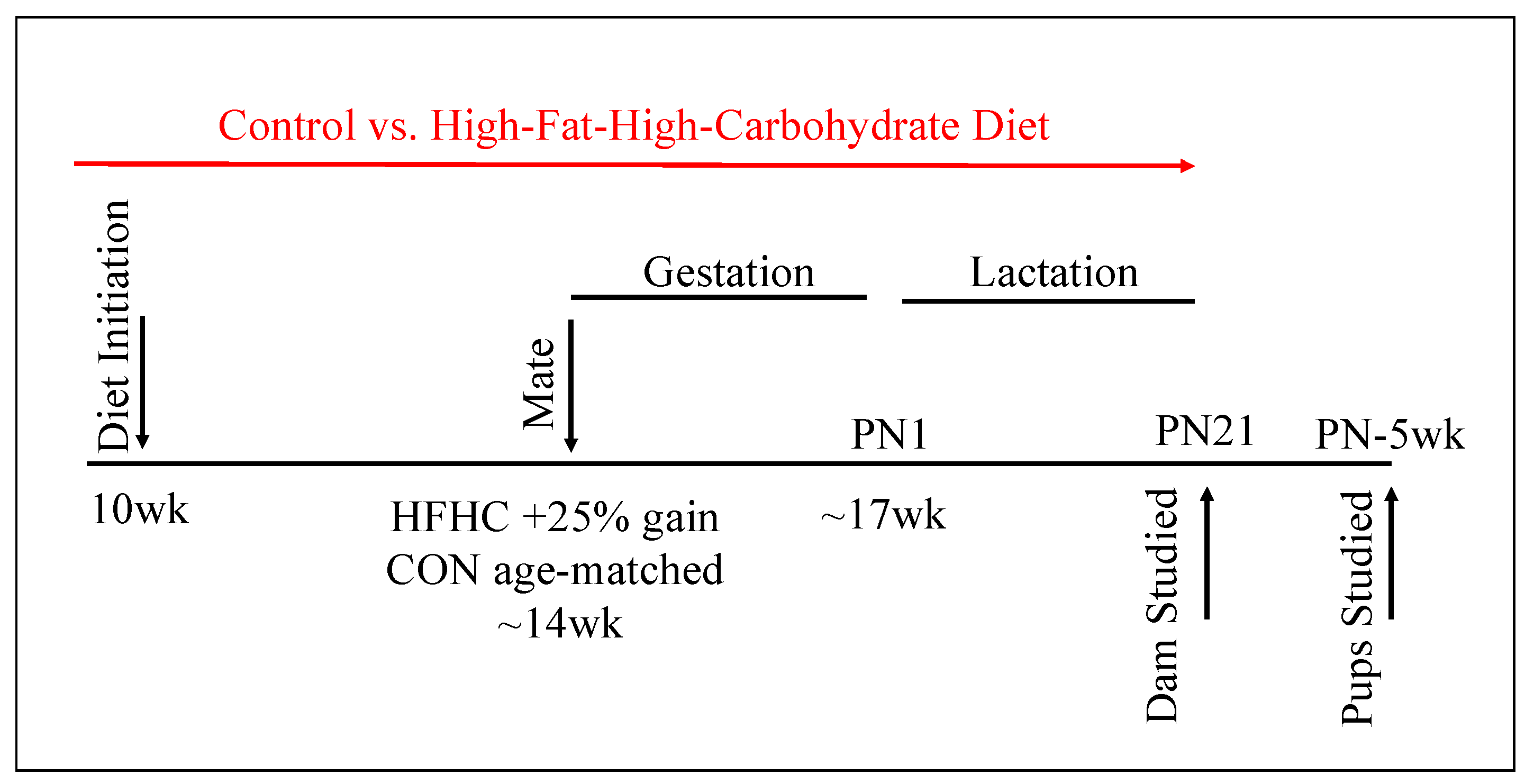
2.1. Animals and Diet
2.2. Collection of Tissue and Blood Samples
2.3. Plasma Biochemical Analyses
2.4. In Vivo Metabolic Analyses
2.5. Gene Expression Analyses
2.6. Data Presentation and Statistical Analyses
3. Results
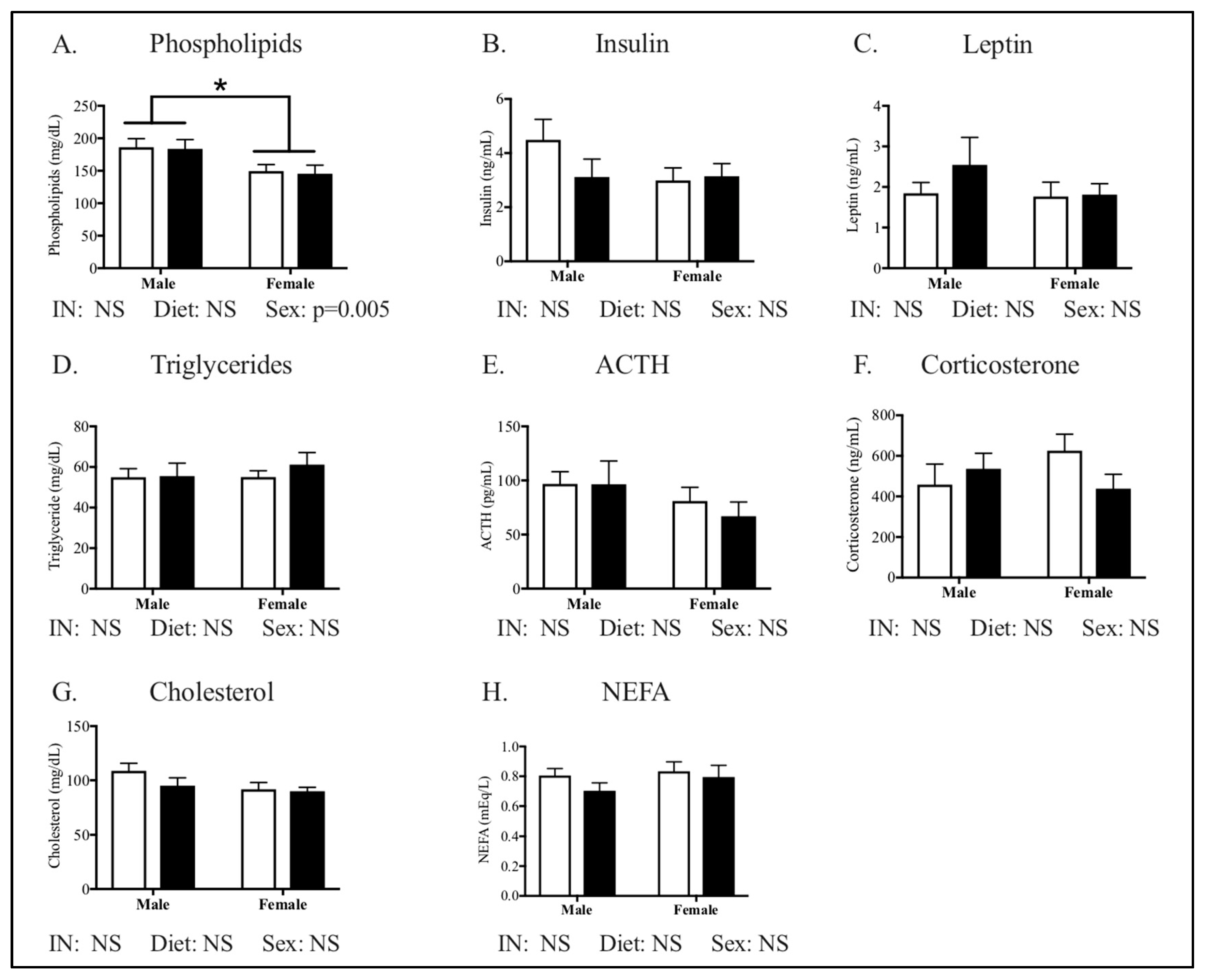
3.1. Maternal Phenotype
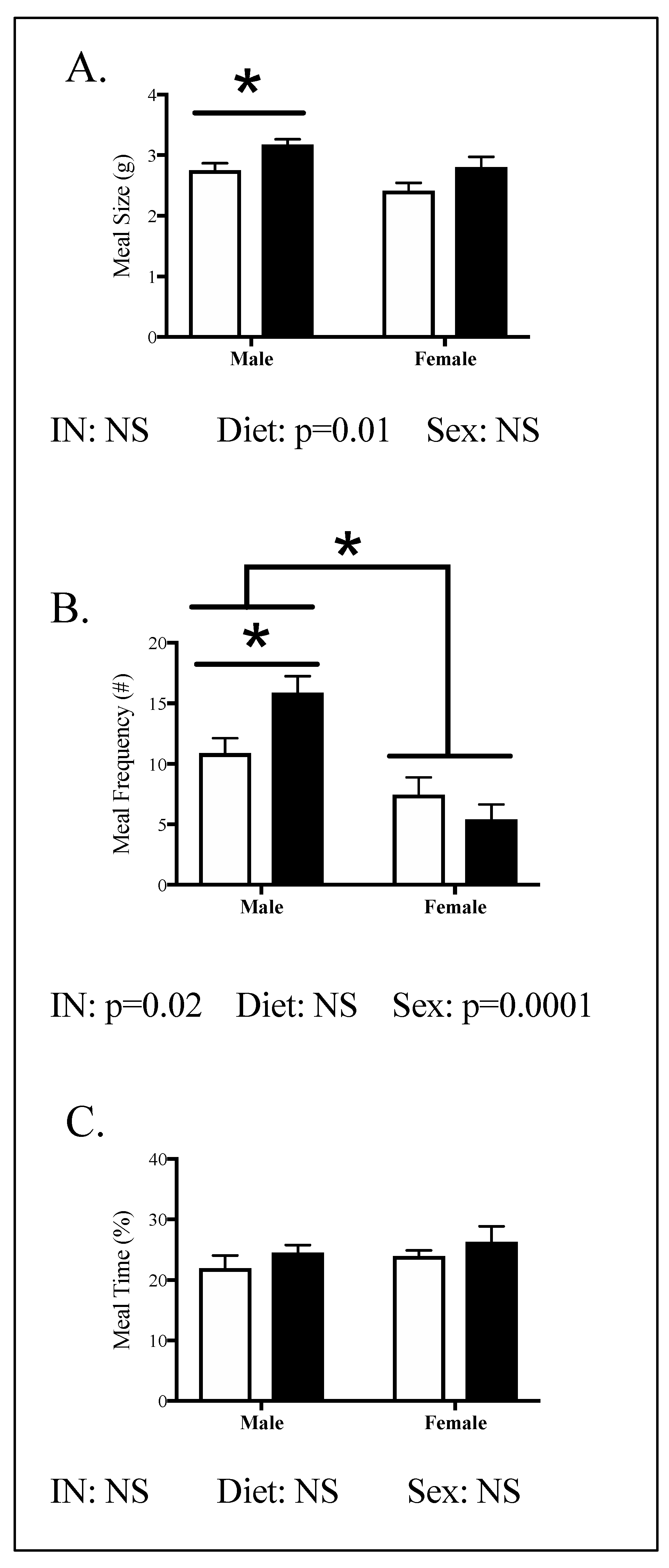
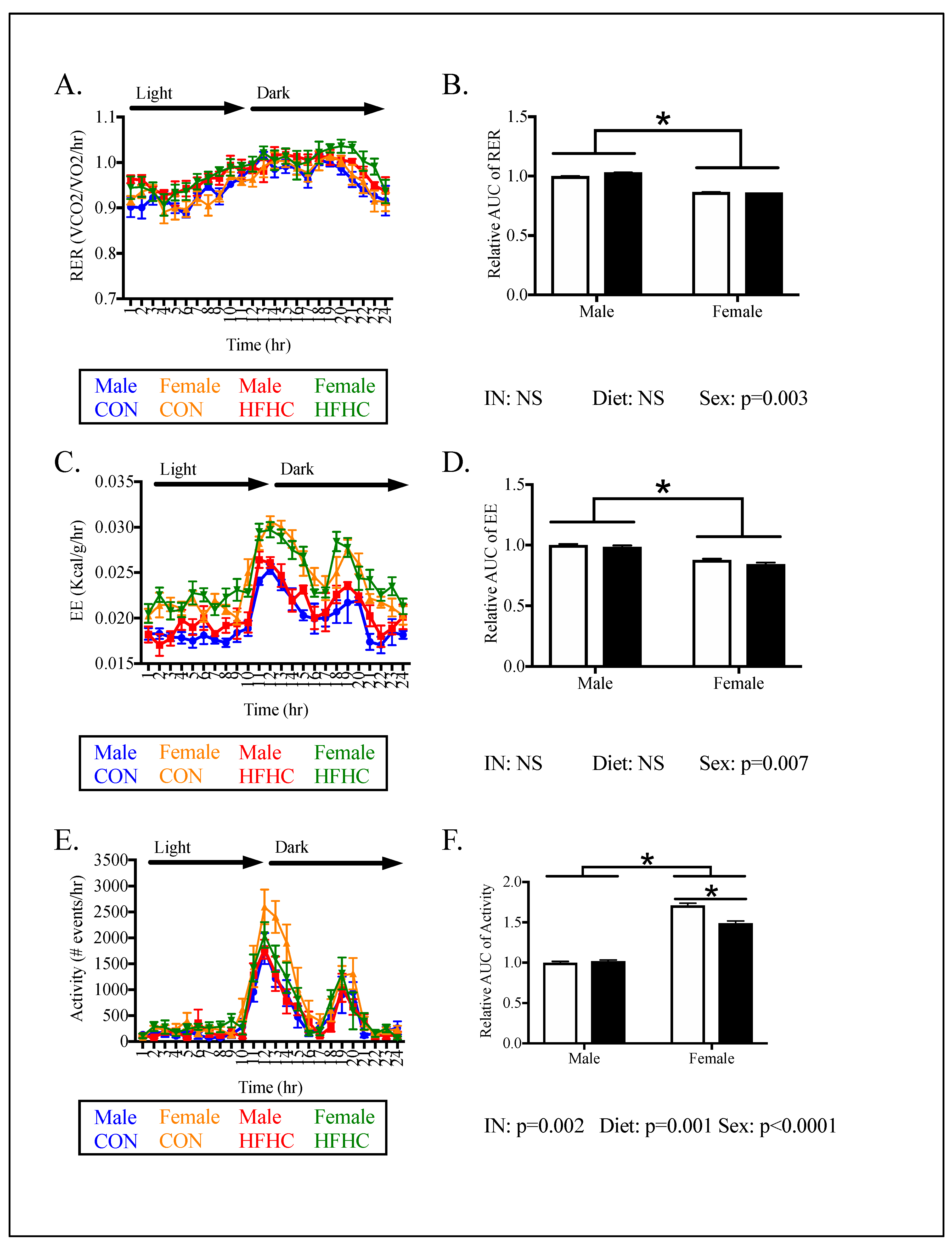
3.2. Offspring Phenotype
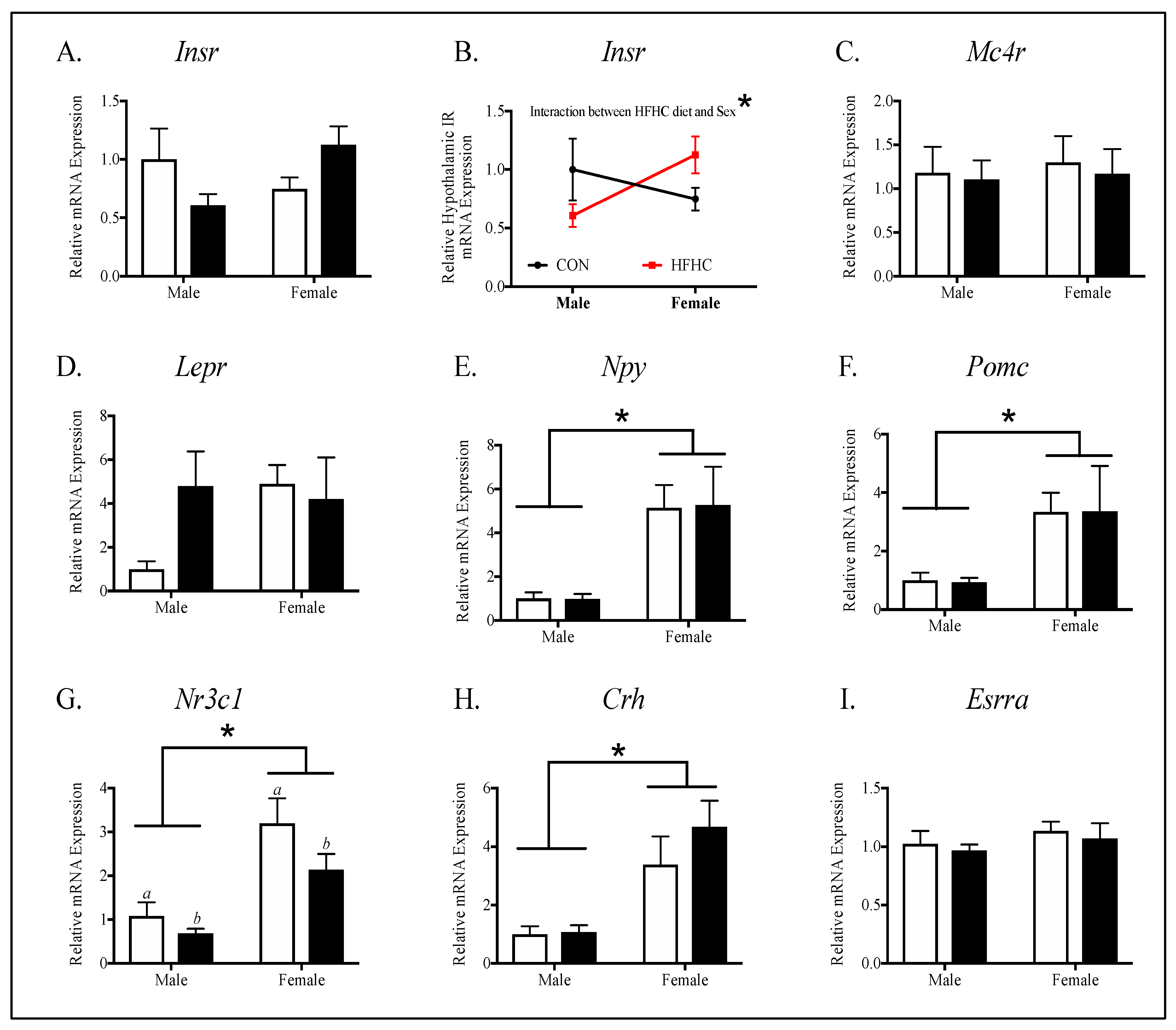
3.3. Offspring Hypothalamic Appetite-Regulating Gene Expression
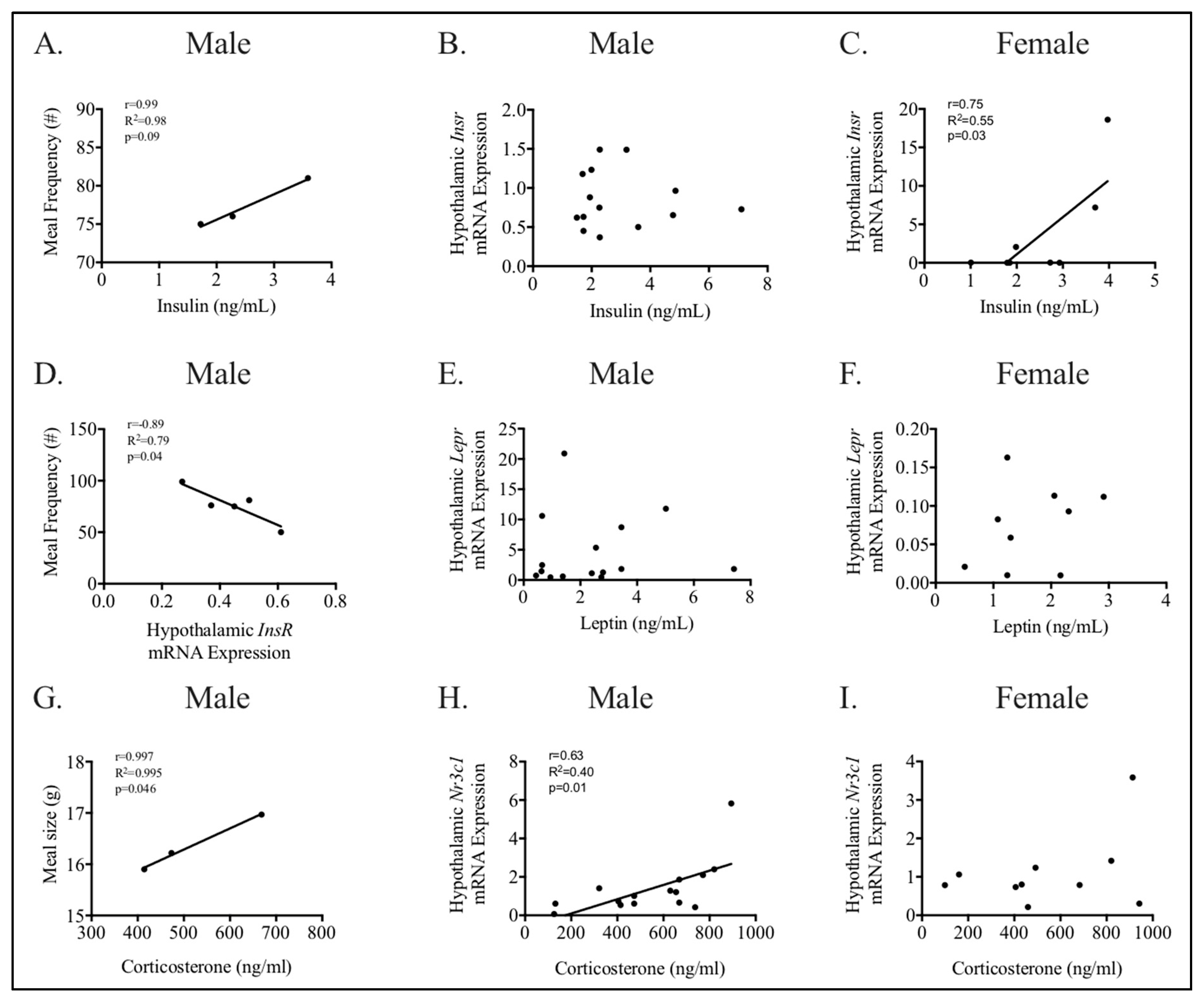
3.4. Relationships between Phenotype and Energy Homeostasis
4. Discussion
4.1. The Effect of Maternal HFHC-Diet-Induced Obesity on Energy Homeostasis
4.2. Sex-Specific Effects of Maternal HFHC-Diet-Induced Obesity on Energy Homeostasis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dearden, L.; Bouret, S.G.; Ozanne, S.E. Sex and gender differences in developmental programming of metabolism. Mol. Metab. 2018, 15, 8–19. [Google Scholar] [CrossRef]
- Alfaradhi, M.Z.; Ozanne, S.E. Developmental programming in response to maternal overnutrition. Front. Genet. 2011, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.G. Nutritional programming of hypothalamic development: Critical periods and windows of opportunity. Int. J. Obes. Suppl. 2012, 2, S19–S24. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.G.; Simerly, R.B. Developmental programming of hypothalamic feeding circuits. Clin. Genet. 2006, 70, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Gali Ramamoorthy, T.; Begum, G.; Harno, E.; White, A. Developmental programming of hypothalamic neuronal circuits: Impact on energy balance control. Front. Neurosci. 2015, 9, 126. [Google Scholar] [CrossRef]
- Penfold, N.C.; Ozanne, S.E. Developmental programming by maternal obesity in 2015, Outcomes, mechanisms, and potential interventions. Horm. Behav. 2015, 76, 143–152. [Google Scholar] [CrossRef]
- Ross, M.G.; Desai, M. Developmental programming of appetite/satiety. Ann. Nutr. Metab. 2014, 64 (Suppl. 1), 36–44. [Google Scholar] [CrossRef]
- Ross, M.G.; Desai, M. Developmental programming of offspring obesity, adipogenesis, and appetite. Clin. Obstet. Gynecol. 2013, 56, 529–536. [Google Scholar] [CrossRef]
- Taylor, P.D.; Poston, L. Developmental programming of obesity in mammals. Exp. Physiol. 2007, 92, 287–298. [Google Scholar] [CrossRef]
- Williams, L.; Seki, Y.; Vuguin, P.M.; Charron, M.J. Animal models of in utero exposure to a high fat diet: A review. Biochim. Biophys. Acta 2014, 1842, 507–519. [Google Scholar] [CrossRef]
- Keleher, M.R.; Zaidi, R.; Shah, S.; Oakley, M.E.; Pavlatos, C.; El Idrissi, S.; Xing, X.; Li, D.; Wang, T.; Cheverud, J.M. Maternal high-fat diet associated with altered gene expression, DNA methylation, and obesity risk in mouse offspring. PLoS ONE 2018, 13, e0192606. [Google Scholar]
- Mischke, M.; Pruis, M.G.; Boekschoten, M.V.; Groen, A.K.; Fitri, A.R.; van de Heijning, B.J.; Verkade, H.J.; Muller, M.; Plosch, T.; Steegenga, W.T. Maternal Western-style high fat diet induces sex-specific physiological and molecular changes in two-week-old mouse offspring. PLoS ONE 2013, 8, e78623. [Google Scholar]
- Khamoui, A.V.; Desai, M.; Ross, M.G.; Rossiter, H.B. Sex-specific effects of maternal and postweaning high-fat diet on skeletal muscle mitochondrial respiration. J. Dev. Orig. Health Dis. 2018, 9, 670–677. [Google Scholar] [PubMed]
- Plagemann, A.; Harder, T.; Janert, U.; Rake, A.; Rittel, F.; Rohde, W.; Dorner, G. Malformations of hypothalamic nuclei in hyperinsulinemic offspring of rats with gestational diabetes. Dev. Neurosci. 1999, 21, 58–67. [Google Scholar] [PubMed]
- Plagemann, A.; Harder, T.; Rake, A.; Janert, U.; Melchior, K.; Rohde, W.; Dorner, G. Morphological alterations of hypothalamic nuclei due to intrahypothalamic hyperinsulinism in newborn rats. Int. J. Dev. Neurosci. 1999, 17, 37–44. [Google Scholar]
- Plagemann, A.; Heidrich, I.; Gotz, F.; Rohde, W.; Dorner, G. Lifelong enhanced diabetes susceptibility and obesity after temporary intrahypothalamic hyperinsulinism during brain organization. Exp. Clin. Endocrinol. 1992, 99, 91–95. [Google Scholar]
- Plagemann, A.; Heidrich, I.; Gotz, F.; Rohde, W.; Dorner, G. Obesity and enhanced diabetes and cardiovascular risk in adult rats due to early postnatal overfeeding. Exp. Clin. Endocrinol. 1992, 99, 154–158. [Google Scholar]
- Sullivan, E.L.; Grove, K.L. Metabolic imprinting in obesity. Forum Nutr. 2010, 63, 186–194. [Google Scholar]
- Muhlhausler, B.S.; Adam, C.L.; Findlay, P.A.; Duffield, J.A.; McMillen, I.C. Increased maternal nutrition alters development of the appetite-regulating network in the brain. FASEB J. 2006, 20, 1257–1259. [Google Scholar]
- Muhlhausler, B.S.; Adam, C.L.; McMillen, I.C. Maternal nutrition and the programming of obesity: The brain. Organogenesis 2008, 4, 144–152. [Google Scholar]
- Reynolds, C.M.; Segovia, S.A.; Vickers, M.H. Experimental Models of Maternal Obesity and Neuroendocrine Programming of Metabolic Disorders in Offspring. Front. Endocrinol. (Lausanne) 2017, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Han, G.; Ross, M.G. Programmed hyperphagia in offspring of obese dams: Altered expression of hypothalamic nutrient sensors, neurogenic factors and epigenetic modulators. Appetite 2016, 99, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Konner, A.C.; Bruning, J.C. CNS-targets in control of energy and glucose homeostasis. Curr. Opin. Pharmacol. 2009, 9, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Konner, A.C.; Klockener, T.; Bruning, J.C. Control of energy homeostasis by insulin and leptin: Targeting the arcuate nucleus and beyond. Physiol. Behav. 2009, 97, 632–638. [Google Scholar] [CrossRef]
- Sanchez-Lasheras, C.; Konner, A.C.; Bruning, J.C. Integrative neurobiology of energy homeostasis-neurocircuits, signals and mediators. Front. Neuroendocrinol. 2010, 31, 4–15. [Google Scholar] [CrossRef]
- Vogt, M.C.; Paeger, L.; Hess, S.; Steculorum, S.M.; Awazawa, M.; Hampel, B.; Neupert, S.; Nicholls, H.T.; Mauer, J.; Hausen, A.C.; et al. Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell 2014, 156, 495–509. [Google Scholar] [CrossRef]
- Rosario, F.J.; Kanai, Y.; Powell, T.L.; Jansson, T. Increased placental nutrient transport in a novel mouse model of maternal obesity with fetal overgrowth. Obesity (Silver Spring) 2015, 23, 1663–1670. [Google Scholar] [CrossRef]
- Bouret, S.G. Development of hypothalamic circuits that control food intake and energy balance. In Appetite and Food Intake: Central Control; RBS H: Boca Raton, FL, USA; CRC Press: Boca Raton, FL, USA; Taylor & Francis: Abingdon, UK, 2017; pp. 1–19. [Google Scholar]
- Kim, D.W.; Glendining, K.A.; Grattan, D.R.; Jasoni, C.L. Maternal Obesity in the Mouse Compromises the Blood-Brain Barrier in the Arcuate Nucleus of Offspring. Endocrinology 2016, 157, 2229–2242. [Google Scholar] [CrossRef]
- Kim, D.W.; Glendining, K.A.; Grattan, D.R.; Jasoni, C.L. Maternal obesity leads to increased proliferation and numbers of astrocytes in the developing fetal and neonatal mouse hypothalamus. Int. J. Dev. Neurosci. 2016, 53, 18–25. [Google Scholar] [CrossRef]
- Chang, G.Q.; Gaysinskaya, V.; Karatayev, O.; Leibowitz, S.F. Maternal high-fat diet and fetal programming: Increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J. Neurosci. 2008, 28, 12107–12119. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Davidson, T.L. Western diet consumption and cognitive impairment: Links to hippocampal dysfunction and obesity. Physiol. Behav. 2011, 103, 59–68. [Google Scholar] [PubMed]
- Moreno-Fernandez, S.; Garces-Rimon, M.; Vera, G.; Astier, J.; Landrier, J.F.; Miguel, M. High Fat/High Glucose Diet Induces Metabolic Syndrome in an Experimental Rat Model. Nutrients 2018, 10, 1502. [Google Scholar]
- Panchal, S.K.; Brown, L. Rodent models for metabolic syndrome research. J. Biomed. Biotechnol. 2011, 2011, 351982. [Google Scholar] [PubMed]
- Panchal, S.K.; Poudyal, H.; Iyer, A.; Nazer, R.; Alam, M.A.; Diwan, V.; Kauter, K.; Sernia, C.; Campbell, F.; Ward, L.; et al. High-carbohydrate, high-fat diet-induced metabolic syndrome and cardiovascular remodeling in rats. J. Cardiovasc. Pharmacol. 2011, 57, 611–624. [Google Scholar]
- Popkin, B.M. Nutrition Transition and the Global Diabetes Epidemic. Curr. Diab. Rep. 2015, 15, 64. [Google Scholar]
- Flegal, K.M.; Kruszon-Moran, D.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Trends in Obesity Among Adults in the United States, 2005 to 2014. JAMA 2016, 315, 2284–2291. [Google Scholar]
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of Obesity and Severe Obesity Among Adults: United States, 2017–2018; NCHS Data Brief; National Center for Health Statistics: Hyattsville, MD, USA, 2020; pp. 1–8. [Google Scholar]
- Hunt, K.J.; Schuller, K.L. The increasing prevalence of diabetes in pregnancy. Obstet. Gynecol. Clin. N. Am. 2007, 34, 173–199, vii. [Google Scholar]
- ACOG Practice Bulletin No 156: Obesity in Pregnancy. Obstet. Gynecol. 2015, 126, e112–e126.
- Bick, D. The challenge of obesity during pregnancy: When to intervene and what could work? Midwifery 2015, 31, 655–656. [Google Scholar]
- Menting, M.D.; Mintjens, S.; van de Beek, C.; Frick, C.J.; Ozanne, S.E.; Limpens, J.; Roseboom, T.J.; Hooijmans, C.R.; van Deutekom, A.W.; Painter, R.C. Maternal obesity in pregnancy impacts offspring cardiometabolic health: Systematic review and meta-analysis of animal studies. Obes. Rev. 2019, 20, 675–685. [Google Scholar]
- Paulsen, M.E.; Rosario, F.J.; Wesolowski, S.R.; Powell, T.L.; Jansson, T. Normalizing adiponectin levels in obese pregnant mice prevents adverse metabolic outcomes in offspring. FASEB J. 2019, 33, 2899–2909. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, O.R.; Rosario, F.J.; Powell, T.L.; Jansson, T. Normalisation of circulating adiponectin levels in obese pregnant mice prevents cardiac dysfunction in adult offspring. Int. J. Obes. 2020, 44, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Aye, I.L.; Rosario, F.J.; Powell, T.L.; Jansson, T. Adiponectin supplementation in pregnant mice prevents the adverse effects of maternal obesity on placental function and fetal growth. Proc. Natl. Acad. Sci. USA 2015, 112, 12858–12863. [Google Scholar] [PubMed]
- Diaz, P.; Harris, J.; Rosario, F.J.; Powell, T.L.; Jansson, T. Increased placental fatty acid transporter 6 and binding protein 3 expression and fetal liver lipid accumulation in a mouse model of obesity in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R1569–R1577. [Google Scholar] [CrossRef] [PubMed]
- Rosario, F.J.; Schumacher, M.A.; Jiang, J.; Kanai, Y.; Powell, T.L.; Jansson, T. Chronic maternal infusion of full-length adiponectin in pregnant mice down-regulates placental amino acid transporter activity and expression and decreases fetal growth. J. Physiol. 2012, 590, 1495–1509. [Google Scholar] [CrossRef]
- Sadagurski, M.; Debarba, L.K.; Werneck-de-Castro, J.P.; Ali Awada, A.; Baker, T.A.; Bernal-Mizrachi, E. Sexual dimorphism in hypothalamic inflammation in the offspring of dams exposed to a diet rich in high fat and branched-chain amino acids. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E526–E534. [Google Scholar]
- Barke, T.L.; Money, K.M.; Du, L.; Serezani, A.; Gannon, M.; Mirnics, K.; Aronoff, D.M. Sex modifies placental gene expression in response to metabolic and inflammatory stress. Placenta 2019, 78, 1–9. [Google Scholar]
- Kanter, R.; Caballero, B. Global gender disparities in obesity: A review. Adv. Nutr. 2012, 3, 491–498. [Google Scholar]
- Mittwoch, U.; Burgess, M.C.; Baker, P.J. Male sexual development in “a sea of oestrogen”. Lancet 1993, 342, 123–124. [Google Scholar] [CrossRef]
- Kurrasch, D.M.; Cheung, C.C.; Lee, F.Y.; Tran, P.V.; Hata, K.; Ingraham, H.A. The neonatal ventromedial hypothalamus transcriptome reveals novel markers with spatially distinct patterning. J. Neurosci. 2007, 27, 13624–13634. [Google Scholar]
- Cero, C.; Razzoli, M.; Han, R.; Sahu, B.S.; Patricelli, J.; Guo, Z.; Zaidman, N.A.; Miles, J.M.; O’Grady, S.M.; Bartolomucci, A. The neuropeptide TLQP-21 opposes obesity via C3aR1-mediated enhancement of adrenergic-induced lipolysis. Mol. Metab. 2017, 6, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Razzoli, M.; Sanghez, V.; Bartolomucci, A. Chronic subordination stress induces hyperphagia and disrupts eating behavior in mice modeling binge-eating-like disorder. Front. Nutr. 2015, 1, 30. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Gavin, T.P.; Kuang, S. Measurement of Resting Energy Metabolism in Mice Using Oxymax Open Circuit Indirect Calorimeter. Bio Protoc. 2015, 5, e1602. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Takeda, K.; Maeda, M.; Ogawa, W.; Sato, T.; Okada, S.; Ohnishi, Y.; Nakajima, H.; Kashiwagi, A. Glucose area under the curve during oral glucose tolerance test as an index of glucose intolerance. Diabetol. Int. 2016, 7, 53–58. [Google Scholar] [CrossRef]
- Tschop, M.H.; Speakman, J.R.; Arch, J.R.; Auwerx, J.; Bruning, J.C.; Chan, L.; Eckel, R.H.; Farese, R.V.; Galgani, J.E., Jr.; Hambly, C.; et al. A guide to analysis of mouse energy metabolism. Nat. Methods 2011, 9, 57–63. [Google Scholar] [CrossRef]
- Maugeri, A.; Kunzova, S.; Medina-Inojosa, J.R.; Agodi, A.; Barchitta, M.; Homolka, M.; Kiacova, N.; Bauerova, H.; Sochor, O.; Lopez-Jimenez, F.; et al. Association between eating time interval and frequency with ideal cardiovascular health: Results from a random sample Czech urban population. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Allison, D.B.; Fontana, L.; Harvie, M.; Longo, V.D.; Malaisse, W.J.; Mosley, M.; Notterpek, L.; Ravussin, E.; Scheer, F.A.; et al. Meal frequency and timing in health and disease. Proc. Natl. Acad. Sci. USA 2014, 111, 16647–16653. [Google Scholar] [CrossRef]
- Paoli, A.; Tinsley, G.; Bianco, A.; Moro, T. The Influence of Meal Frequency and Timing on Health in Humans: The Role of Fasting. Nutrients 2019, 11, 719. [Google Scholar] [CrossRef]
- St-Onge, M.P.; Ard, J.; Baskin, M.L.; Chiuve, S.E.; Johnson, H.M.; Kris-Etherton, P.; Varady, K.; American Heart Association Obesity Committee of the Council on Lifestyle and Cardiometabolic Health; Council on Cardiovascular Disease in the Young; Council on Clinical Cardiology; et al. Meal Timing and Frequency: Implications for Cardiovascular Disease Prevention: A Scientific Statement From the American Heart Association. Circulation 2017, 135, e96–e121. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Tovar, S. Editorial: Crosstalk of Mitochondria With Brain Insulin and Leptin Signaling. Front. Endocrinol. 2018, 9, 761. [Google Scholar]
- Lin, X.; Taguchi, A.; Park, S.; Kushner, J.A.; Li, F.; Li, Y.; White, M.F. Dysregulation of insulin receptor substrate 2 in beta cells and brain causes obesity and diabetes. J. Clin. Investig. 2004, 114, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Bruning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Muller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [PubMed]
- Kimura, K.; Tanida, M.; Nagata, N.; Inaba, Y.; Watanabe, H.; Nagashimada, M.; Ota, T.; Asahara, S.; Kido, Y.; Matsumoto, M.; et al. Central Insulin Action Activates Kupffer Cells by Suppressing Hepatic Vagal Activation via the Nicotinic Alpha 7 Acetylcholine Receptor. Cell Rep. 2016, 14, 2362–2374. [Google Scholar]
- Stevens, A.; Begum, G.; Cook, A.; Connor, K.; Rumball, C.; Oliver, M.; Challis, J.; Bloomfield, F.; White, A. Epigenetic changes in the hypothalamic proopiomelanocortin and glucocorticoid receptor genes in the ovine fetus after periconceptional undernutrition. Endocrinology 2010, 151, 3652–3664. [Google Scholar]
- Laryea, G.; Arnett, M.; Muglia, L.J. Ontogeny of hypothalamic glucocorticoid receptor-mediated inhibition of the hypothalamic-pituitary-adrenal axis in mice. Stress 2015, 18, 400–407. [Google Scholar] [PubMed]
- Laryea, G.; Muglia, L.; Arnett, M.; Muglia, L.J. Dissection of glucocorticoid receptor-mediated inhibition of the hypothalamic-pituitary-adrenal axis by gene targeting in mice. Front. Neuroendocrinol. 2015, 36, 150–164. [Google Scholar]
- Laryea, G.; Schutz, G.; Muglia, L.J. Disrupting hypothalamic glucocorticoid receptors causes HPA axis hyperactivity and excess adiposity. Mol. Endocrinol. 2013, 27, 1655–1665. [Google Scholar]
- Choi, J.S. Effects of Maternal and Post-Weaning High-Fat Diet on Leptin Resistance and Hypothalamic Appetite Genes in Sprague Dawley Rat Offspring. Clin. Nutr. Res. 2018, 7, 276–290. [Google Scholar]
- Chen, H.; Simar, D.; Lambert, K.; Mercier, J.; Morris, M.J. Maternal and postnatal overnutrition differentially impact appetite regulators and fuel metabolism. Endocrinology 2008, 149, 5348–5356. [Google Scholar]
- Chen, H.; Simar, D.; Morris, M.J. Hypothalamic neuroendocrine circuitry is programmed by maternal obesity: Interaction with postnatal nutritional environment. PLoS ONE 2009, 4, e6259. [Google Scholar] [CrossRef] [PubMed]
- Mehran, A.E.; Templeman, N.M.; Brigidi, G.S.; Lim, G.E.; Chu, K.Y.; Hu, X.; Botezelli, J.D.; Asadi, A.; Hoffman, B.G.; Kieffer, T.J.; et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012, 16, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Balthasar, N.; Coppari, R.; McMinn, J.; Liu, S.M.; Lee, C.E.; Tang, V.; Kenny, C.D.; McGovern, R.A.; Chua, S.C.; Elmquist, J.K., Jr.; et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 2004, 42, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Belgardt, B.F.; Bruning, J.C. CNS leptin and insulin action in the control of energy homeostasis. Ann. N. Y. Acad. Sci. 2010, 1212, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.G. Neurodevelopmental actions of leptin. Brain Res. 2010, 1350, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.G.; Draper, S.J.; Simerly, R.B. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 2004, 304, 108–110. [Google Scholar] [CrossRef]
- Pankey, C.L.; Walton, M.W.; Odhiambo, J.F.; Smith, A.M.; Ghnenis, A.B.; Nathanielsz, P.W.; Ford, S.P. Intergenerational impact of maternal overnutrition and obesity throughout pregnancy in sheep on metabolic syndrome in grandsons and granddaughters. Domest. Anim. Endocrinol. 2017, 60, 67–74. [Google Scholar] [CrossRef]
- Fuente-Martin, E.; Garcia-Caceres, C.; Granado, M.; de Ceballos, M.L.; Sanchez-Garrido, M.A.; Sarman, B.; Liu, Z.W.; Dietrich, M.O.; Tena-Sempere, M.; Argente-Arizon, P.; et al. Leptin regulates glutamate and glucose transporters in hypothalamic astrocytes. J. Clin. Investig. 2012, 122, 3900–3913. [Google Scholar] [CrossRef]
- Dearden, L.; Balthasar, N. Sexual dimorphism in offspring glucose-sensitive hypothalamic gene expression and physiological responses to maternal high-fat diet feeding. Endocrinology 2014, 155, 2144–2154. [Google Scholar] [CrossRef]
- Wang, C.; Xu, Y. Mechanisms for Sex Differences in Energy Homeostasis. J. Mol. Endocrinol 2019, 62, R129–R143. [Google Scholar] [CrossRef]
- Schellong, K.; Melchior, K.; Ziska, T.; Ott, R.; Henrich, W.; Rancourt, R.C.; Plagemann, A. Hypothalamic insulin receptor expression and DNA promoter methylation are sex-specifically altered in adult offspring of high-fat diet (HFD)-overfed mother rats. J. Nutr. Biochem. 2019, 67, 28–35. [Google Scholar] [PubMed]
- Chin, E.H.; Schmidt, K.L.; Martel, K.M.; Wong, C.K.; Hamden, J.E.; Gibson, W.T.; Soma, K.K.; Christians, J.K. A maternal high-fat, high-sucrose diet has sex-specific effects on fetal glucocorticoids with little consequence for offspring metabolism and voluntary locomotor activity in mice. PLoS ONE 2017, 12, e0174030. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CON | HFHC | p Value | |
|---|---|---|---|
| Pregnancy | |||
| Body weight (conception), g | 26.17 ± 0.40 | 33.51 ± 0.41 | <0.0001 |
| Body weight (delivery), g | 38.38 ± 0.80 | 41.63 ± 0.64 | 0.006 |
| Body weight (weaning), g | 26.87 ± 0.40 | 30.68 ± 1.40 | 0.004 |
| Litter size, # | 7.0 ± 0.5 | 6.0 ± 0.4 | NS |
| Male offspring/litter, % | 54 ± 4.4 | 43 ± 5.8 | NS |
| Consumption | |||
| Energy intake (pregnancy), kcal/day | 21.17 ± 6.07 | 59.87 ± 3.61 | <0.0001 |
| Energy intake (lactation), kcal/day | 27.17 ± 0.88 | 99.80 ± 7.05 | <0.0001 |
| Metabolic | |||
| Glucose, mg/dL | 89.4 ± 3.6 | 83.7 ± 4.0 | NS |
| Glucose tolerance test, AUC | 25.43 ± 1.94 | 22.22 ± 1.16 | NS |
| Cholesterol, mg/dL | 138.3 ± 16.5 | 182.3 ± 9.7 | 0.02 |
| Insulin, ng/mL | 1.62 ± 0.5 | 2.05 ± 0.2 | NS |
| Leptin, ng/dL | 4.13 ± 0.49 | 10.2 ± 0.9 | <0.0001 |
| NEFA, mEq/L | 0.74 ± 0.05 | 0.83 ± 0.04 | NS |
| Phospholipids, mg/dL | 209.8 ± 15.1 | 239.1 ± 12.4 | NS |
| Triglycerides, mg/dL | 63.4 ± 5.7 | 69.8 ± 4.1 | NS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulhanek, D.; Weigel, R.; Paulsen, M.E. Maternal High-Fat–High-Carbohydrate Diet-Induced Obesity Is Associated with Increased Appetite in Peripubertal Male but Not Female C57Bl/6J Mice. Nutrients 2020, 12, 2919. https://doi.org/10.3390/nu12102919
Kulhanek D, Weigel R, Paulsen ME. Maternal High-Fat–High-Carbohydrate Diet-Induced Obesity Is Associated with Increased Appetite in Peripubertal Male but Not Female C57Bl/6J Mice. Nutrients. 2020; 12(10):2919. https://doi.org/10.3390/nu12102919
Chicago/Turabian StyleKulhanek, Debra, Rachel Weigel, and Megan E. Paulsen. 2020. "Maternal High-Fat–High-Carbohydrate Diet-Induced Obesity Is Associated with Increased Appetite in Peripubertal Male but Not Female C57Bl/6J Mice" Nutrients 12, no. 10: 2919. https://doi.org/10.3390/nu12102919
APA StyleKulhanek, D., Weigel, R., & Paulsen, M. E. (2020). Maternal High-Fat–High-Carbohydrate Diet-Induced Obesity Is Associated with Increased Appetite in Peripubertal Male but Not Female C57Bl/6J Mice. Nutrients, 12(10), 2919. https://doi.org/10.3390/nu12102919