1. Introduction
Phenylketonuria (PKU; OMIM 261600) is a rare inherited metabolic disease characterized by the absence of the liver enzyme phenylalanine (Phe) hydroxylase (PAH; EC 1.14.16.1) that converts Phe into tyrosine. The absence of this enzyme leads to elevation of blood Phe levels. If left untreated, irreversible neurological damage may occur due to the accumulation of Phe and its metabolites in the brain [
1,
2].
Based on pre-treatment blood Phe levels, patients are classified into three different phenotypes that require treatment: mild PKU with pre-treatment Phe levels of 360–600 μmol/L, moderate PKU with pre-treatment Phe >600–1200 μmol/L, and classical PKU with pre-treatment Phe >1200 μmol/L [
2]. Patients presenting with a pre-treatment blood Phe level <360 µmol/L do not require treatment and are described as mild hyperphenylalaninemia (mHPA) [
2,
3]. A low Phe diet with natural protein restriction and supplementation with a synthetic Phe-free/low-Phe protein substitute is the standard treatment in PKU, although more recent alternative or adjunct therapies such as the use of tetrahydrobiopterin (BH4), or large neutral amino acids (LNAA’s) are prescribed for certain subgroups of patients. Dietary treatment is commenced following detection by newborn screening, preferably in the first two weeks of life in order to maintain blood Phe levels within a safe target range and to achieve optimal neurological development [
3]. The severity of dietary restriction varies according to the residual activity of PAH enzyme (or phenotype), which influences individual Phe tolerance. Most patients with classical PKU tolerate less than 10 g natural protein (<20 mg/kg/day Phe), and most permitted natural protein sources are derived from plant sources such as fruits and vegetables [
3,
4,
5]. For nutritional adequacy, the remaining protein requirements are usually provided by a Phe-free/low-Phe protein substitute supplemented with vitamins, minerals, and essential fatty acids [
4]. Special low-protein foods (SLPF) provide energy and aid adherence by adding variety [
5,
6]. Milder phenotypes are likely to tolerate more natural protein (20–50 mg/kg/day Phe) and may respond to BH4 treatment, which allows some relaxation of natural protein intake.
There has been a considerable progress in dietary practices of PKU since its first introduction by Bickel and colleagues in 1951 [
7]. In the early years of treatment, protein requirements were not fully understood, and the use of unpalatable low-Phe protein hydrolysates with inadequate nutritional composition resulted in poor adherence and unfavorable outcomes [
8]. As the main objective of the treatment was the protection of brain from harmful effects of increased blood Phe levels, early treatment protocols used very restrictive diets, particularly during infancy and early childhood [
9,
10]. In the 1960s and 1970s, many PKU centers stopped dietary treatment as early as age 4 to 8 years in children, when it was believed that the brain development was essentially complete [
11]. The age of continuing either a strict or relaxed diet was gradually extended, and in the early 1990s, lifetime dietary treatment was recommended in the UK, which was later reinforced by the European PKU Guidelines [
3,
12].
Lifelong treatment prevents neurocognitive impairment and abnormal executive functioning and helps maintaining mental health [
3,
13]. Over time, recommendations on target Phe levels and dietary protein intakes have changed [
3]. There have also been efforts to improve the palatability and increase the availability of protein substitutes and SLPFs. The first Phe-free L-amino acid supplements were introduced in the 1970s, and since then, more acceptable protein substitutes with different presentations (e.g., powder, tablets, shakes), supplemented with micronutrients and essential fatty acids, have been developed [
14,
15,
16,
17]. However, both the nutritional composition and availability of these products significantly vary between countries [
17].
Despite the improvements in dietary treatment of PKU, lifelong adherence is challenging, and there are concerns regarding the long-term use of a semi-synthetic low-Phe diet [
18]. Early reports have suggested that initial treatment protocols caused growth impairment in children with PKU compared to healthy controls [
19,
20,
21,
22,
23,
24,
25]. Although normal growth has been documented in more recent studies, which were mostly conducted in patients who were born after 1990s and who had good metabolic control [
26,
27,
28,
29], there are still some reports of reduced final height or suboptimal growth following adolescence, which was influenced by gender or disease phenotype [
30,
31,
32].
Overall, there is ongoing debate about the impact of disease and treatment on long-term growth in PKU. The aims of this systematic review were: (1) to investigate if a Phe-restricted diet affects long-term growth in patients with PKU compared to normal populations, (2) to compare growth of patients with PKU treated by a Phe-restricted diet with mHPA patients who did not require dietary treatment, and (3) to determine if there are any growth gender differences between males and females.
2. Materials and Methods
This study was conducted by using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) current guidelines [
33]. The protocol was developed by authors and registered to PROSPERO with the record number CRD42018110779.
2.1. Literature Search
A systematic literature review was performed in 4 electronic databases including PubMed, Web of Science, Scopus, and Central Cochrane Library. The following keywords were used in the PubMed search query: (“Phenylketonurias” [MeSH Terms] OR “Phenylketonurias” [All Fields] OR (“Phenylketonuria” [All Fields] AND “PKU” [All Fields]) OR “Phenylketonuria PKU” [All Fields]) AND (“Phenylketonurias” [MeSH Terms] OR “Phenylketonurias” [All Fields] OR “Hyperphenylalaninemia” [All Fields]) AND (“Growth and development” [Subheading] OR (“Growth” [All Fields] AND “Development” [All Fields]) OR “Growth and development” [All Fields] OR “Growth” [All Fields] OR “Growth” [MeSH Terms]). For the remaining three databases, these main terms were customized. We limited our search to English, Spanish, Italian, Portuguese, and French languages. The last search was completed on the 21 September 2018.
2.2. Study Selection
The PICO (population, intervention, comparison, outcomes) method was applied to formulate the review question, and to determine the eligibility criteria. All retrospective and prospective longitudinal studies, randomized-controlled trials, and case-control studies conducted in patients with PKU being treated with a Phe-restricted diet from all age groups, and with a minimum of two years of follow up were included. Preclinical studies (in vitro and in vivo studies conducted on cell cultures or animals), cross-sectional studies, reviews, case reports, abstracts and thesis, and studies without a clear definition of the dietary treatment or with insufficient growth data were excluded. Studies were also considered for exclusion if the study population included the following: (1) patients with maternal PKU, (2) patients with a late diagnosis of PKU, (3) untreated PKU patients whose dietary treatment was not started within the first two months of age, (4) patients with a diagnosis of tetrahydrobiopterin (BH4) deficiency, and (5) patients treated by sapropterin or Pegvaliase.
Two independent reviewers (F.I. and A.P.) screened titles and abstracts according to eligibility criteria. All potentially relevant articles were identified for full-text review. Disagreements were resolved by consensus or through discussion with a third author (A.M.).
2.3. Outcome Measures
The primary outcomes were anthropometric measurements or indexes related to physical growth including body weight, height/recumbent length, and body mass index (BMI). Secondary outcome measures were birth weight, head circumference, and measures of metabolic control (e.g., blood Phe levels).
2.4. Data Extraction
Data was collected by two independent authors (F.I. and A.P.) using a standardized data extraction form. Information obtained from all included studies was (1) study characteristics (authors, publication year, country, duration, and design of the study), (2) description of population (sample size, gender, age, and ethnic origin), (3) description of dietary treatment (time of diet initiation, level of Phe-restriction, types of Phe-free/low-Phe protein substitutes, dietary natural protein and Phe intakes, total protein intake, and duration of follow-up), and (4) outcomes (weight, height or length, BMI, birth weight, head circumference, body composition, blood Phe control, and parental growth). Growth data, expressed as both age-specific z-scores and/or as mean (±SD) value, was extracted from tables. If the growth data was only available in figures, open source software Plot Digitizer (version 2.6.8, General Public License, Ankit Rohatgi, Austin, TX, USA) was used. We corresponded with five authors of papers to obtain further information.
2.5. Quality Appraisal
Two authors (F.I. and E.K.) independently assessed the quality of evidence of the included studies by using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach [
34]. The GRADE ranks for risk of bias, inconsistency, indirectness and imprecision were “not serious,” “serious,” and “very serious,” and for publication bias as “not likely,” “likely,” and “very likely.”
2.6. Risk of Bias Assessment
Two independent reviewers (F.I. and E.K.) assessed the selected articles for risk of bias by using “The Risk of Bias in Non-Randomised Studies of Interventions (ROBINS-I)” assessment tool [
35]. This tool was developed by the Cochrane Bias Methods Group and assesses internal validity. There are seven specific bias domains in the tool, including (1) confounding, (2) selection of participants, (3) classification of interventions, (4) deviations from intended interventions, (5) missing data, (6) measurement of outcomes, and (7) selection of reported results. Signaling questions were provided to help assessors decide the overall assessment for each domain. Risk of bias was rated as 0—no information; 1—low risk; 2—moderate risk; 3—serious risk; and 4—critical risk.
2.7. Data Analysis
The main objective of this systematic review was to assess how growth differs between PKU children compared with non-PKU control groups (e.g., healthy children, healthy siblings, or mHPA patients who have a normal diet). From 13 included articles, all studies measured weight and height or length (in children <2 years of age), except one study by Hoeksma et al. [
36] that did not evaluate weight. Five studies also measured BMI, and head circumference was reported in seven studies. Body composition was investigated in only one study.
There was heterogeneity between studies in terms of presentation of growth data, but the most frequently used method was the z-score system [
19,
20,
22,
26,
27,
28,
31,
32,
36,
37]. Growth data was presented only with mean anthropometric values in three studies [
8,
38,
39], and only two studies provided both [
20,
31]. As most of the included studies used z-scores, which is widely accepted for presentation and interpretation of anthropometric data with several advantages (e.g., evaluation of growth by combining gender and age groups) [
40], the studies only showing mean values were excluded from this meta-analysis. We also excluded six articles due to missing or insufficient data (e.g., standard deviations, sample size too small). Overall, the meta-analysis was performed with the remaining three articles [
20,
31,
32], and the excluded studies were evaluated qualitatively. For the interpretation of growth results, a mean z-score value of “zero” or “close to zero” was considered as “similar growth” between patients with PKU or mHPA and healthy population. The values between the two cut-off points of “−2SDs” and “+2SDs” were interpreted as “normal” range for growth [
40].
The secondary objective was to determine the differences between growth of male and female patients with PKU. Two studies [
20,
31] reported genders separately. However, growth data was only limited to height-for-age z-scores (HAZ) in the first study [
20] since body weight was expressed as “weight-for-height z-scores.” Therefore, gender difference could not be evaluated due to insufficient data.
BMI-for-age z-scores were only available in one study [
31], as well as head circumference data [
20]. We, therefore, only compared weight-for-age (WAZ) and HAZ of patients with PKU and control groups. The duration and the frequency of follow-up were also different between studies. In the study by Schaefer et al. [
20], patients with PKU were followed from birth to six years of age with six-month intervals. The remaining two studies [
31,
32] evaluated patients with PKU from birth until 18 years of age, and the frequency of measurements ranged between six months to one year. In Thiele et al. study [
32], dietary treatment was interrupted in 27 patients at a median age of seven years (range: 6.0–15.0). Since this could be a confounding factor, WAZ data between the ages of six and 18 were not included in the meta-analysis. Growth data of mHPA patients were only available in two studies [
31,
32]. Clinical visits for this group were scheduled less frequently after 12 years in one study [
31], so WAZ and HAZ were evaluated in the meta-analysis between 1 to 12 years of age only. A pooled analysis could not be conducted on BMI, head circumference, and body composition due to lack of data.
Heterogeneity between studies was calculated by I
2 statistic. I
2 value of 25%, 50% and 75% were considered as low, medium and high heterogeneity, respectively. Given the high heterogeneity level between studies, a random-effects model was used to calculate pooled estimates with the “metafor” package of R software (version 3.5.1, R foundation for statistical computer, Vienna, Austria) [
41]. A
p value less than 0.05 was considered statistically significant.
4. Discussion
This is the first systematic review and meta-analysis investigating the impact of a Phe-restricted diet on long term growth in patients with PKU. The primary aim was to determine if children with PKU could achieve a normal growth similar to healthy children from birth until adulthood. The effects of gender, disease severity (phenotype), metabolic control, and nutritional intake on growth were also evaluated.
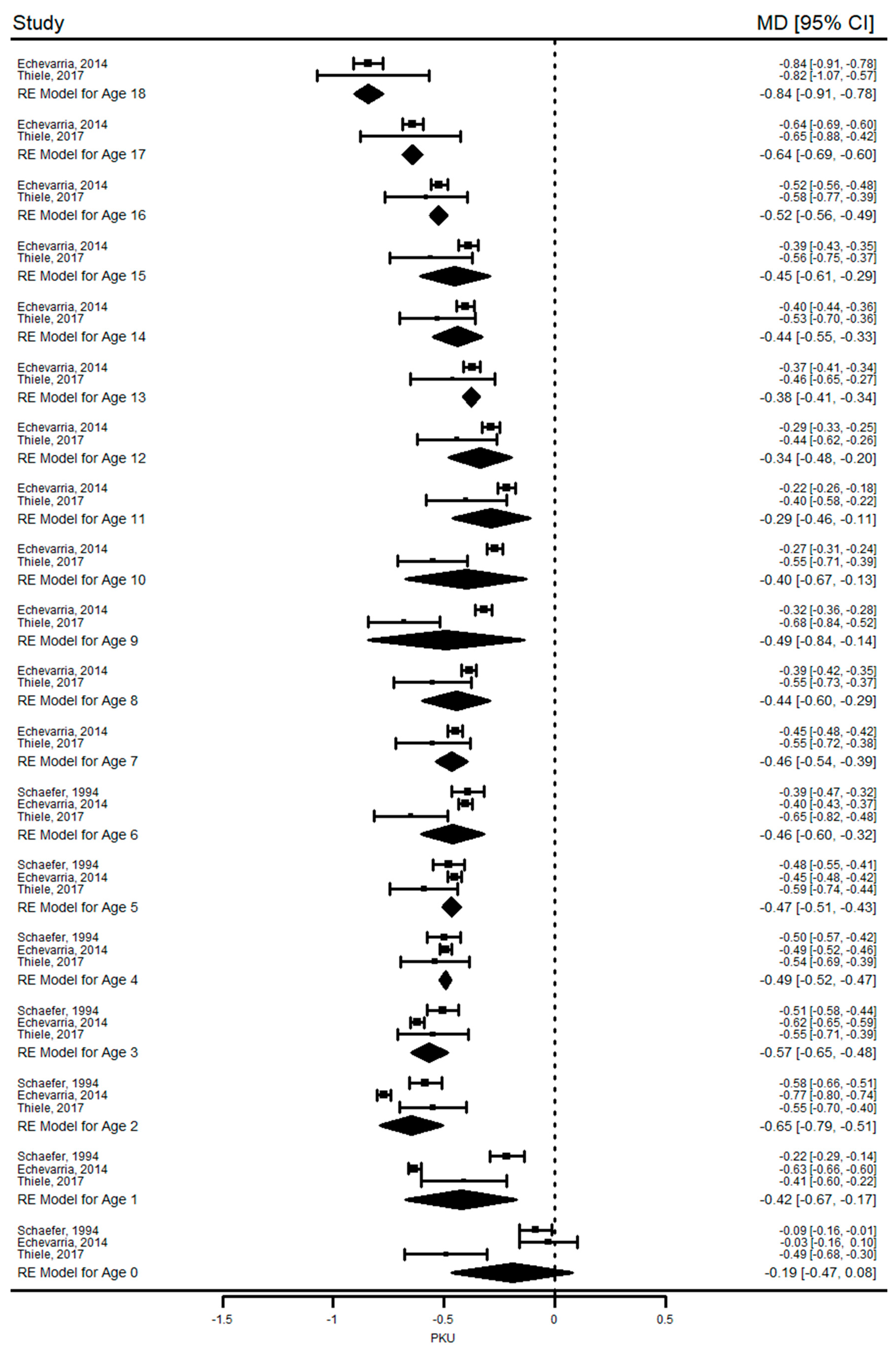
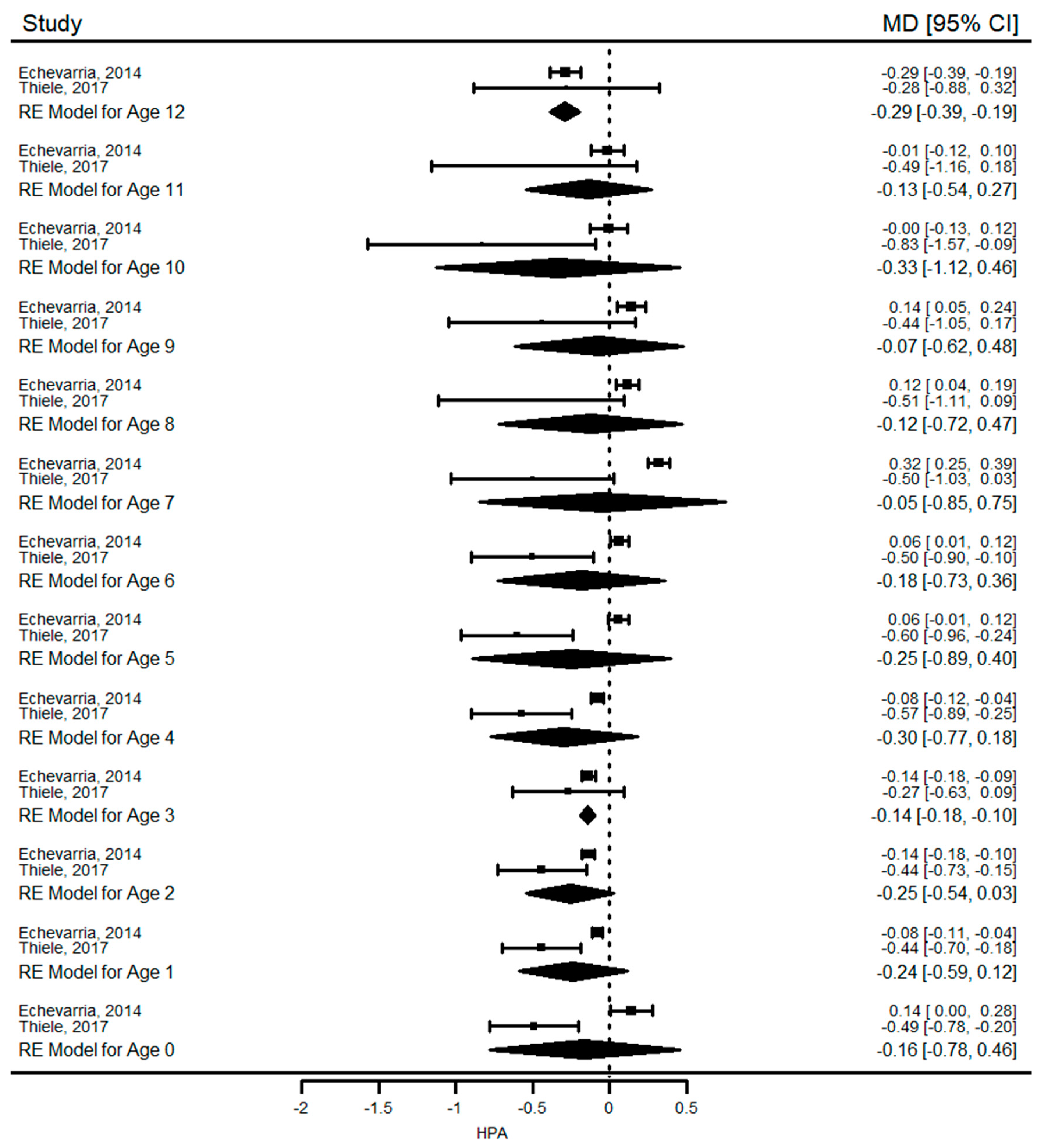
Our results show that growth (height and weight) in PKU was similar to healthy children at birth and during infancy but they were significantly shorter and had lower weight for age than reference population during the first four years of life. We could not perform a long-term analysis for weight, but reduced height growth was observed in PKU until the end of 18 years of age when the mean difference in HAZ between PKU patients and controls reached its highest value of −0.8 SD. In children with mHPA, a pooled analysis of two studies revealed that growth was not significantly affected. Another finding of this systematic review was that mean HAZ and WAZ of children with PKU and mHPA were within the normal ranges in almost all included studies (mean difference between the z-scores was consistently close to or less than 1 SD). Overall, these results suggest that children with PKU have not attained their growth potential compared with healthy peers.
This meta-analysis and previous studies [
8,
19,
20,
21,
22,
23,
31,
32,
36,
37] consistently showed an impairment of growth in children with PKU during the first three years of life, when growth is mainly determined by nutritional factors. Later, genetics or hormonal status (e.g., growth hormone, thyroid hormones, insulin-like growth factor I, insulin-like growth factor binding protein, sex hormones) impact growth during later childhood and adolescence [
42,
43]. Several hypotheses have been suggested for the possible causes of suboptimal growth outcomes in PKU but studies addressing the effects of hormonal status, bone age, or genetics (parental growth) on growth in PKU failed to show any associations [
20,
32,
44]. Therefore, faltering growth in young children with PKU has been mostly attributed to the use of a semi-synthetic low-Phe diet, commenced within the first few weeks of life. This is further supported by studies demonstrating no impact on growth in children with mHPA.
Despite substantial data on growth in PKU, the effects of dietary factors or metabolic control on growth outcomes have been little studied [
8,
20,
23,
28,
30,
31,
32,
36,
44,
45,
46]. No relationship was found between blood Phe levels and anthropometric measurements [
8,
20,
23,
31,
37,
44,
46]. Higher Phe intake was associated with better growth in one study [
31], but this was not confirmed by others [
8,
44]. Inadequate energy intake was eliminated as a cause of impaired growth, as a Phe restricted diet does not limit energy from carbohydrates and fats [
44]. There was also inconsistency between studies examining the effects of total protein, natural protein and protein equivalent intake from protein substitutes on growth outcomes. Considering earlier studies demonstrated unsatisfactory growth when total protein intake met the recommended dietary allowance (RDA) for general population [
19,
20], a higher protein intake (largely from Phe-free substitutes) was recommended to help achieving optimal growth [
47]. This increment in total protein intake was associated with normal growth in most [
26,
27,
30,
45,
48], but not all studies [
31,
32,
44].
In our systematic review, we observed that mean total protein intake for a given age was higher in studies that showed optimal growth in children with PKU [
26,
27,
28,
36] compared to studies with suboptimal growth results (
Supplementary Materials Table S2) [
20,
31,
32]. One possible reason may be that protein prescriptions were different, as shown in a recent study comparing total protein prescriptions from different countries in Europe [
49]. The authors concluded that there was variation according to region, with the highest median amount prescribed in Northern and Southern Europe, followed by Eastern and finally Western Europe [
49]. Our results were similar, such that the mean total protein intakes were lower in studies conducted in Western Europe that showed suboptimal growth [
20,
32] compared to studies in Southern Europe with optimal outcomes [
26,
27]. Interestingly, the results of two recent studies from different centers in Southern Europe (Spain) presented inconsistent results. Growth was similar to healthy Spanish population in one study [
26], but lower in the other [
31]. Since the mean total protein intake in the first study showing adequate growth was 2–3 times higher than the latter study with impaired growth results (
Supplementary Materials Table S2), one could speculate that different protein prescriptions may have led to the contrasting growth outcomes even in the same country or population.
Protein quality, rather than enhancement of total protein intake alone, may result in better growth outcomes or body composition. The effect of the type of protein on growth has been explored. Most studies failed to demonstrate an association between anthropometric measurements, natural protein [
28,
31,
44] or protein substitute intake [
28,
31,
36]. However, there has been evidence of a positive correlation between natural protein intake and head circumference (but not height) within the first three years of life [
36]. Moreover, higher natural protein intake was positively associated with fat-free mass, and negatively correlated with fat-mass in two other studies [
28,
45]. We did not include studies conducted on patients treated with BH4, which is also associated with increased natural protein intake according to patient’s tolerance. However, evidence from literature about the effects of BH4 treatment on growth has shown inconsistent results [
50,
51,
52]. Some of these studies included very small sample sizes and short follow-up periods. Therefore, it is too early to draw conclusions about the long-term effects of BH4 treatment and concomitant increase in natural protein intake on growth in PKU.
The impact of low biological efficiency of protein substitutes has also been explored. Low-Phe/Phe-free protein substitutes constitute the majority of dietary protein intake in children with PKU. It is well-established that ingestion of L-amino acids leads to rapid absorption, increased oxidation and poor nitrogen retention compared to intact proteins [
53], especially when taken in large single doses rather than small frequent doses [
54,
55]. Low biological efficiency of L-amino acids in protein substitutes, poor adherence to the timing and dosage recommendations may compromise growth in young children who have increased protein turnover due to faster growth rates and who are more susceptible to any protein depletion than adults [
56]. However, this has not been investigated in the studies included in this systematic review, so it is not possible to associate the metabolic efficiency of protein substitutes with growth outcomes.
Poor adherence is also an important issue which may possibly affect growth in PKU [
57]. It has been well-established that adherence to the low-Phe diet deteriorates with age, especially during adolescence and adulthood [
10,
57,
58,
59,
60]. Interestingly, this is the period in our study (11 to 18 years) in which patients were significantly shorter compared with reference values after catching up on differences from early childhood. It was not possible to establish an association between dietary adherence and growth from the results of this systematic review and meta-analysis due to lack of patient adherence rates (the mean percentage of patients with blood Phe levels within the target ranges). Additionally, the results were conflicting; there was still evidence of impaired growth despite good metabolic control, whereas better growth was achieved with poor adherence due to relaxation of the diet.
Being overweight is very common in the general population worldwide. In this systematic review, BMI in both children with PKU and mHPA was similar to healthy controls during the follow-up, and overweight rates were actually lower than the normal population levels. Several studies have suggested that rates would be even higher in PKU, particularly in female patients [
26,
61,
62], but more recent studies showed no significant difference in body composition or overweight rates between PKU and healthy populations [
45,
63,
64]. The etiology of overweight and obesity is very complex, with several contributing factors, but it is possible that in PKU this is related to unhealthy eating habits and lack of exercise similar to the general population [
65].
The impact of phenotype was also investigated. One study found that physical growth was normal in PKU regardless of the phenotype but studied twice the number of patients with a mild-moderate phenotype compared with severe PKU [
26]. Severe phenotype was more dominant than milder types in three studies that showed suboptimal growth [
8,
31,
32]. Kennedy et al. [
8] also found that length was normal during the first two years of life in milder PKU, compared to reduced length in severe PKU. The phenotype distribution was not provided in all studies; however, these findings may partly explain the discrepancy between the studies in terms of growth results. As the dietary Phe tolerance, and hence the restrictions of diet therapy, depend on the severity of the disease, phenotype distribution should always be considered in future studies assessing growth in PKU.
One of our main objectives was to evaluate the effects of gender on growth in patients with PKU, but we were unable to perform a meta-analysis examining impact of gender due to insufficient data. Although some evidence in this systematic review indicated a delayed catch-up in height and head circumference in boys [
20], further studies are needed to confirm if there is an impact of gender on growth in PKU.
There are several limitations to this systematic review. Firstly, most studies with suboptimal growth outcomes presented results from patients born before 1990s [
8,
19,
20,
22,
36,
37]. In contrast, studies with normal growth outcomes were mostly published after 2010, and data was collected more recently (1980 to 2014) [
26,
27,
28]. A similar cohort effect was supported by others [
29]. Additionally, Thiele et al. [
32] showed that growth impairment was more pronounced in children who received a casein hydrolysate during childhood, which was the major protein substitute of early years with a poor nutritional composition. Therefore, the suboptimal growth outcomes in early years of treatment can be attributed to the effects of outdated dietary practices and poor quality of early protein substitutes, rather than the effects of the Phe restricted diet or the disorder itself. More recent studies, following newer guidelines, using improved and more palatable protein substitutes provide evidence of adequate growth in PKU. Another important limitation was the high heterogeneity between studies probably arising from difference in phenotype distribution, country of study, time of data recruitment and sample sizes. Factors underlying the growth impairment in PKU were complicated due to lack of data, particularly on dietary intakes, metabolic control, and patient adherence. The duration of follow-up in majority of the studies was too short to examine the metabolic control during the whole developmental period. There was also limited information on early feeding practices (e.g., breastfeeding status, the type of protein substitutes used in infancy), parental growth, the use of adjunct treatments (such as LNAAs) or presence of any other chronic diseases which may all affect growth.
The methods of presenting growth data were variable (e.g., z-scores vs. mean values of anthropometric data, weight-for-age vs. weight-for-height, tables vs. figures). This heterogeneity led to inability to include all data in the meta-analysis. These challenges highlight the importance of using a standard data presentation. National growth standards of healthy children or healthy siblings were used for comparison of growth results with PKU, rather than using of a global reference (e.g., growth reference of World Health Organization). In future studies, interpretation of growth outcomes in PKU by using a standard growth reference as a control would allow for a valid comparison between studies conducted in different countries.


 ,
, 




{kind=link}
{kind=link}
{kind=link}