Lycopene Inhibits Activation of Epidermal Growth Factor Receptor and Expression of Cyclooxygenase-2 in Gastric Cancer Cells



Abstract
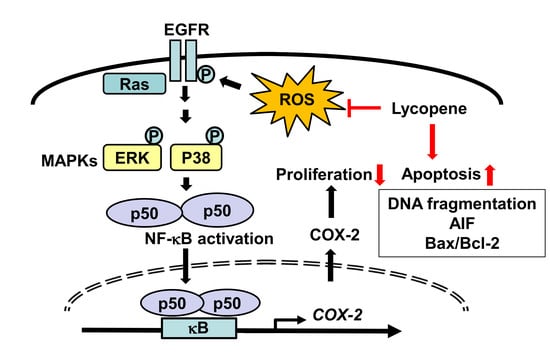
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Line and Culture Condition
2.2. Lycopene Treatment
2.3. Determination of Cell Viability
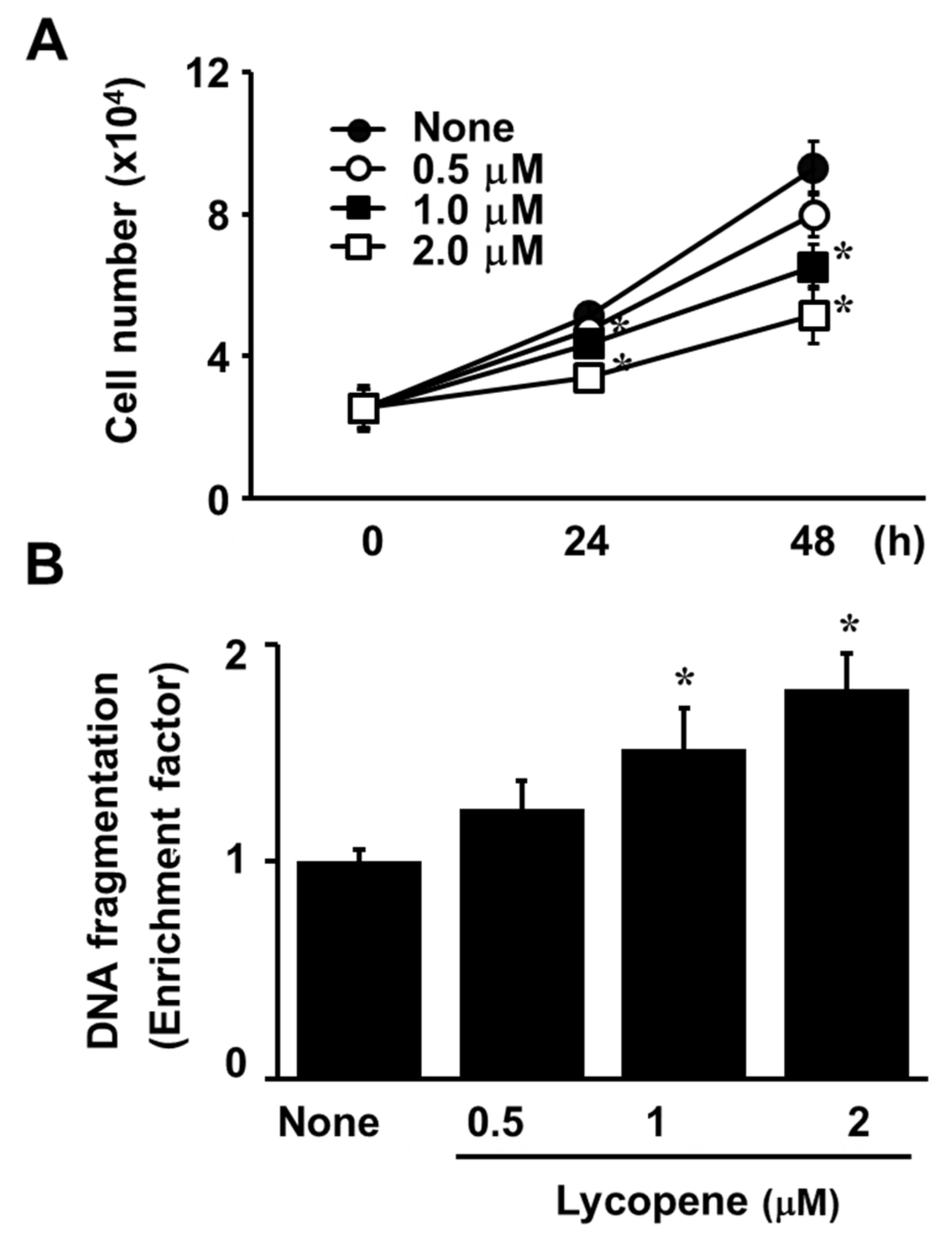
2.4. Assessment of DNA Fragmentation
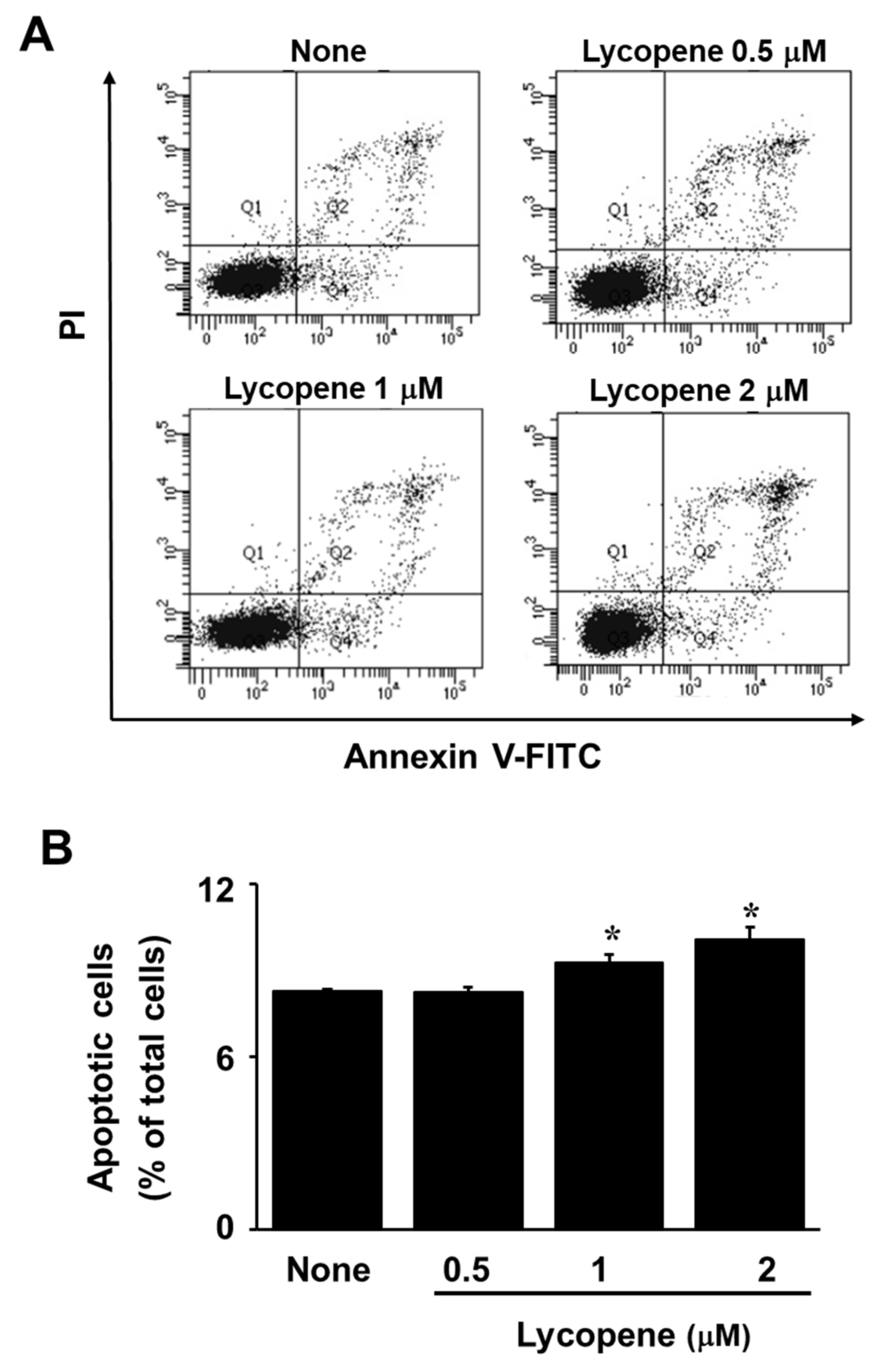
2.5. Annexin V/Propidium Iodide (PI)—Staining Assay
2.6. Measurement of Intracellular and Mitochondrial ROS Levels
2.7. Preparation of Whole-Cell Extracts, Membrane Extracts, and Nuclear Extracts
2.8. Western Blot Analysis
2.9. GST-Fusion Pull-Down Assay for Ras Activation
2.10. Immunoprecipitation for Detection of Phosphorylated Tyrosine and EGFR Interaction
2.11. Electrophoretic Mobility Shift Assay (EMSA)
2.12. Statistical Analysis
3. Results
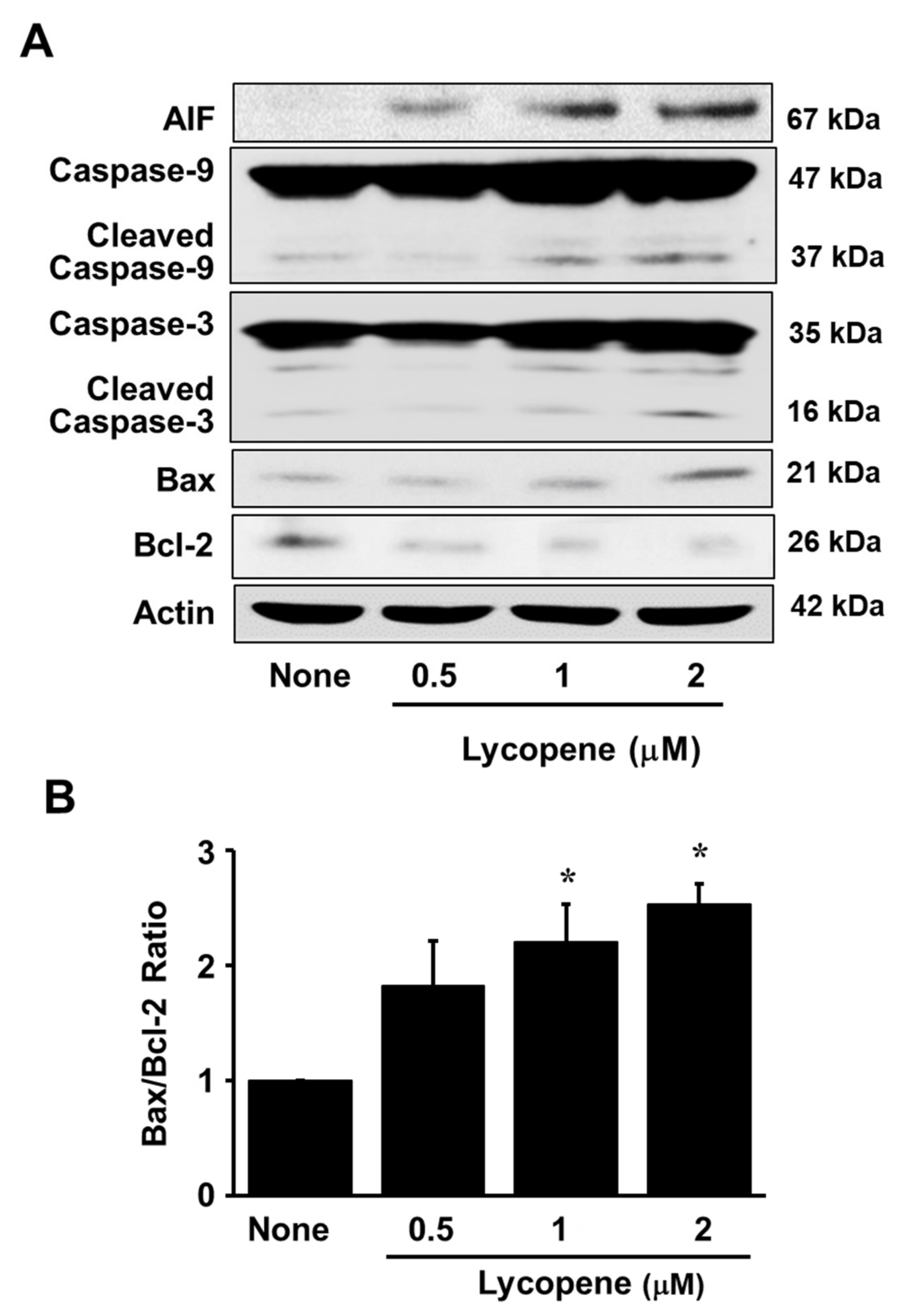
3.1. Lycopene Decreases Cell Viability and Induces Apoptosis in AGS Cells
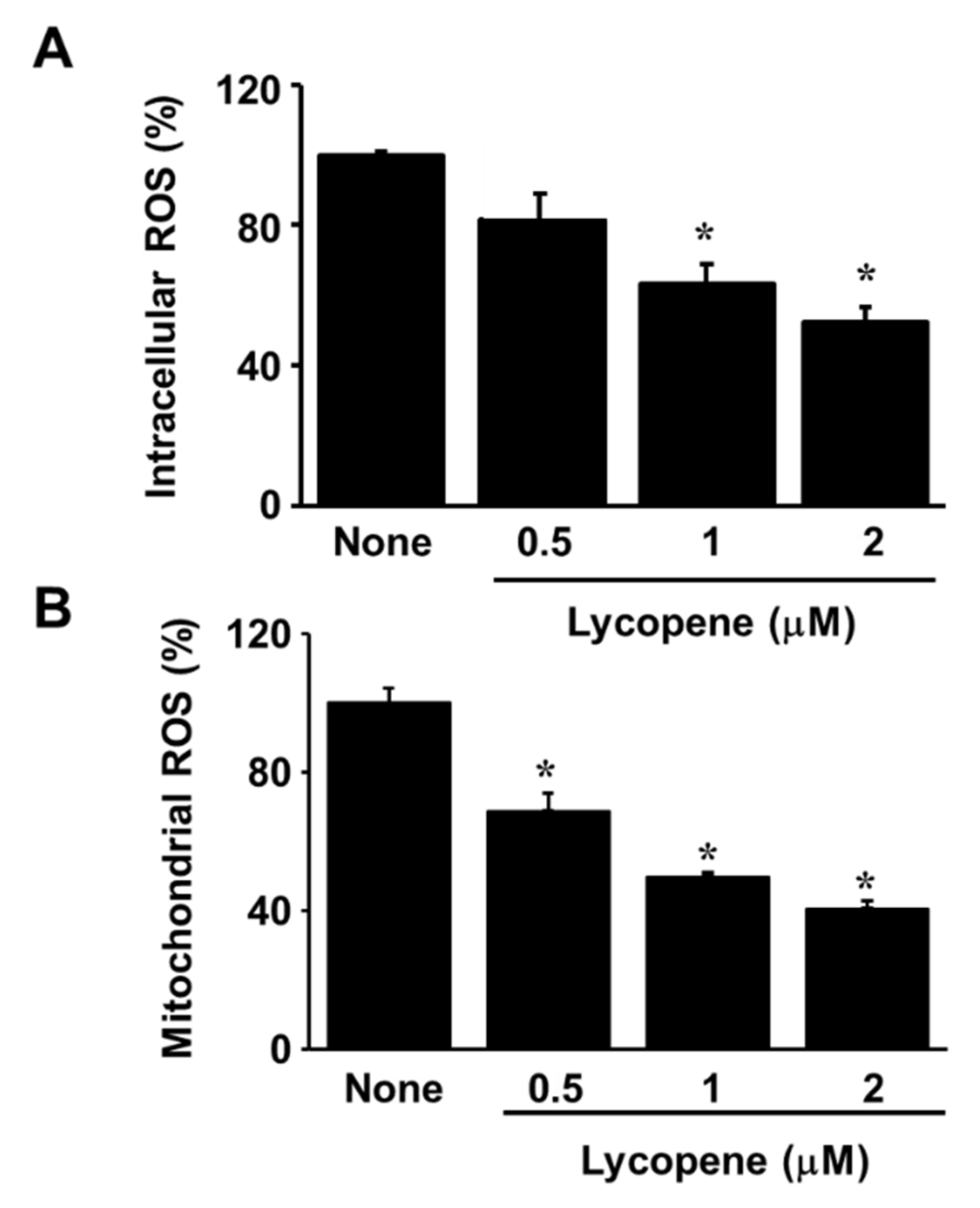
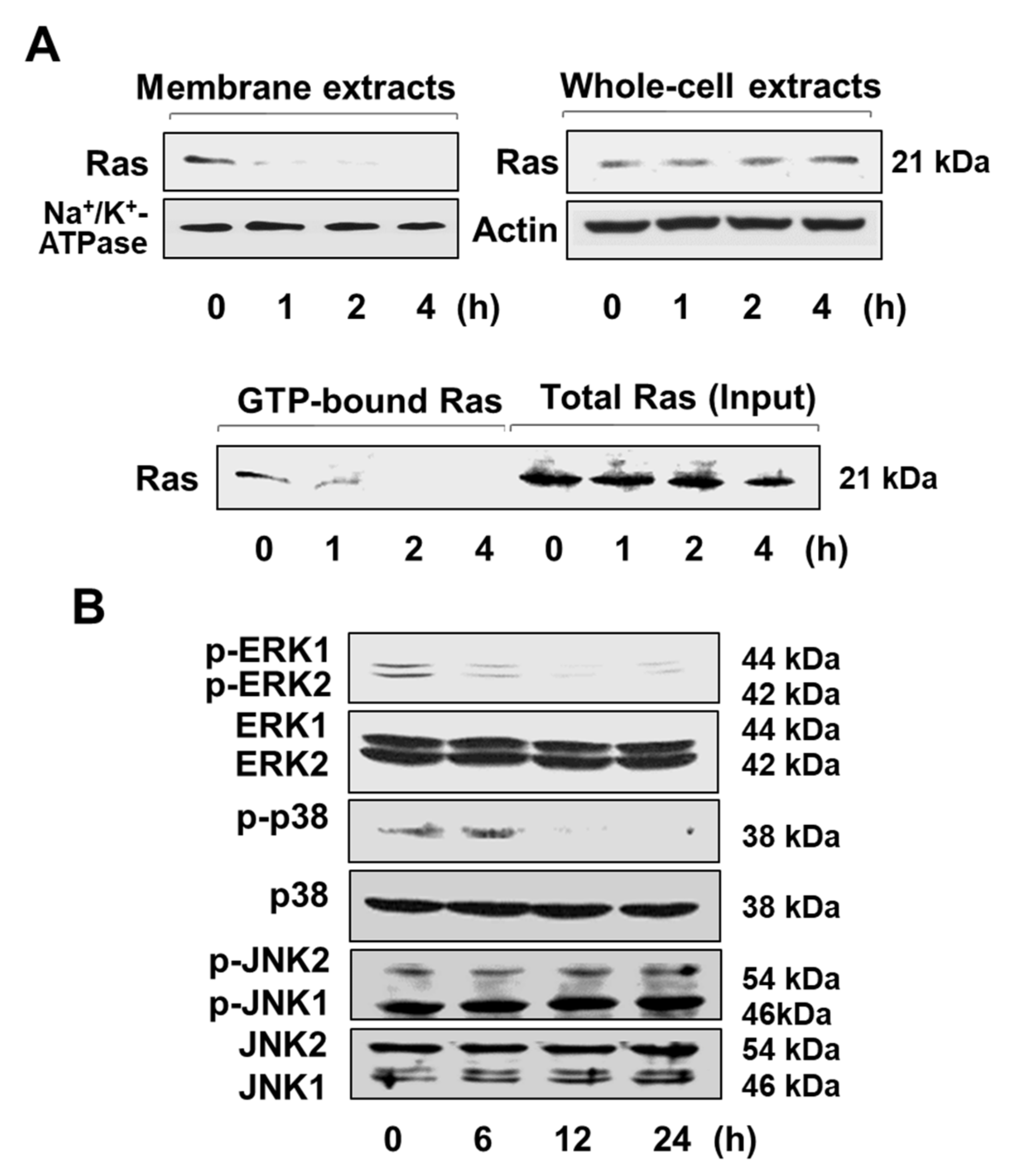
3.2. Lycopene Reduces ROS Levels and Inhibits EGFR/Ras/ERK and p38 MAPK Signaling in AGS Cells
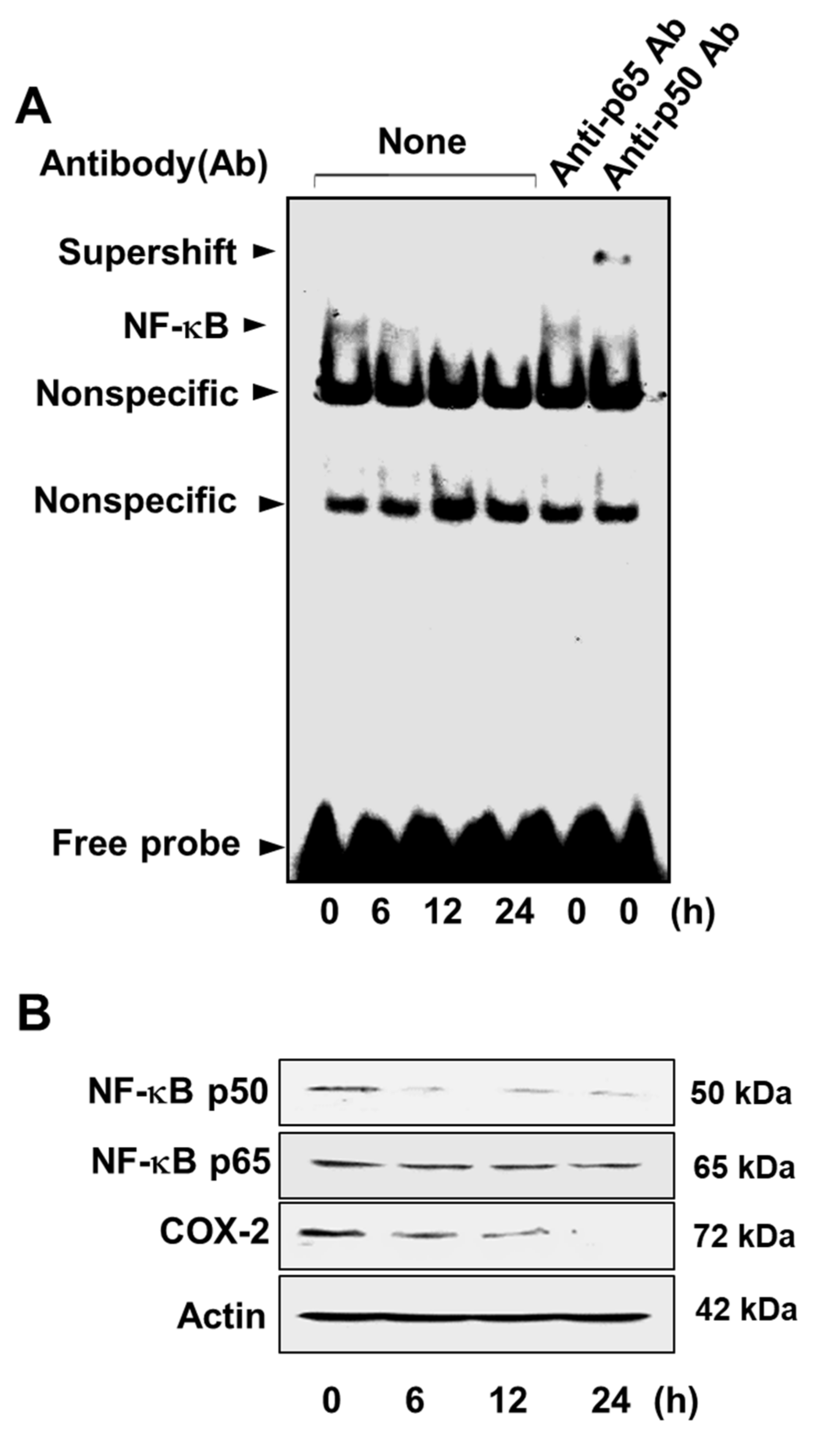
3.3. Lycopene Attenuates NF-κB p50/p50 Activity and COX-2 Gene Expression
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Møller, H.; Heseltine, E.; Vainio, H. Working group report on schistosomes, liver flukes and helicobacter pylori. Meeting held at IARC, Lyon, 7–14 June 1994. Int. J. Cancer 1995, 60, 587–589. [Google Scholar] [CrossRef]
- D’Elia, L.; Rossi, G.; Ippolito, R.; Cappuccio, F.P.; Strazzullo, P. Habitual salt intake and risk of gastric cancer: A meta-analysis of prospective studies. Clin. Nutr. 2012, 31, 489–498. [Google Scholar] [CrossRef]
- Ladeiras-Lopes, R.; Pereira, A.K.; Nogueira, A.; Pinheiro-Torres, T.; Pinto, I.; Santos-Pereira, R.; Lunet, N. Smoking and gastric cancer: Systematic review and meta-analysis of cohort studies. Cancer Causes Control 2008, 19, 689–701. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Pelicano, H.; Carney, D.; Huang, P. Ros stress in cancer cells and therapeutic implications. Drug Resist. Updat. 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Futagami, S.; Hiratsuka, T.; Shindo, T.; Horie, A.; Hamamoto, T.; Suzuki, K.; Kusunoki, M.; Miyake, K.; Gudis, K.; Crowe, S.E. Expression of apurinic/apyrimidinic endonuclease-1 (ape-1) in h. Pylori-associated gastritis, gastric adenoma, and gastric cancer. Helicobacter 2008, 13, 209–218. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, L.; Rong, S.; Qu, H.; Zhang, Y.; Chang, D.; Pan, H.; Wang, W. Relation between gastric cancer and protein oxidation, DNA damage, and lipid peroxidation. Oxid. Med. Cell Longev. 2013, 2013, 543760. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Hiraoka, S.; Tsutsui, S.; Kitamura, S.; Shinomura, Y.; Matsuzawa, Y. Epidermal growth factor receptor mediates stress-induced expression of its ligands in rat gastric epithelial cells. Gastroenterology 2001, 120, 108–116. [Google Scholar] [CrossRef]
- Gamou, S.; Shimizu, N. Hydrogen peroxide preferentially enhances the tyrosine phosphorylation of epidermal growth factor receptor. FEBS Lett. 1995, 357, 161–164. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef]
- Kim, M.A.; Lee, H.S.; Lee, H.E.; Jeon, Y.K.; Yang, H.K.; Kim, W.H. EGFR in gastric carcinomas: Prognostic significance of protein overexpression and high gene copy number. Histopathology 2008, 52, 738–746. [Google Scholar] [CrossRef]
- Takehana, T.; Kunitomo, K.; Suzuki, S.; Kono, K.; Fujii, H.; Matsumoto, Y.; Ooi, A. Expression of epidermal growth factor receptor in gastric carcinomas. Clin. Gastroenterol. Hepatol. 2003, 1, 438–445. [Google Scholar] [CrossRef]
- Gao, M.; Liang, X.J.; Zhang, Z.S.; Ma, W.; Chang, Z.W.; Zhang, M.Z. Relationship between expression of EGFR in gastric cancer tissue and clinicopathological features. Asian Pac. J. Trop. Med. 2013, 6, 260–264. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. Ras proteins and their regulators in human disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Jeong, W.J.; Ro, E.J.; Choi, K.Y. Interaction between Wnt/β -catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of beta-catenin and RAS by targeting the Wnt/β -catenin pathway. NPJ Precis. Oncol. 2018, 2, 5. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Hsu, T.C.; Young, M.R.; Cmarik, J.; Colburn, N.H. Activator protein 1 (ap-1)–and nuclear factor κb (nf-κb)–dependent transcriptional events in carcinogenesis. Free Radic. Biol. Med. 2000, 28, 1338–1348. [Google Scholar] [CrossRef]
- Kurland, J.F.; Voehringer, D.W.; Meyn, R.E. The MEK/ERK pathway acts upstream of NFκB1 (p50) homodimer activity and bcl-2 expression in a murine b-cell lymphoma cell line. MEK inhibition restores radiation-induced apoptosis. J. Biol. Chem. 2003, 278, 32465–32470. [Google Scholar] [CrossRef]
- Kim, H.; Seo, J.Y.; Kim, K.H. Effects of mannitol and dimethylthiourea on helicobacter pylori-induced il-8 production in gastric epithelial cells. Pharmacology 1999, 59, 201–211. [Google Scholar] [CrossRef]
- Sasaki, N.; Morisaki, T.; Hashizume, K.; Yao, T.; Tsuneyoshi, M.; Noshiro, H.; Nakamura, K.; Yamanaka, T.; Uchiyama, A.; Tanaka, M.; et al. Nuclear factor-kappaB p65 (rela) transcription factor is constitutively activated in human gastric carcinoma tissue. Clin. Cancer Res. 2001, 7, 4136–4142. [Google Scholar]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-kappaB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. [Google Scholar] [CrossRef]
- Tanabe, T.; Tohnai, N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002, 68, 95–114. [Google Scholar] [CrossRef]
- Thomas, B.; Berenbaum, F.; Humbert, L.; Bian, H.; Béréziat, G.; Crofford, L.; Olivier, J.L. Critical role of C/EBPδ and C/EBPβ factors in the stimulation of the cyclooxygenase-2 gene transcription by interleukin-1β in articular chondrocytes. Eur. J. Biochem. 2000, 267, 6798–6809. [Google Scholar] [CrossRef]
- Zha, S.; Yegnasubramanian, V.; Nelson, W.G.; Isaacs, W.B.; De Marzo, A.M. Cyclooxygenases in cancer: Progress and perspective. Cancer Lett. 2004, 215, 1–20. [Google Scholar] [CrossRef]
- Lim, H.Y.; Joo, H.J.; Choi, J.H.; Yi, J.W.; Yang, M.S.; Cho, D.Y.; Kim, H.S.; Nam, D.K.; Lee, K.B.; Kim, H.C. Increased expression of cyclooxygenase-2 protein in human gastric carcinoma. Clin. Cancer Res. 2000, 6, 519–525. [Google Scholar]
- Ristimaki, A.; Honkanen, N.; Jankala, H.; Sipponen, P.; Harkonen, M. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res. 1997, 57, 1276–1280. [Google Scholar]
- Sun, W.H.; Zhu, F.; Chen, G.S.; Su, H.; Luo, C.; Zhao, Q.S.; Zhang, Y.; Shao, Y.; Sun, J.; Zhou, S.M.; et al. Blockade of cholecystokinin-2 receptor and cyclooxygenase-2 synergistically induces cell apoptosis, and inhibits the proliferation of human gastric cancer cells in vitro. Cancer Lett. 2008, 263, 302–311. [Google Scholar] [CrossRef]
- Di Mascio, P.; Kaiser, S.; Sies, H. Lycopene as the most efficient biological carotenoid singlet oxygen quencher. Arch. Biochem. Biophys. 1989, 274, 532–538. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.; Choi, I.J.; Kim, Y.I.; Kwon, O.; Kim, H.; Kim, J. Dietary carotenoids intake and the risk of gastric cancer: A case-control study in Korea. Nutrients 2018, 10, 1031. [Google Scholar] [CrossRef]
- De Stefani, E.; Boffetta, P.; Brennan, P.; Deneo-Pellegrini, H.; Carzoglio, J.C.; Ronco, A.; Mendilaharsu, M. Dietary carotenoids and risk of gastric cancer: A case-control study in Uruguay. Eur. J. Cancer Prev. 2000, 9, 329–334. [Google Scholar] [CrossRef]
- Assar, E.A.; Vidalle, M.C.; Chopra, M.; Hafizi, S. Lycopene acts through inhibition of ikappaB kinase to suppress NFκB1 signaling in human prostate and breast cancer cells. Tumour Biol. 2016, 37, 9375–9385. [Google Scholar] [CrossRef]
- Palozza, P.; Colangelo, M.; Simone, R.; Catalano, A.; Boninsegna, A.; Lanza, P.; Monego, G.; Ranelletti, F.O. Lycopene induces cell growth inhibition by altering mevalonate pathway and ras signaling in cancer cell lines. Carcinogenesis 2010, 31, 1813–1821. [Google Scholar] [CrossRef]
- Park, Y.O.; Hwang, E.S.; Moon, T.W. The effect of lycopene on cell growth and oxidative DNA damage of Hep3B human hepatoma cells. Biofactors 2005, 23, 129–139. [Google Scholar] [CrossRef]
- Jang, S.H.; Lim, J.W.; Morio, T.; Kim, H. Lycopene inhibits helicobacter pylori-induced ATM/ATR-dependent DNA damage response in gastric epithelial AGS cells. Free Radic. Biol. Med. 2012, 52, 607–615. [Google Scholar] [CrossRef]
- Park, B.; Lim, J.W.; Kim, H. Lycopene treatment inhibits activation of Jak1/Stat3 and Wnt/β -catenin signaling and attenuates hyperproliferation in gastric epithelial cells. Nutr. Res. 2018. [Google Scholar] [CrossRef]
- Hantz, H.L.; Young, L.F.; Martin, K.R. Physiologically attainable concentrations of lycopene induce mitochondrial apoptosis in LNCaP human prostate cancer cells. Exp. Biol. Med. 2005, 230, 171–179. [Google Scholar] [CrossRef]
- Livny, O.; Kaplan, I.; Reifen, R.; Polak-Charcon, S.; Madar, Z. Lycopene inhibits proliferation and enhances gap-junctional communication on KB-1 human oral tumor cells. J. Nutr. 2002, 132, 3754–3759. [Google Scholar] [CrossRef]
- Burgess, L.C.; Rice, E.; Fischer, T.; Seekins, J.R.; Burgess, T.P.; Sticka, S.J.; Klatt, K. Lycopene has limited effect on cell proliferation in only two of seven human cell lines (both cancerous and noncancerous) in an in vitro system with doses across the physiological range. Toxicol. In Vitro 2008, 22, 1297–1300. [Google Scholar] [CrossRef]
- Luo, C.; Wu, X.G. Lycopene enhances antioxidant enzyme activities and immunity function in n-methyl-n’-nitro-n-nitrosoguanidine-enduced gastric cancer rats. Int. J. Mol. Sci. 2011, 12, 3340–3351. [Google Scholar] [CrossRef]
- Velmurugan, B.; Nagini, S. Combination chemoprevention of experimental gastric carcinogenesis by s-allylcysteine and lycopene: Modulatory effects on glutathione redox cycle antioxidants. J. Med. Food 2005, 8, 494–501. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, W.; Tao, G.Q.; Sun, J.; Shi, L.P. Study on the proliferation of human gastric cancer cells by activation of EGFR in H2O2. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1006–1012. [Google Scholar]
- Denu, J.M.; Tanner, K.G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef]
- Hernes, E.; Fossa, S.D.; Berner, A.; Otnes, B.; Nesland, J.M. Expression of the epidermal growth factor receptor family in prostate carcinoma before and during androgen independence. Br. J. Cancer 2004, 90, 449–454. [Google Scholar] [CrossRef]
- Rafi, M.M.; Kanakasabai, S.; Reyes, M.D.; Bright, J.J. Lycopene modulates growth and survival associated genes in prostate cancer. J. Nutr. Biochem. 2013, 24, 1724–1734. [Google Scholar] [CrossRef]
- Suyun, B.I.; Li, L.I.; Song, X.U.; Zhang, M.; Heng, G.U.; Zhou, Z.; Chen, X. Regulatory effects of lycopene on the key signaling receptors in human cutaneous squamous cell carcinoma cell line COLO16. Chin. J. Dermatol. 2018, 51, 421–424. [Google Scholar]
- Willumsen, B.M.; Christensen, A.; Hubbert, N.L.; Papageorge, A.G.; Lowy, D.R. The p21 ras c-terminus is required for transformation and membrane association. Nature 1984, 310, 583–586. [Google Scholar] [CrossRef]
- Zhang, B.; Gu, Y. Low expression of ERK signaling pathway affecting proliferation, cell cycle arrest and apoptosis of human gastric HGC-27 cells line. Mol. Biol. Rep. 2014, 41, 3659–3669. [Google Scholar] [CrossRef]
- Lim, J.W.; Kim, H.; Kim, K.H. Nuclear factor-kappaB regulates cyclooxygenase-2 expression and cell proliferation in human gastric cancer cells. Lab. Invest. 2001, 81, 349–360. [Google Scholar] [CrossRef]
- Kang, Y.J.; Wingerd, B.A.; Arakawa, T.; Smith, W.L. Cyclooxygenase-2 gene transcription in a macrophage model of inflammation. J. Immunol. 2006, 177, 8111–8122. [Google Scholar] [CrossRef]
- Song, J.; Su, H.; Zhou, Y.Y.; Guo, L.L. Cyclooxygenase-2 expression is associated with poor overall survival of patients with gastric cancer: A meta-analysis. Dig. Dis. Sci. 2014, 59, 436–445. [Google Scholar] [CrossRef]
- Tsujii, M.; DuBois, R.N. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell 1995, 83, 493–501. [Google Scholar] [CrossRef]
- Sawaoka, H.; Kawano, S.; Tsuji, S.; Tsujii, M.; Gunawan, E.S.; Takei, Y.; Nagano, K.; Hori, M. Cyclooxygenase-2 inhibitors suppress the growth of gastric cancer xenografts via induction of apoptosis in nude mice. Am. J. Physiol. 1998, 274, G1061–G1067. [Google Scholar] [CrossRef]
- Li, H.L.; Chen, D.D.; Li, X.H.; Zhang, H.W.; Lu, Y.Q.; Ye, C.L.; Ren, X.D. Changes of NF-kB, p53, Bcl-2 and caspase in apoptosis induced by jte-522 in human gastric adenocarcinoma cell line AGS cells: Role of reactive oxygen species. World J. Gastroenterol. 2002, 8, 431–435. [Google Scholar] [CrossRef]
- Couillard, C.; Lemieux, S.; Vohl, M.C.; Couture, P.; Lamarche, B. Carotenoids as biomarkers of fruit and vegetable intake in men and women. Br. J. Nutr. 2016, 116, 1206–1215. [Google Scholar] [CrossRef]
- Hodge, A.M.; Simpson, J.A.; Fridman, M.; Rowley, K.; English, D.R.; Giles, G.G.; Su, Q.; O’Dea, K. Evaluation of an ffq for assessment of antioxidant intake using plasma biomarkers in an ethnically diverse population. Public Health Nutr. 2009, 12, 2438–2447. [Google Scholar] [CrossRef]
- Chen, J.; Song, Y.; Zhang, L. Effect of lycopene supplementation on oxidative stress: An exploratory systematic review and meta-analysis of randomized controlled trials. J. Med. Food 2013, 16, 361–374. [Google Scholar] [CrossRef]
- Allen, C.M.; Schwartz, S.J.; Craft, N.E.; Giovannucci, E.L.; De Groff, V.L.; Clinton, S.K. Changes in plasma and oral mucosal lycopene isomer concentrations in healthy adults consuming standard servings of processed tomato products. Nutr. Cancer 2003, 47, 48–56. [Google Scholar] [CrossRef]
- Mellert, W.; Deckardt, K.; Gembardt, C.; Schulte, S.; Van Ravenzwaay, B.; Slesinski, R. Thirteen-week oral toxicity study of synthetic lycopene products in rats. Food Chem. Toxicol. 2002, 40, 1581–1588. [Google Scholar] [CrossRef]
- Trumbo, P.R. Are there adverse effects of lycopene exposure? J. Nutr. 2005, 135, 2060S–2061S. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, H.; Lim, J.W.; Kim, H. Lycopene Inhibits Activation of Epidermal Growth Factor Receptor and Expression of Cyclooxygenase-2 in Gastric Cancer Cells. Nutrients 2019, 11, 2113. https://doi.org/10.3390/nu11092113
Han H, Lim JW, Kim H. Lycopene Inhibits Activation of Epidermal Growth Factor Receptor and Expression of Cyclooxygenase-2 in Gastric Cancer Cells. Nutrients. 2019; 11(9):2113. https://doi.org/10.3390/nu11092113
Chicago/Turabian StyleHan, Hwana, Joo Weon Lim, and Hyeyoung Kim. 2019. "Lycopene Inhibits Activation of Epidermal Growth Factor Receptor and Expression of Cyclooxygenase-2 in Gastric Cancer Cells" Nutrients 11, no. 9: 2113. https://doi.org/10.3390/nu11092113
APA StyleHan, H., Lim, J. W., & Kim, H. (2019). Lycopene Inhibits Activation of Epidermal Growth Factor Receptor and Expression of Cyclooxygenase-2 in Gastric Cancer Cells. Nutrients, 11(9), 2113. https://doi.org/10.3390/nu11092113