Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation


 , and
, and
Abstract
1. Introduction
1.1. Aging and Oxidative Stress
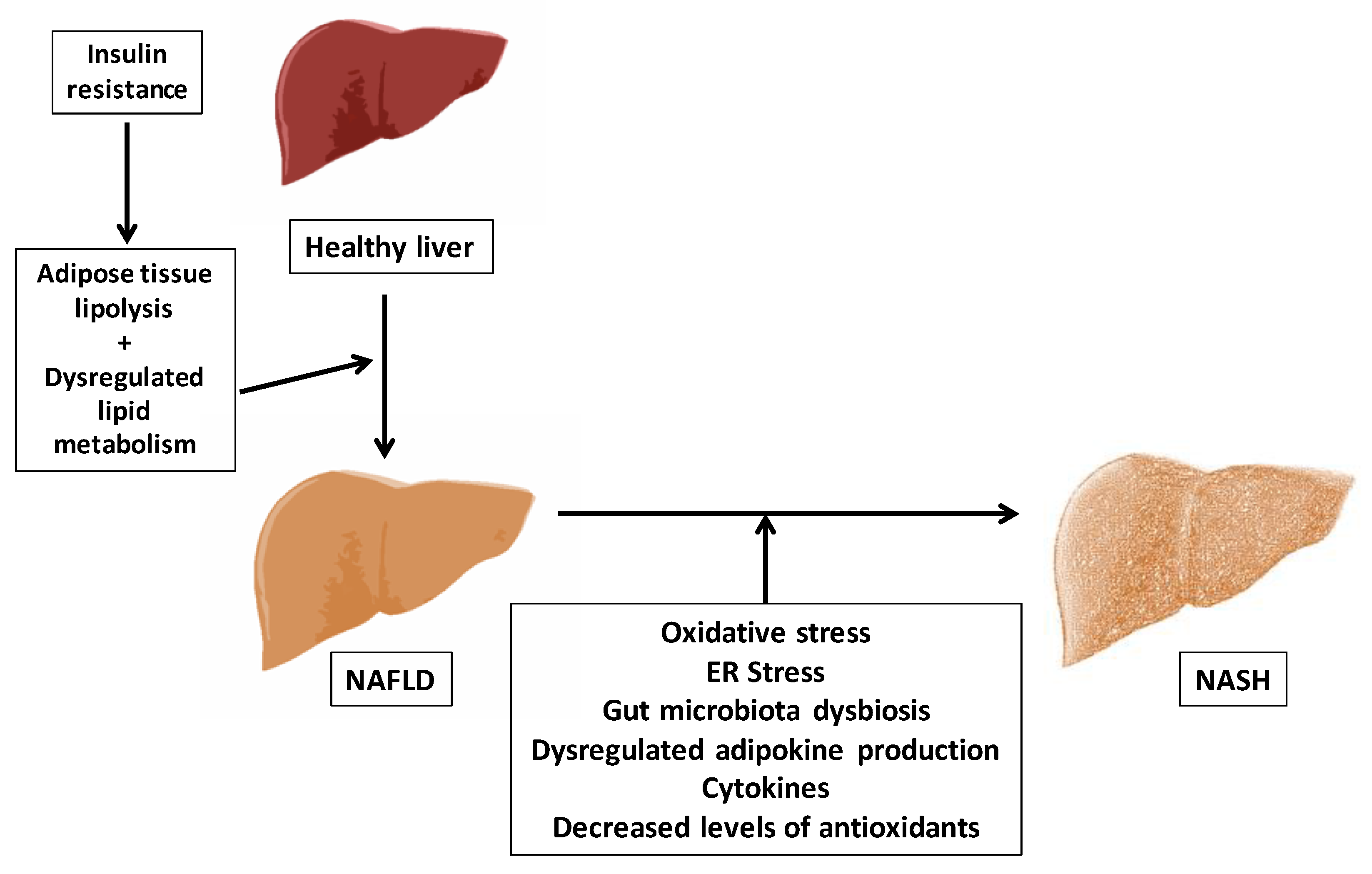
1.2. Aging and the Pathophysiology of Non-Alcoholic Fatty Liver Disease (NAFLD)
1.2.1. Concept and Pathogenesis of NAFLD
1.2.2. Genetic Variances Susceptibility for NASH and Oxidative Stress
1.2.3. The Diagnosis of NAFLD
2. Nutrients, Oxidative Stress and NAFLD/NASH
3. n-3 PUFA in Oxidative Stress and NAFLD/NASH
3.1. n-3 PUFA (Dietary Sources and Metabolism)
3.2. n-3 PUFA and Oxidative Stress
3.3. n-3 PUFA Supplementation in NAFLD and NASH Adult Patients
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Da Costa, J.P.; Vitorino, R.; Silva, G.M.; Vogel, C.; Duarte, A.C.; Rocha-Santos, T. A synopsis on aging—Theories, mechanisms and future prospects. Ageing Res. Rev. 2016, 29, 90–112. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.L. Systems biology of ageing and longevity. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Fanelli, F.; Calvani, R.; Mulè, G.; Pesce, V.; Sisto, A.; Pantanelli, C.; Bernabei, R.; Landi, F.; Marzetti, E. Gut Dysbiosis and Muscle Aging: Searching for Novel Targets against Sarcopenia. Mediat. Inflamm. 2018, 2018, 7026198. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, U.; Gannon, M. Type 2 Diabetes and the Aging Pancreatic Beta Cell. Aging 2011, 3, 565–575. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- He, F.; Zuo, L. Redox roles of reactive oxygen species in cardiovascular diseases. Int. J. Mol. Sci. 2015, 16, 27770–27780. [Google Scholar] [CrossRef]
- Zuo, L.; Pannell, B.K. Redox characterization of functioning skeletal muscle. Front. Physiol. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Pomatto, L.C.D.; Davies, K.J.A. Adaptive homeostasis and the free radical theory of ageing. Free Radic. Biol. Med. 2018, 124, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Peeva, V.; Kotlyar, A.; Kunz, W.S. Is there still any role for oxidative stress in mitochondrial DNA-dependent aging? Genes 2018, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.S.; Yoshida, Y.; Erickson, K.L. Do n-3 polyunsaturated fatty acids increase or decrease lipid peroxidation in humans? Metab. Syndr. Relat. Disord. 2014, 12, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.A.; Rokach, J.; FitzGerald, G.A. Isoprostanes: Formation, analysis and use as indices of lipid peroxidation in vivo. J. Biol. Chem. 1999, 274, 24441–24444. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; MY Lee, V.; Trojanowski, J.Q.; Rokach, J.; Fitzgerald, G.A. Increased F2-isoprostanes in Alzheimer’s disease: Evidence for enhanced lipid peroxidation in vivo. FASEB J. 1998, 12, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef]
- Romá-Mateo, C.; Seco-Cervera, M.; Ibáñez-Cabellos, J.S.; Pérez, G.; Berenguer-Pascual, E.; Rodríguez, L.R.; García-Giménez, J.L. Oxidative stress and the epigenetics of cell senescence: Insights from progeroid syndromes. Curr. Pharm. Des. 2018, 24, 4755–4770. [Google Scholar] [CrossRef]
- Schmucker, D.L. Age-related changes in liver structure and function: Implications for disease? Exp. Gerontol. 2005, 40, 650–659. [Google Scholar] [CrossRef]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Angulo, P. GI epidemiology: Nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2007, 25, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, E.; Taniai, M.; Tokushige, K. Characteristics and diagnosis of NAFLD/NASH. J. Gastroenterol. Hepatol. 2013, 28, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Bugianesi, E.; McCullough, A.J.; Marchesini, G. Insulin Resistance: A Metabolic Pathway to Chronic Liver Disease. Hepatology 2005, 42, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- Parekh, S.; Anania, F.A. Abnormal Lipid and Glucose Metabolism in Obesity: Implications for Nonalcoholic Fatty Liver Disease. Gastroenterology 2007, 132, 2191–2207. [Google Scholar] [CrossRef] [PubMed]
- Chitturi, S.; Farrell, G.C.; Hashimoto, E.; Saibara, T.; Lau, G.K.; Sollano, J.D. Non-alcoholic fatty liver disease in the Asia–Pacific region: Definitions and overview of proposed guidelines. J. Gastroenterol. Hepatol. 2007, 22, 778–787. [Google Scholar] [CrossRef]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37, 81–84. [Google Scholar] [CrossRef]
- Koehler, E.M.; Schouten, J.N.L.; Hansen, B.E.; Van Rooij, F.J.A.; Hofman, A.; Stricker, B.H.; Janssen, H.L.A. Prevalence and risk factors of non-alcoholic fatty liver disease in the elderly: Results from the Rotterdam study. J. Hepatol. 2012, 57, 1305–1311. [Google Scholar] [CrossRef]
- Souza, M.R.; Diniz Mde, F.; Medeiros Filho, J.E.; Araújo, M.S. Metabolic syndrome and risk factors for non-alcoholic fatty liver disease. Arq. Gastroenterol. 2012, 49, 89–96. [Google Scholar] [CrossRef]
- Klair, J.S.; Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Yates, K.; Unalp Arida, A.; Lavine, J.E.; Clark, J.M.; Diehl, A.M.; et al. A longer duration of estrogen deficiency increases fibrosis risk among postmenopausal women with nonalcoholic fatty liver disease. Hepatology 2016, 64, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Vehmas, A.P.; Adam, M.; Laajala, T.D.; Kastenmüller, G.; Prehn, C.; Rozman, J.; Ohlsson, C.; Fuchs, H.; Hrabě de Angelis, M.; Gailus Durner, V.; et al. Liver lipid metabolism is altered by increased circulating estrogen to androgen ratio in male mouse. J. Proteom. 2016, 133, 66–75. [Google Scholar] [CrossRef]
- Panic, A.; Stanimirovic, J.; Obradovic, M.; Sudar Milovanovic, E.; Perovic, M.; Lackovic, M.; Petrovic, N.; Isenovic, E.R. Estradiol-mediated regulation of hepatic iNOS in obese rats: Impact of Src, ERK1/2, AMPKα, and miR-221. Biotechnol. Appl. Biochem. 2018, 65, 797–806. [Google Scholar] [CrossRef]
- Veronese, N.; Notarnicola, M.; Osella, A.R.; Cisternino, A.M.; Reddavide, R.; Inguaggiato, R.; Guerra, V.; Rotolo, O.; Zinzi, I.; Chiloiro, M.; et al. Menopause Does Not Affect Fatty Liver Severity in Women: A Population Study in a Mediterranean Area. Endocr. Metab. Immune Disord. Drug Targets 2018, 18, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Frith, J.; Jones, D.; Newton, J.L. Chronic liver disease in an ageing population. Age Ageing 2009, 38, 11–18. [Google Scholar] [CrossRef]
- Wynne, H.A.; Cope, L.H.; Mutch, E.; Rawlins, M.D.; Woodhouse, K.W.; James, O.F.W. The effect of age upon liver volume and apparent liver blood flow in healthy man. Hepatology 1989, 9, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Tietz, N.W.; Shuey, D.F.; Wekstein, D.R. Laboratory values in fit aging individuals—Sexagenarians through centenarians. Clin. Chem. 1992, 38, 1167–1185. [Google Scholar] [PubMed]
- Ericsson, S.; Eriksson, M.; Vitols, S.; Einarsson, K.; Berglund, L.; Angelin, B. Influence of age on the metabolism of plasma low density lipoproteins in healthy males. J. Clin. Investig. 1991, 87, 591–596. [Google Scholar] [CrossRef]
- Bussel, J.B.; Berkowitz, R.L.; McFarland, J.G.; Lynch, L.; Chitkara, U. Influence of age on secretion of cholesterol and synthesis of bile acids by the liver. N. Engl. J. Med. 1985, 319, 1374–1378. [Google Scholar] [CrossRef]
- Conlon, B.A.; Beasley, J.M.; Aebersold, K.; Jhangiani, S.S.; Wylie-Rosett, J. Nutritional management of insulin resistance in nonalcoholic fatty liver disease (NAFLD). Nutrients 2013, 11, 4093–4114. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Paglialunga, S.; Dehn, C.A. Clinical assessment of hepatic de novo lipogenesis in non-alcoholic fatty liver disease. Lipids Health Dis. 2016, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef] [PubMed]
- Koliwad, S.K.; Streeper, R.; Monetti, M.; Cornelissen, I.; Chan, L.; Terayama, K.; Naylor, S.; Rao, M.; Hubbard, B.; Farese, R.V., Jr. DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J. Clin. Investig. 2010, 120, 756–767. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Amaro, A.; Fabbrini, E.; Kars, M.; Yue, P.; Schechtman, K.; Schonfeld, G.; Klein, S. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology 2010, 139, 149–153. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar] [CrossRef]
- Novo, E.; Busletta, C.; di Bonzo, L.V.; Povero, D.; Paternostro, C.; Mareschi, K.; Ferrero, I.; David, E.; Bertolani, C.; Caligiuri, A.; et al. Intracellular reactive oxygen species are required for directional migration of resident and bone marrow-derived hepatic pro-fibrogenic cells. J. Hepatol. 2011, 54, 964–974. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of Inflammation in Nonalcoholic Fatty Liver Disease: The Multiple Parallel Hits Hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Cancello, R.; Tordjman, J.; Poitou, C.; Guilhem, G.; Bouillot, J.L.; Hugol, D.; Coussieu, C.; Basdevant, A.; Hen, A.B.; Bedossa, P.; et al. Increased Infiltration of Macrophages in Omental Adipose Tissue Is Associated with Marked Hepatic Lesions in Morbid Human Obesity. Diabetes 2006, 55, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Canfora, E.E.; Meex, R.C.R.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferriéres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008, 87, 1219–1223. [Google Scholar] [CrossRef]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef]
- Vernia, P.; Marcheggiano, A.; Caprilli, R.; Frieri, G.; Corrao, G.; Valpiani, D. Short-chain fatty acid topical treatment in distal ulcerative colitis. Aliment. Pharmacol. Ther. 1995, 9, 309–313. [Google Scholar] [CrossRef]
- Purohit, V.; Bode, J.C.; Bode, C.; Brenner, D.A.; Choudhry, M.A.; Hamilton, F.; Kang, Y.J.; Keshavarzian, A.; Rao, R.; Sartor, R.B.; et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: Summary of a symposium. Alcohol 2008, 42, 349–361. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef]
- Su, G.L. Lipopolysaccharides in liver injury: Molecular mechanisms of Kupffer cell activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G256–G265. [Google Scholar] [CrossRef]
- Jones, R.M.; Neish, A.S. Redox signaling mediated by the gut microbiota. Free Radic. Biol. Med. 2017, 105, 41–47. [Google Scholar] [CrossRef]
- Gustot, T.; Lemmers, A.; Moreno, C.; Nagy, N.; Quertinmont, E.; Nicaise, C.; Franchimont, D.; Louis, H.; Devière, J.; Le Moine, O. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology 2006, 43, 989–1000. [Google Scholar] [CrossRef]
- Mardinoglu, S.; Shoaie, M.; Bergentall, P.; Ghaffari, C.; Zhang, E.L.; Nielsen, F.B.J. The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol. Syst. Biol. 2015, 11, 834. [Google Scholar] [CrossRef]
- Kojer, K.; Bien, M.; Gangel, H.; Morgan, B.; Dick, T.P.; Riemer, J. Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 2012, 31, 3169–3182. [Google Scholar] [CrossRef]
- Morgan, B.; Ezeriņa, D.; Amoako, T.N.; Riemer, J.; Seedorf, M.; Dick, T.P. Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nat. Chem. Biol. 2013, 9, 119–125. [Google Scholar] [CrossRef]
- Oliveira, C.P.; Stefano, J.T. Genetic polymorphisms and oxidative stress in non-alcoholic steatohepatitis (NASH): A mini review. Clin. Res. Hepatol. Gastroenterol. 2015, 39 (Suppl. 1), S35–S40. [Google Scholar] [CrossRef]
- Cobbina, E.; Akhlaghi, F. Non-alcoholic fatty liver disease (NAFLD)—Pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 2017, 49, 197–211. [Google Scholar] [CrossRef]
- Dai, G.; Liu, P.; Li, X.; Zhou, X.; He, S. Association between PNPLA3 rs738409 polymorphism and nonalcoholic fatty liver disease (NAFLD) susceptibility and severity: A meta-analysis. Medicine 2019, 98, e14324. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Gao, Y.; Ma, H.; Zhan, S.; Yang, Y.; Xin, Y.; Xuan, S. Association of TM6SF2 rs58542926 gene polymorphism with the risk of non-alcoholic fatty liver disease and colorectal adenoma in Chinese Han population. BMC Biochem. 2019, 20, 3. [Google Scholar] [CrossRef]
- Wang, J.K.; Feng, Z.W.; Li, Y.C.; Li, Q.Y.; Tao, X.Y. Association of tumornecrosis factor-gene promoter polymorphism at sites -308 and -238 with non-alcoholic fatty liver disease: A meta-analysis. J. Gastroenterol. Hepatol. 2012, 27, 670–676. [Google Scholar] [CrossRef]
- Nelson, J.E.; Handa, P.; Aouizerat, B.; Wilson, L.; Vemulakonda, L.A.; Yeh, M.M.; Kowdley, K.V. Increased parenchymal damage and steatohepatitis in Caucasian non-alcoholic fatty liver disease patients with common IL1B and IL6 polymorphisms. Aliment. Pharmacol. Ther. 2016, 44, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Kurbatova, I.V.; Topchieva, L.V.; Dudanova, O.P. Gene TNF Polymorphism -308G>A (rs1800629) and Its Relationship with the Efficiency of Ursodeoxycholic Acid Therapy in Patients with Nonalcoholic Stetohepatitis. Bull. Exp. Biol. Med. 2017, 164, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Kiziltas, S.; Ata, P.; Colak, Y.; Mesçi, B.; Senates, E.; Enc, F.; Ulasoglu, C.; Tuncer, I.; Oguz, A. TLR4 gene polymorphism in patients with nonalcoholic fatty liver disease in comparison to healthy controls. Metab. Syndr. Relat. Disord. 2014, 12, 165–170. [Google Scholar] [CrossRef]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Genetic polymorphism in CD14 gene, a co-receptor of TLR4 associated with non-alcoholic fatty liver disease. World J. Gastroenterol. 2016, 14, 9346–9355. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Donati, B.; Panera, N.; Vongsakulyanon, A.; Alisi, A.; Dallapiccola, B.; Valenti, L. A 4-polymorphism risk score predicts steatohepatitis in children with nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 632–636. [Google Scholar] [CrossRef]
- Al-Serri, A.; Anstee, Q.M.; Valenti, L.; Nobili, V.; Leathart, J.B.; Dongiovanni, P.; Patch, J.; Fracanzani, A.; Fargion, S.; Day, C.P.; et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case-control and intra-familial allele association studies. J. Hepatol. 2012, 56, 448–454. [Google Scholar] [CrossRef]
- Xu, Y.P.; Liang, L.; Wang, C.L.; Fu, J.F.; Liu, P.N.; Lv, L.Q.; Zhu, Y.M. Association between UCP3 gene polymorphisms and nonalcoholic fatty liver disease in Chinese children. World J. Gastroenterol. 2013, 19, 5897–5903. [Google Scholar] [CrossRef]
- Aller, R.; De Luis, D.A.; Izaola, O.; González Sagrado, M.; Conde, R.; Alvarez, T.; Pacheco, D.; Velasco, M.C. Role of -55CT polymorphism of UCP3 gene on non-alcoholic fatty liver disease and insulin resistance in patients with obesity. Nutr. Hosp. 2010, 25, 572–576. [Google Scholar]
- Hashemi, M.; Hoseini, H.; Yaghmaei, P.; Moazeni-Roodi, A.; Bahari, A.; Hashemzehi, N.; Shafieipour, S. Association of polymorphisms in glutamate-cysteine ligase catalytic subunit and microsomal triglyceride transfer protein genes with nonalcoholic fatty liver disease. DNA Cell Biol. 2011, 308, 569–575. [Google Scholar] [CrossRef]
- Oliveira, C.P.; Stefano, J.T.; Cavaleiro, A.M.; Zanella Fortes, M.A.; Vieira, S.M.; Rodrigues Lima, V.M.; Santos, T.E.; Santos, V.N.; de Azevedo Salgado, A.L.; Parise, E.R.; et al. Association of polymorphisms of glutamate-cystein ligase and microsomal triglyceride transfer protein genes in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2010, 25, 357–361. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Sanyal, A.J. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology 2002, 123, 1705–1725. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.; Caldwell, S.H. Nonalcoholic steatohepatitis: Summary of an AASLD Single Topic Conference. Hepatology 2003, 37, 1202–1219. [Google Scholar] [CrossRef]
- Joseph, A.E.; Saverymuttu, S.H.; Al-Sam, S.; Cook, M.G.; Maxwell, J.D. Comparison of liver histology with ultrasonography in assessing diffuse parenchymal liver disease. Clin. Radiol. 1991, 43, 26–31. [Google Scholar] [CrossRef]
- Preiss, D.; Sattar, N. Non-alcoholic fatty liver disease: An overview of prevalence, diagnosis, pathogenesis and treatment considerations. Clin. Sci. 2008, 115, 141–150. [Google Scholar] [CrossRef]
- Matteoni, C.; Younossi, Z.; Gramlich, T.; Boparai, N.; Liu, Y.; Mccullough, A. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Jung, C.H.; Kang, Y.M.; Jang, J.E.; Hwang, J.Y.; Kim, E.H.; Park, J.Y.; Kim, H.K.; Lee, W.J. Fatty liver index is a risk determinant of incident type 2 diabetes in a metabolically healthy population with obesity. Obesity 2016, 24, 1373–1379. [Google Scholar] [CrossRef]
- Huh, J.H.; Ahn, S.V.; Koh, S.B.; Choi, E.; Kim, J.Y.; Sung, K.C.; Kim, E.J.; Park, J.B. A prospective study of fatty liver index and incident hypertension: The KoGES-ARIRANG study. PLoS ONE 2015, 10, e0143560. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, D.; Kim, H.J.; Lee, C.H.; Yang, J.I.; Kim, W.; Kim, Y.J.; Yoon, J.H.; Cho, S.H.; Sung, M.W.; et al. Hepatic steatosis index: A simple screening tool reflecting nonalcoholic fatty liver disease. Dig. Liver Dis. 2010, 42, 503–508. [Google Scholar] [CrossRef]
- Wang, J.; Xu, C.; Xun, Y.; Lu, Z.; Shi, J.; Yu, C.; Li, Y. ZJU index: A novel model for predicting nonalcoholic fatty liver disease in a Chinese population. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Ruffillo, G.; Fassio, E.; Alvarez, E.; Landeira, G.; Longo, C.; Domínguez, N.; Gualano, G. Comparison of NAFLD fibrosis score and BARD score in predicting fibrosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 54, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Hui, J.M.; Marchesini, G.; Bugianesi, E.; George, J.; Farrell, G.C.; Enders, F.; Saksena, S.; Burt, A.D.; Bida, J.P.; et al. The NAFLD fibrosis score: A noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007, 45, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Wai, C.T.; Greenson, J.K.; Fontana, R.J.; Kalbfleisch, J.D.; Marrero, J.A.; Conjeevaram, H.S.; Lok, A.S.F. Simple Noninvasive Index Can Predict Both Significant Fibrosis and Cirrhosis in Patients with Chronic Hepatitis C. Hepatology 2003, 38, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Cui, H.; Li, N.; Wei, Y.; Lai, S.; Yang, Y.; Yin, X.; Chen, D.F. Comparison of FIB-4 index, NAFLD fibrosis score and BARD score for prediction of advanced fibrosis in adult patients with non-alcoholic fatty liver disease: A meta-analysis study. Hepatol. Res. 2016, 46, 862–870. [Google Scholar] [CrossRef]
- Bedogni, G.; Bellentani, S.; Miglioli, L.; Masutti, F.; Passalacqua, M.; Castiglione, A.; Tiribelli, C. The fatty liver index: A simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2006, 6, 1–7. [Google Scholar] [CrossRef]
- Kahn, H.S. The “lipid accumulation product” performs better than the body mass index for recognizing cardiovascular risk: A population-based comparison. BMC Cardiovasc. Disord. 2005, 5, 1–10. [Google Scholar] [CrossRef]
- Dai, H.; Wang, W.; Chen, R.; Chen, Z.; Lu, Y.; Yuan, H. Lipid accumulation product is a powerful tool to predict non-alcoholic fatty liver disease in Chinese adults. Nutr. Metab. 2017, 14, 1–9. [Google Scholar] [CrossRef]
- Rotter, I.; Rył, A.; Szylińska, A.; Pawlukowska, W.; Lubkowska, A.; Laszczyńska, M. Lipid Accumulation Product (LAP) as an Index of Metabolic and Hormonal Disorders in Aging Men. Exp. Clin. Endocrinol. Diabetes 2017, 125, 176–182. [Google Scholar] [CrossRef]
- Cuthbertson, D.J.; Weickert, M.O.; Lythgoe, D.; Sprung, V.S.; Dobson, R.; Shoajee-Moradie, F.; Umpleby, M.; Pfeiffer, A.F.H.; Thomas, E.L.; Bell, J.D.; et al. External validation of the fatty liver index and lipid accumulation product indices, using1H-magnetic resonance spectroscopy, to identify hepatic steatosis in healthy controls and obese, insulin-resistant individuals. Eur. J. Endocrinol. 2014, 171, 561–569. [Google Scholar] [CrossRef]
- Harrison, S.A.; Oliver, D.; Arnold, H.L.; Gogia, S.; Neuschwander-Tetri, B.A. Development and validation of a simple NAFLD clinical scoring system for identifying patients without advanced disease. Gut 2008, 57, 1441–1447. [Google Scholar] [CrossRef]
- Sterling, R.K.; Lissen, E.; Clumeck, N.; Sola, R.; Correa, M.C.; Montaner, J.; Sulkowski, M.S.; Torriani, F.J.; Dieterich, D.T.; Thomas, D.L.; et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006, 43, 1317–1325. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Human Requirement for N-3 Polyunsaturated Fatty Acids. Poult. Sci. 2000, 79, 961–970. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Patel, M.K.; Riley, M.A.; Hobbs, S.; Cortez-Cooper, M.; Robinson, V.J. Can α-Lipoic Acid Mitigate Progression of Aging-Related Decline Caused by Oxidative Stress? South. Med. J. 2014, 107, 780–787. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Bartosz, G. Effect of antioxidants supplementation on aging and longevity. BioMed Res. Int. 2014, 2014, 1–17. [Google Scholar] [CrossRef]
- Lorente-Cebrián, S.; Costa, A.G.V.; Navas-Carretero, S.; Zabala, M.; Martínez, J.A.; Moreno-Aliaga, M.J. Role of omega-3 fatty acids in obesity, metabolic syndrome, and cardiovascular diseases: A review of the evidence. J. Physiol. Biochem. 2013, 69, 633–651. [Google Scholar] [CrossRef]
- Lorente-Cebrián, S.; Costa, A.G.V.; Navas-Carretero, S.; Zabala, M.; Laiglesia, L.M.; Martínez, J.A.; Moreno-Aliaga, M.J. An update on the role of omega-3 fatty acids on inflammatory and degenerative diseases. J. Physiol. Biochem. 2015, 71, 341–349. [Google Scholar] [CrossRef]
- Martínez-Fernández, L.; Laiglesia, L.M.; Huerta, A.E.; Martínez, J.A.; Moreno-Aliaga, M.J. Omega-3 fatty acids and adipose tissue function in obesity and metabolic syndrome. Prostaglandins Other Lipid Mediat. 2015, 121, 24–41. [Google Scholar] [CrossRef]
- Richard, D.; Kefi, K.; Barbe, U.; Bausero, P.; Visioli, F. Polyunsaturated fatty acids as antioxidants. Pharmacol. Res. 2008, 57, 451–455. [Google Scholar] [CrossRef]
- Pettinelli, P.; del Pozo, T.; Araya, J.; Rodrigo, R.; Araya, A.V.; Smok, G.; Csendes, A.; Gutierrez, L.; Rojas, J.; Korn, O.; et al. Enhancement in liver SREBP-1c/PPAR-α ratio and steatosis in obese patients: Correlations with insulin resistance and n-3 long-chain polyunsaturated fatty acid depletion. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 1080–1086. [Google Scholar] [CrossRef]
- Spadaro, L.; Magliocco, O.; Spampinato, D.; Piro, S.; Oliveri, C.; Alagona, C.; Papa, G.; Rabuazzo, A.M.; Purrello, F. Effects of n-3 polyunsaturated fatty acids in subjects with nonalcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 194–199. [Google Scholar] [CrossRef]
- Zúñiga, J.; Cancino, M.; Medina, F.; Varela, P.; Vargas, R.; Tapia, G. N-3 PUFAS supplementation triggers PPAR-α activation and PPAR-α/NF-κB interaction: Anti-inflammatory implications in liver ischemia-reperfusion injury. PLoS ONE 2011, 6, e28502. [Google Scholar] [CrossRef]
- Nenseter, M.S.; Drevon, C.A. Dietary polyunsaturates and peroxidation of low density lipoprotein. Curr. Opin. Lipidol. 1996, 7, 8–13. [Google Scholar] [CrossRef]
- Liu, J.; Yeo, H.C.; Doniger, S.J.; Ames, B.N. Assay of aldehydes from lipid peroxidation: Gas chromatography-mass spectrometry compared to thiobarbituric acid. Anal. Biochem. 1997, 245, 161–166. [Google Scholar] [CrossRef]
- Moreno-Aliaga, M.J.; Lorente-Cebrián, S.; Martínez, J.A. Regulation of adipokine secretion by n-3 fatty acids. Proc. Nutr. Soc. 2010, 69, 324–332. [Google Scholar] [CrossRef]
- Hernandez-Rodas, M.C.; Valenzuela, R.; Videla, L.A. Relevant aspects of nutritional and dietary interventions in non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2015, 16, 25168–25198. [Google Scholar] [CrossRef]
- Softic, S.; Gupta, M.K.; Wang, G.-X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Investig. 2017, 127, 4059–4074. [Google Scholar] [CrossRef]
- Yang, Z.H.; Miyahara, H.; Takeo, J.; Katayama, M. Diet high in fat and sucrose induces rapid onset of obesity-related metabolic syndrome partly through rapid response of genes involved in lipogenesis, insulin signalling and inflammation in mice. Diabetol. Metab. Syndr. 2012, 4, 1–10. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Ter Horst, K.W.; Serlie, M.J. Fructose consumption, lipogenesis, and non-alcoholic fatty liver disease. Nutrients 2017, 9, 981. [Google Scholar] [CrossRef]
- Crescenzo, R.; Bianco, F.; Falcone, I.; Coppola, P.; Liverini, G.; Iossa, S. Increased hepatic de novo lipogenesis and mitochondrial efficiency in a model of obesity induced by diets rich in fructose. Eur. J. Nutr. 2013, 52, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Aigner, E.; Theurl, I.; Haufe, H.; Seifert, M.; Hohla, F.; Scharinger, L.; Stickel, F.; Mourlane, F.; Weiss, G.; Datz, C. Copper Availability Contributes to Iron Perturbations in Human Nonalcoholic Fatty Liver Disease. Gastroenterology 2008, 135, 680–688. [Google Scholar] [CrossRef]
- Aigner, E.; Strasser, M.; Haufe, H.; Sonnweber, T.; Hohla, F.; Stadlmayr, A.; Solioz, M.; Tilg, H.; Patsch, W.; Weiss, G.; et al. A Role for Low Hepatic Copper Concentrations in Nonalcoholic Fatty Liver Disease. Am. J. Gastroenterol. 2010, 105, 1978–1985. [Google Scholar] [CrossRef]
- Aigner, E.; Weiss, G.; Datz, C. Dysregulation of iron and copper homeostasis in nonalcoholic fatty liver. World J. Hepatol. 2014, 7, 177. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.H.; Guan, B.J.; Gao, H.Y.; Peng, X.E. Omega-3 polyunsaturated fatty acid supplementation and non-alcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Medicine 2018, 97, e12271. [Google Scholar] [CrossRef] [PubMed]
- Musa-Veloso, K.; Venditti, C.; Lee, H.Y.; Darch, M.; Floyd, S.; West, S.; Simon, R. Systematic review and meta-analysis of controlled intervention studies on the effectiveness of long-chain omega-3 fatty acids in patients with nonalcoholic fatty liver disease. Nutr. Rev. 2018, 76, 581–602. [Google Scholar] [CrossRef]
- Yu, L.; Yuan, M.; Wang, L. The effect of omega-3 unsaturated fatty acids on non-alcoholic fatty liver disease: A systematic review and meta-analysis of RCTs. Pak. J. Med. Sci. 2017, 33, 1022–1028. [Google Scholar] [CrossRef]
- He, X.X.; Wu, X.L.; Chen, R.P.; Chen, C.; Liu, X.G.; Wu, B.J.; Huang, Z.M. Effectiveness of Omega-3 Polyunsaturated Fatty Acids in Non-Alcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2016, 11, e0162368. [Google Scholar] [CrossRef]
- Lu, W.; Li, S.; Li, J.; Wang, J.; Zhang, R.; Zhou, Y.; Yin, Q.; Zheng, Y.; Wang, F.; Xia, Y.; et al. Effects of Omega-3 Fatty Acid in Nonalcoholic Fatty Liver Disease: A Meta-Analysis. Gastroenterol. Res. Pract. 2016, 2016, 1459790. [Google Scholar] [CrossRef]
- Scorletti, E.; Byrne, C.D. Omega-3 fatty acids and non-alcoholic fatty liver disease: Evidence of efficacy and mechanism of action. Mol. Asp. Med. 2018, 64, 135–146. [Google Scholar] [CrossRef]
- Kris-Etherton, P.; Taylor, D.S.; Yu-Poth, S.; Huth, P.; Moriarty, K.; Fishell, V.; Hargrove, R.L.; Zhao, G.; Etherton, T.D. Polyunsaturated fatty acids in the food chain in the United States. Am. J. Clin. Nutr. 2000, 71, 179S–188S. [Google Scholar] [CrossRef]
- Gerster, H. Can adults adequately convert alpha-linolenic acid (18:3n-3) to eicosapentaenoic acid (20:5n-3) and docosahexaenoic acid (22:6n-3)? Int. J. Vitam. Nutr. Res. 1998, 68, 159–173. [Google Scholar] [PubMed]
- Meyer, B.J.; Mann, N.J.; Lewis, J.L.; Milligan, G.C.; Sinclair, A.J.; Howe, P.R.C. Dietary intakes and food sources of omega-6 and omega-3 polyunsaturated fatty acids. Lipids 2003, 38, 391–398. [Google Scholar] [CrossRef]
- Elmadfa, I.; Kornsteiner, M. Fats and fatty acid requirements for adults. Ann. Nutr. Metab. 2009, 55, 56–75. [Google Scholar] [CrossRef] [PubMed]
- Kris-Etherton, P.M.; Harris, W.S.; Appel, L.J. Fish Consumption, Fish Oil, Omega-3 Fatty Acids, and Cardiovascular Disease. Circulation 2002, 106, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. Omega-3 fatty acids in health and disease and in growth and development. Am. J. Clin. Nutr. 1991, 54, 438–463. [Google Scholar] [CrossRef]
- Sperling, R.I.; Austen, K.F. Effects of Omega-3 Polyunsaturated Fatty Acids on Human Leukocyte Function and Biochemistry. In Health Effects of Polyunsaturated Fatty Acids in Seafoods; Springer: Berlin/Heidelberg, Germany, 1987; pp. 227–238. [Google Scholar]
- Seki, H.; Tani, Y.; Arita, M. Omega-3 PUFAS derived anti-inflammatory lipid mediator resolvin E1. Prostaglandins Other Lipid Mediat. 2009, 89, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L. Dietary n-6 and n-3 polyunsaturated fatty acids: From biochemistry to clinical implications in cardiovascular prevention. Biochem. Pharmacol. 2009, 77, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B.; Lytle, K.A.; Depner, C.M.; Tripathy, S. Omega-3 polyunsaturated fatty acids as a treatment strategy for nonalcoholic fatty liver disease. Pharmacol. Ther. 2018, 181, 108–125. [Google Scholar] [CrossRef]
- George, E.S.; Forsyth, A.; Itsiopoulos, C.; Nicoll, A.J.; Ryan, M.; Sood, S.; Roberts, S.K.; Tierney, A.C. Practical dietary recommendations for the prevention andmanagement of nonalcoholic fatty liver disease in adults. Adv. Nutr. 2018, 9, 30–40. [Google Scholar] [CrossRef] [PubMed]
- De Lorgeril, M.; Renaud, S.; Salen, P.; Monjaud, I.; Mamelle, N.; Martin, J.L.; Guidollet, J.; Touboul, P.; Delaye, J. Mediterranean alpha-linolenic acid-rich diet in secondary prevention of coronary heart disease. Lancet 1994, 343, 1454–1459. [Google Scholar] [CrossRef]
- Ambring, A.; Johansson, M.; Axelsen, M.; Gan, L.M.; Strandvik, B.; Friberg, P. Mediterranean-inspired diet lowers the ratio of serum phospholipid n-6 to n-3 fatty acids, the number of leukocytes and platelets, and vascular endothelial growth factor in healthy subjects. Am. J. Clin. Nutr. 2006, 83, 575–581. [Google Scholar] [CrossRef]
- Leclercq, I.A.; Molendi-Coste, O.; Legry, V. Why and how meet n-3 PUFA dietary recommendations? Gastroenterol. Res. Pract. 2011, 2011, 364040. [Google Scholar]
- Simopoulos, A.P. An increase in the Omega-6/Omega-3 fatty acid ratio increases the risk for obesity. Nutrients 2016, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, R.; Videla, L.A. The importance of the long-chain polyunsaturated fatty acid n-6/n-3 ratio in development of non-alcoholic fatty liver associated with obesity. Food Funct. 2011, 2, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Systems approach to inflammation resolution: Identification of novel anti-inflammatory and pro-resolving mediators. J. Thromb. Haemost. 2009, 7, 44–48. [Google Scholar] [CrossRef]
- Rius, B.; Duran-Güell, M.; Flores-Costa, R.; López-Vicario, C.; Lopategi, A.; Alcaraz-Quiles, J.; Casulleras, M.; José Lozano, J.; Titos, E.; Clària, J. The specialized proresolving lipid mediator maresin 1 protects hepatocytes from lipotoxic and hypoxia-induced endoplasmic reticulum stress. FASEB J. 2017, 31, 5384–5398. [Google Scholar] [CrossRef]
- Martínez-Fernández, L.; González-Muniesa, P.; Laiglesia, L.M.; Sáinz, N.; Prieto-Hontoria, P.L.; Escoté, X.; Odriozola, L.; Corrales, F.J.; Arbones-Mainar, J.M.; Martínez, J.A.; et al. Maresin 1 improves insulin sensitivity and attenuates adipose tissue inflammation in ob/ob and diet-induced obese mice. FASEB J. 2017, 31, 2135–2145. [Google Scholar] [CrossRef]
- Laiglesia, L.M.; Lorente-Cebrián, S.; Martínez-Fernández, L.; Sáinz, N.; Prieto-Hontoria, P.L.; Burrell, M.A.; Rodríguez-Ortigosa, C.M.; Martínez, J.A.; Moreno-Aliaga, M.J. Maresin 1 mitigates liver steatosis in ob/ob and diet-induced obese mice. Int. J. Obes. 2018, 42, 572–579. [Google Scholar] [CrossRef]
- Rius, B.; Titos, E.; Morán-Salvador, E.; López-Vicario, C.; García-Alonso, V.; González-Périz, A.; Arroyo, V.; Clària, J. Resolvin D1 primes the resolution process initiated by calorie restriction in obesity-induced steatohepatitis. FASEB J. 2014, 28, 836–848. [Google Scholar] [CrossRef]
- Barden, A.; Mas, E.; Croft, K.D.; Phillips, M.; Mori, T.A. Short-term n-3 fatty acid supplementation but not aspirin increases plasma proresolving mediators of inflammation. J. Lipid Res. 2014, 55, 2401–2407. [Google Scholar] [CrossRef]
- Jung, T.W.; Kim, H.C.; El-Aty, A.M.A.; Jeong, J.H. Maresin 1 attenuates NAFLD by suppression of endoplasmic reticulum stress via AMPK-SERCA2b pathway. J. Biol. Chem. 2018, 293, 3981–3988. [Google Scholar] [CrossRef]
- González-Périz, A.; Planagumà, A.; Gronert, K.; Miquel, R.; López-Parra, M.; Titos, E.; Horrillo, R.; Ferré, N.; Deulofeu, R.; Arroyo, V.; et al. Docosahexaenoic acid (DHA) blunts liver injury by conversion to protective lipid mediators: Protectin D1 and 17S-hydroxy-DHA. FASEB J. 2006, 20, 2537–2539. [Google Scholar] [CrossRef]
- González-Périz, A.; Horrillo, R.; Ferré, N.; Gronert, K.; Dong, B.; Morán-Salvador, E.; Titos, E.; Martínez-Clemente, M.; López-Parra, M.; Arroyo, V.; et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by ω-3 fatty acids: A role for resolvins and protectins. FASEB J. 2009, 23, 1946–1957. [Google Scholar] [CrossRef]
- Neuhofer, A.; Zeyda, M.; Mascher, D.; Itariu, B.K.; Murano, I.; Leitner, L.; Hochbrugger, E.E.; Fraisl, P.; Cinti, S.; Serhan, C.N.; et al. Impaired local production of proresolving lipid mediators in obesity and 17-HDHA as a potential treatment for obesity-associated inflammation. Diabetes 2013, 62, 1945–1956. [Google Scholar] [CrossRef]
- Itariu, B.K.; Zeyda, M.; Hochbrugger, E.E.; Neuhofer, A.; Prager, G.; Schindler, K.; Bohdjalian, A.; Mascher, D.; Vangala, S.; Schranz, M.; et al. Long-chain n-3 PUFAs reduce adipose tissue and systemic inflammation in severely obese nondiabetic patients: A randomized controlled trial. Am. J. Clin. Nutr. 2012, 96, 1137–1149. [Google Scholar] [CrossRef]
- Mori, T.A.; Dunstan, D.W.; Burke, V.; Croft, K.D.; Rivera, J.H.; Beilin, L.J.; Puddey, I.B. Effect of dietary fish and exercise training on urinary F2-isoprostane excretion in non-insulin-dependent diabetic patients. Metabolism 1999, 48, 1402–1408. [Google Scholar] [CrossRef]
- Higdon, J.V.; Liu, J.; Du, S.-H.; Morrow, J.D.; Ames, B.N.; Wander, R.C. Supplementation of postmenopausal women with fish oil rich in eicosapentaenoic acid and docosahexaenoic acid is not associated with greater in vivo lipid peroxidation compared with oils rich in oleate and linoleate as assessed by plasma malondialdehyde and F2-isoprostanes. Am. J. Clin. Nutr. 2000, 72, 714–722. [Google Scholar]
- Fayh, A.P.T.; Borges, K.; Cunha, G.S.; Krause, M.; Rocha, R.; de Bittencourt, P.I.H.; Moreira, J.C.F.; Friedman, R.; da Silva Rossato, J.; Fernandes, J.R.; et al. Effects of n-3 fatty acids and exercise on oxidative stress parameters in type 2 diabetic: A randomized clinical trial. J. Int. Soc. Sports Nutr. 2018, 15, 1–9. [Google Scholar] [CrossRef]
- Mori, T.A.; Puddey, I.B.; Burke, V.; Croft, K.D.; Dunstan, D.W.; Rivera, J.H.; Beilin, L.J. Effect of ω3 fatty acids on oxidative stress in humans: GC–MS measurement of urinary F2-isoprostane excretion. Redox Rep. 2000, 5, 45–46. [Google Scholar] [CrossRef]
- Cazzola, R.; Russo-Volpe, S.; Miles, E.A.; Rees, D.; Banerjee, T.; Roynette, C.E.; Wells, S.J.; Goua, M.; Wahle, K.W.J.; Calder, P.C.; et al. Age- and dose-dependent effects of an eicosapentaenoic acid-rich oil on cardiovascular risk factors in healthy male subjects. Atherosclerosis 2007, 193, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Mas, E.; Woodman, R.J.; Burke, V.; Puddey, I.B.; Beilin, L.J.; Durand, T.; Mori, T.A. The omega-3 fatty acids EPA and DHA decrease plasma F2- isoprostanes: Results from two placebo-controlled interventions. Free Radic. Res. 2010, 44, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Stahl, F.; Mutz, K.-O.; Scheper, T.; Hahn, A.; Schuchardt, J.P. Transcriptome-based identification of antioxidative gene expression after fish oil supplementation in normo- and dyslipidemic men. Nutr. Metab. 2012, 9, 45. [Google Scholar] [CrossRef]
- Kiecolt-Glaser, J.K.; Epel, E.S.; Belury, M.A.; Andridge, R.; Lin, J.; Glaser, R.; Malarkey, W.B.; Hwang, B.S.; Blackburn, E. Omega-3 fatty acids, oxidative stress, and leukocyte telomere length: A randomized controlled trial. Brain Behav. Immun. 2013, 28, 16–24. [Google Scholar] [CrossRef]
- Hajianfar, H.; Paknahad, Z.; Bahonar, A. The Effect of Omega-3 Supplements on Antioxidant Capacity in Patients with Type 2 Diabetes. Int. J. Prev. Med. 2013, 4, S234–S238. [Google Scholar] [PubMed]
- Véricel, E.; Colas, R.; Calzada, C.; Lê, Q.H.; Feugier, N.; Cugnet, C.; Vidal, H.; Laville, M.; Moulin, P.; Lagarde, M. Moderate oral supplementation with docosahexaenoic acid improves platelet function and oxidative stress in type 2 diabetic patients. Thromb. Haemost. 2015, 114, 289–296. [Google Scholar] [PubMed]
- Berge, R.K.; Ramsvik, M.S.; Bohov, P.; Svardal, A.; Nordrehaug, J.E.; Rostrup, E.; Bruheim, I.; Bjørndal, B. Krill oil reduces plasma triacylglycerol level and improves related lipoprotein particle concentration, fatty acid composition and redox status in healthy young adults—A pilot study. Lipids Health Dis. 2015, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Meydani, M.; Natiello, F.; Goldin, B.; Free, N.; Woods, M.; Schaefer, E.; Blumberg, J.B.; Gorbach, S.L. Effect of long-term fish oil supplementation on vitamin E status and lipid peroxidation in women. J. Nutr. 1991, 121, 484–491. [Google Scholar] [CrossRef]
- Harats, D.; Dabach, Y.; Hollander, G.; Ben-Naim, M.; Schwartz, R.; Berry, E.M.; Stein, O.; Stein, Y. Fish oil ingestion in smokers and nonsmokers enhances peroxidation of plasma lipoproteins. Atherosclerosis 1991, 90, 127–139. [Google Scholar] [CrossRef]
- Wander, R.C.; Du, S.-H. Oxidation of plasma proteins is not increased after supplementation with eicosapentaenoic and docosahexaenoic acids. Am. J. Clin. Nutr. 2000, 72, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Egert, S.; Somoza, V.; Kannenberg, F.; Fobker, M.; Krome, K.; Erbersdobler, H.F.; Wahrburg, U. Influence of three rapeseed oil-rich diets, fortified with alpha-linolenic acid, eicosapentaenoic acid or docosahexaenoic acid on the composition and oxidizability of low-density lipoproteins: Results of a controlled study in healthy volunteers. Eur. J. Clin. Nutr. 2007, 61, 314–325. [Google Scholar] [CrossRef]
- Bloomer, R.J.; Larson, D.E.; Fisher-Wellman, K.H.; Galpin, A.J.; Schilling, B.K. Effect of eicosapentaenoic and docosahexaenoic acid on resting and exercise-induced inflammatory and oxidative stress biomarkers: A randomized, placebo controlled, cross-over study. Lipids Health Dis. 2009, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Alves Luzia, L.; Mendes Aldrighi, J.; Teixeira Damasceno, N.R.; Rodrigues Sampaio, G.; Aparecida Manólio Soares, R.; Tande Silva, I.; De Queiroz Mello, A.P.; Ferreira Carioca, A.A.; Ferraz da Silva Torres, E.A. Fish Oil and Vitamin E Change Lipid Profiles and Anti-Ldl-Antibodies in Two Different Ethnic Groups of Women Transitioning through Menopause. Nutr. Hosp. 2015, 32, 165–174. [Google Scholar] [PubMed]
- Nenseter, M.S.; Rustan, A.C.; Lund-Katz, S.; Søyland, E.; Maelandsmo, G.; Phillips, M.C.; Drevon, C.A. Effect of dietary supplementation with n-3 polyunsaturated fatty acids on physical properties and metabolism of low density lipoprotein in humans. Arterioscler. Thromb. 1992, 12, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Frankel, E.N.; Parks, E.J.; Xu, R.; Schneeman, B.O.; Davis, P.A.; German, J.B. Effect of n-3 fatty acid-rich fish oil supplementation on the oxidation of low density lipoproteins. Lipids 1994, 29, 233–236. [Google Scholar] [CrossRef]
- Brude, I.R.; Drevon, C.A.; Hjermann, I.; Seljeflot, I.; Lund-Katz, S.; Saarem, K.; Sandstad, B.; Solvoll, K.; Halvorsen, B.; Arnesen, H.; et al. Peroxidation of LDL from combined-hyperlipidemic male smokers supplied with omega-3 fatty acids and antioxidants. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2576–2588. [Google Scholar] [CrossRef]
- Wu, W.H.; Lu, S.C.; Wang, T.F.; Jou, H.J.; Wang, T.A. Effects of docosahexaenoic acid supplementation on blood lipids, estrogen metabolism, and in vivo oxidative stress in postmenopausal vegetarian women. Eur. J. Clin. Nutr. 2006, 60, 386–392. [Google Scholar] [CrossRef]
- Hanwell, H.E.C.; Kay, C.D.; Lampe, J.W.; Holub, B.J.; Duncan, A.M. Acute Fish Oil and Soy Isoflavone Supplementation Increase Postprandial Serum (n-3) Polyunsaturated Fatty Acids and Isoflavones but Do Not Affect Triacylglycerols or Biomarkers of Oxidative Stress in Overweight and Obese Hypertriglyceridemic Men. J. Nutr. 2009, 139, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Petersson, H.; Risérus, U.; McMonagle, J.; Gulseth, H.L.; Tierney, A.C.; Morange, S.; Helal, O.; Shaw, D.I.; Ruano, J.A.; López-Miranda, J.; et al. Effects of dietary fat modification on oxidative stress and inflammatory markers in the LIPGENE study. Br. J. Nutr. 2010, 104, 1357–1362. [Google Scholar] [CrossRef][Green Version]
- Ulven, S.M.; Kirkhus, B.; Lamglait, A.; Basu, S.; Elind, E.; Haider, T.; Berge, K.; Vik, H.; Pedersen, J.I. Metabolic effects of krill oil are essentially similar to those of fish oil but at lower dose of EPA and DHA, in healthy volunteers. Lipids 2011, 46, 37–46. [Google Scholar] [CrossRef]
- Ottestad, I.; Vogt, G.; Retterstøl, K.; Myhrstad, M.C.; Haugen, J.E.; Nilsson, A.; Ravn-Haren, G.; Nordvi, B.; Brønner, K.W.; Andersen, L.F.; et al. Oxidised fish oil does not influence established markers of oxidative stress in healthy human subjects: A randomised controlled trial. Br. J. Nutr. 2012, 108, 315–326. [Google Scholar] [CrossRef]
- Egert, S.; Lindenmeier, M.; Harnack, K.; Krome, K.; Erbersdobler, H.F.; Wahrburg, U.; Somoza, V. Margarines fortified with α-linolenic acid, eicosapentaenoic acid, or docosahexaenoic acid alter the fatty acid composition of erythrocytes but do not affect the antioxidant status of healthy adults. J. Nutr. 2012, 142, 1638–1644. [Google Scholar] [CrossRef] [PubMed]
- Kirkhus, B.; Lamglait, A.; Eilertsen, K.E.; Falch, E.; Haider, T.; Vik, H.; Hoem, N.; Hagve, T.A.; Basu, S.; Olsen, E.; et al. Effects of similar intakes of marine n-3 fatty acids from enriched food products and fish oil on cardiovascular risk markers in healthy human subjects. Br. J. Nutr. 2012, 107, 1339–1349. [Google Scholar] [CrossRef]
- Knasmüller, S.; Nersesyan, A.; Misík, M.; Gerner, C.; Mikulits, W.; Ehrlich, V.; Hoelzl, C.; Szakmary, A.; Wagner, K.H. Use of conventional and -omics based methods for health claims of dietary antioxidants: A critical overview. Br. J. Nutr. 2008, 99, ES3–ES52. [Google Scholar] [CrossRef] [PubMed]
- Dreissigacker, U.; Suchy, M.T.; Maassen, N.; Tsikas, D. Human plasma concentrations of malondialdehyde (MDA) and the F2-isoprostane 15(S)-8-iso-PGF2α may be markedly compromised by hemolysis: Evidence by GC-MS/MS and potential analytical and biological ramifications. Clin. Biochem. 2010, 43, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, J.; Barden, A.; Mori, T.A.; Burke, V.; Croft, K.D.; Beilin, L.J.; Puddey, I.B. Measurement of urinary F2-Isoprostanes as markers of in vivo lipid peroxidation—A comparison of enzyme immunoassay with gas chromatography/mass spectrometry. Anal. Biochem. 1999, 272, 209–215. [Google Scholar] [CrossRef]
- Smith, K.A.; Shepherd, J.; Wakil, A.; Kilpatrick, E.S. A comparison of methods for the measurement of 8-iso- PGF(2alpha): A marker of oxidative stress. Ann. Clin. Biochem. 2011, 48, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef]
- Verhoye, E.; Langlois, M.R.; Asklepios Investigators. Circulating oxidized low-density lipoprotein: A biomarker of atherosclerosis and cardiovascular risk? Clin. Chem. Lab. Med. 2009, 47, 128–137. [Google Scholar] [CrossRef]
- Kalousová, M.; Skrha, J.; Zima, T. Advanced glycation end-products and advanced oxidation protein products in patients with diabetes mellitus. Physiol. Res. 2002, 51, 597–604. [Google Scholar] [PubMed]
- Du, S.L.; Zeng, X.Z.; Tian, J.W.; Ai, J.; Wan, J.; He, J.X. Advanced oxidation protein products in predicting acute kidney injury following cardiac surgery. Biomarkers 2015, 20, 206–211. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Tsikas, D.; Rossi, R. Oxidative stress and human diseases: Origin, link, measurement, mechanisms, and biomarkers. Crit. Rev. Clin. Lab. Sci. 2009, 46, 241–281. [Google Scholar] [CrossRef] [PubMed]
- Tsikas, D.; Rothmann, S.; Schneider, J.Y.; Suchy, M.T.; Trettin, A.; Modun, D.; Stuke, N.; Maassen, N.; Frölich, J.C. Development, validation and biomedical applications of stable-isotope dilution GC-MS and GC-MS/MS techniques for circulating malondialdehyde (MDA) after pentafluorobenzyl bromide derivatization: MDA as a biomarker of oxidative stress and its relation to 15(S)-8-iso-prostaglandin F2α and nitric oxide (NO). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1019, 95–111. [Google Scholar]
- Fenton, J.I.; Hord, N.G.; Ghosh, S.G. Long chain omega-3 fatty acid immunomodulation and the potencial for adverse healthoutcomes. Prostaglandins Leukot. Essent. Fat. Acids 2014, 89, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Robinson, K.A.; Gabbita, S.P.; Salsman, S.; Floyd, R.A. Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 2000, 28, 1456–1462. [Google Scholar] [CrossRef]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative Stress in Obesity: A Critical Component in Human Diseases. Int. J. Mol. Sci. 2014, 16, 378–400. [Google Scholar] [CrossRef]
- Adkins, Y.; Kelley, D.S. Mechanisms underlying the cardioprotective effects of omega-3 polyunsaturated fatty acids. J. Nutr. Biochem. 2010, 21, 781–792. [Google Scholar] [CrossRef]
- Capanni, M.; Calella, F.; Biagini, M.R.; Genise, S.; Raimondi, L.; Bedogni, G.; Svegliati-Baroni, G.; Sofi, F.; Milani, S.; Abbate, R.; et al. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: A pilot study. Aliment. Pharmacol. Ther. 2006, 23, 1143–1151. [Google Scholar] [CrossRef]
- Zhu, F.S.; Liu, S.; Chen, X.M.; Huang, Z.G.; Zhang, D.W. Effects of n-3 polyunsaturated fatty acids from seal oils on nonalcoholic fatty liver disease associated with hyperlipidemia. World J. Gastroenterol. 2008, 14, 6395–6400. [Google Scholar] [CrossRef]
- Tanaka, N.; Sano, K.; Horiuchi, A.; Tanaka, E.; Kiyosawa, K.; Aoyama, T. Highly purified eicosapentaenoic acid treatment improves nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2008, 42, 413–418. [Google Scholar] [CrossRef]
- Sofi, F.; Giangrandi, I.; Cesari, F.; Corsani, I.; Abbate, R.; Gensini, G.F.; Casini, A. Effects of a 1-year dietary intervention with n-3 polyunsaturated fatty acid-enriched olive oil on non-alcoholic fatty liver disease patients: A preliminary study. Int. J. Food Sci. Nutr. 2010, 61, 792–802. [Google Scholar] [CrossRef]
- Scorletti, E.; Bhatia, L.; Mccormick, K.G.; Clough, G.F.; Nash, K.; Hodson, L.; Moyses, H.E.; Calder, P.C.; Byrne, C.D. Effects of purified eicosapentaenoic and docosahexaenoic acids in nonalcoholic fatty liver disease: Results from the WELCOME* study. Hepatology 2014, 60, 1211–1221. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Abdelmalek, M.F.; Suzuki, A.; Cummings, O.W.; Chojkier, M. No significant effects of ethyl-eicosapentanoic acid on histologic features of nonalcoholic steatohepatitis in a phase 2 triaL. Gastroenterology 2014, 147, 377–384. [Google Scholar] [CrossRef]
- Li, Y.H.; Yang, L.H.; Sha, K.H.; Liu, T.G.; Zhang, L.G.; Liu, X.X. Efficacy of poly-unsaturated fatty acid therapy on patients with nonalcoholic steatohepatitis. World J. Gastroenterol. 2015, 21, 7008–7013. [Google Scholar] [CrossRef]
- Argo, C.K.; Patrie, J.T.; Lackner, C.; Henry, T.D.; de Lange, E.E.; Weltman, A.L.; Shah, N.L.; Al-Osaimi, A.M.; Pramoonjago, P.; Jayakumar, S.; et al. Effects of N-3 Fish Oil on Metabolic and Histological Parameters in NASH: A Double-Blind, Randomized, Placebo-Controlled Trial. J. Hepatol. 2015, 62, 190–197. [Google Scholar] [CrossRef]
- Qin, Y.; Zhou, Y.; Chen, S.H.; Zhao, X.L.; Ran, L.; Zeng, X.L.; Wu, Y.; Chen, J.L.; Kang, C.; Shu, F.R.; et al. Fish oil supplements lower serum lipids and glucose in correlation with a reduction in plasma fibroblast growth factor 21 and prostaglandin E2 in nonalcoholic fatty liver disease associated with hyperlipidemia: A randomized clinical trial. PLoS ONE 2015, 10, e0133496. [Google Scholar] [CrossRef]
- Dasarathy, S.; Dasarathy, J.; Khiyami, A.; Yerian, L.; Hawkins, C.; Sargent, R.; McCullough, A.J. Double-blind randomized placebo-controlled clinical trial of omega 3 fatty acids for the treatment of diabetic patients with nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2015, 49, 137–144. [Google Scholar] [CrossRef]
- Nogueira, M.A.; Oliveira, C.P.; Ferreira Alves, V.A.; Stefano, J.T.; Rodrigues, L.S.; Torrinhas, R.S.; Cogliati, B.; Barbeiro, H.; Carrilho, F.J.; Waitzberg, D.L. Omega-3 polyunsaturated fatty acids in treating non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled trial. Clin. Nutr. 2016, 35, 578–586. [Google Scholar] [CrossRef]
- Tobin, D.; Brevik-Andersen, M.; Qin, Y.; Innes, J.K.; Calder, P.C. Evaluation of a High Concentrate Omega-3 for Correcting the Omega-3 Fatty Acid Nutritional Deficiency in Non-Alcoholic Fatty Liver Disease (CONDIN). Nutrients 2018, 10, 1126. [Google Scholar] [CrossRef]
- Carpentier, Y.A.; Portois, L.; Malaisse, W.J. n-3 fatty acids and the metabolic syndrome. Am. J. Clin. Nutr. 2006, 83, 1499S–1504S. [Google Scholar] [CrossRef]
- Satapathy, S.K.; Garg, S.; Chauhan, R.; Sakhuja, P.; Malhotra, V.; Sharma, B.C.; Sarin, S.K. Beneficial effects of tumor necrosis factor-α inhibition by pentoxifylline on clinical, biochemical, and metabolic parameters of patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2004, 99, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Jacobs, D.R. Association between serum gamma-glutamyltransferase and C-reactive protein. Atherosclerosis 2005, 178, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Marcellini, M.; Giannone, G.; Nobili, V. Correlation of serum TNF-α levels and histologic liver injury scores in pediatric nonalcoholic fatty liver disease. Am. J. Clin. Pathol. 2007, 127, 954–960. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Candelaresi, C.; Saccomanno, S.; Ferretti, G.; Bachetti, T.; Marzioni, M.; De Minicis, S.; Nobili, L.; Salzano, R.; Omenetti, A.; et al. A model of insulin resistance and nonalcoholic steatohepatitis in rats: Role of peroxisome proliferator-activated receptor-α and n-3 polyunsaturated fatty acid treatment on liver injury. Am. J. Pathol. 2006, 169, 846–860. [Google Scholar] [CrossRef]
- Maratos-Flier, E. Fatty liver and FGF21 physiology. Exp. Cell Res. 2017, 360, 2–5. [Google Scholar] [CrossRef]
- Scorletti, E.; Byrne, C.D. Omega-3 Fatty Acids, Hepatic Lipid Metabolism, and Nonalcoholic Fatty Liver Disease. Annu. Rev. Nutr. 2013, 33, 231–248. [Google Scholar] [CrossRef]
- Nobili, V.; Carpino, G.; Alisi, A.; De Vito, R.; Franchitto, A.; Alpini, G.; Onori, P.; Gaudio, E. Role of docosahexaenoic acid treatment in improving liver histology in pediatric nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e88005. [Google Scholar] [CrossRef]
- Pacifico, L.; Bonci, E.; Di Martino, M.; Versacci, P.; Andreoli, G.; Silvestri, L.M.; Chiesa, C. A double-blind, placebo-controlled randomized trial to evaluate the efficacy of docosahexaenoic acid supplementation on hepatic fat and associated cardiovascular risk factors in overweight children with nonalcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 734–741. [Google Scholar] [CrossRef]
- Wang, J.Z.; Cao, H.X.; Chen, J.N.; Pan, Q. PNPLA3 rs738409 underlies treatment response in nonalcoholic fatty liver disease. World J. Clin. Cases 2018, 6, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Okada, L.S.D.R.R.; Oliveira, C.P.; Stefano, J.T.; Nogueira, M.A.; Silva, I.D.C.G.D.; Cordeiro, F.B.; Alves, V.A.F.; Torrinhas, R.S.; Carrilho, F.J.; Puri, P.; et al. Omega-3 PUFA modulate lipogenesis, ER stress, and mitochondrial dysfunction markers in NASH—Proteomic and lipidomic insight. Clin. Nutr. 2018, 37, 1474–1484. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| NAFLD Index | Predictors | Hallmarks | Interpretation |
|---|---|---|---|
| Fatty liver index (FLI) | Fatty Liver Index (FLI) = ey / (1 + ey) × 100. Where y = 0.953 × ln(triglycerides, mg/dL) + 0.139 × BMI, kg/m2 + 0.718 × ln (GGT, U/L) + 0.053 × WC, cm − 15.745) [95] | Identified NAFLD and the optimal cut-off point with accuracy. | FLI < 30, no FL (with a negative likelihood ratio of up to 0.2); 60 < FLI < 30, Inconclusive FLI ≥ 60, FL present (with a likelihood ratio starting from 4.3) |
| Lipid accumulation product (LAP) | LAP for men = (WC [cm]–65) × (TG concentration [mmol/L]) LAP for women = (WC [cm] − 58) × (TG concentration [mmol/L]) [96] | Associated with the presence and severity of NAFLD, among young and aged population [97,98]. NOT able to predict liver fat content [99]. | The optimal cut-off value for LAP was 31.6 with sensitivity of 88% (95% CI, 77–96%), specificity of 82% (95% CI, 76–87%) for males and with a sensitivity of 66% (95% CI, 52–78%), specificity of 93% (95% CI, 88–96%) for females. |
| Hepatic steatosis index (HSI) | Hepatic steatosis index (HSI) = 8 × (ALT/AST ratio) + BMI (+2, if female; +2, if diabetes mellitus) [89]. | A simple, efficient screening tool for NAFLD, used for selecting individuals for liver ultrasonography [89]. | At values of < 30.0 or > 36.0, HSI ruled out NAFLD with a sensitivity of 93.1%, or detected NAFLD with a specificity of 92.4%, respectively [89]. |
| The ZJU (Zhejiang University) index | ZJU index = BMI (Kg/m2) + FPG (mmol/L) + TG (mmol/L) + 3 × ALT (IU/L)/AST (IU/L) ratio (+2, if female) [90]. | Confirmed to have significance in terms of diagnosing NAFLD [90]. | At a value of <32.0, the ZJU index could rule out NAFLD with a sensitivity of 92.2%, and at a value of >38.0, the ZJU index could detect NAFLD with a specificity of 93.4% [90]. |
| NAFLD fibrosis score | NAFLD Score = −1.675 + (0.037 × age [years]) + (0.094 × BMI [kg/m2]) + (1.13 × IFG/diabetes [yes = 1, no = 0]) + (0.99 × AST/ALT ratio) − (0.013 × platelet count [×109/L]) − (0.66 × albumin [g/dL]) [92] | Identifies patients without severe fibrosis, comparatively more difficult to estimate [91]. | Low cut-off score (−1.455): advanced fibrosis ruled out with high accuracy (negative predictive value of 93% and 88% in the estimation and validation groups, respectively). High cut-off score (0.676), advanced fibrosis diagnosed with high accuracy (positive predictive value of 90% and 82% in the estimation and validation groups, respectively) [92]. |
| BARD score | Based on AST/ALT ratio, presence of diabetes and BMI [100]. | Identifies patients without severe fibrosis, but easier to estimate and does not have indeterminate results [91]. | BMI ≥28 = 1 point, AAR of ≥0.8 = 2 points, DM = 1 point A score of 2–4 was associated with an OR for advanced fibrosis of 17 (confidence interval 9.2 to 31.9) and a negative predictive value of 96% [100]. |
| FIB-4 index | FIB-4 Score = age ([yr] × AST [U/L])/((PLT [109/L]) × (ALT [U/L])1/2) [101]. | In patients <35 or >65 years old, the score has been shown to be less reliable [94,101]. | At a cut-off of <1.45 in the validation set, the negative predictive value to exclude advanced fibrosis (stage 4–6) was 90% with a sensitivity of 70%. A cut-off of >3.25 had a positive predictive value of 65% and a specificity of 97%. Using these cut-offs, 87% of the 198 patients with FIB-4 values outside 1.45–3.25 would be correctly classified [101]. |
| Reference | Study Design | Population | Intervention | Outcome Measurements | Comments |
|---|---|---|---|---|---|
| Meydani et al., 1991 [170] | Randomized intervention before and after comparison | Young females, aged 51–71; n = 14. Old females, aged 51–71. n = 9 | 1680 mg EPA + 720 mg DHA per day for 3 months | Plasma MDA level | ↑Plasma MDA level |
| Harats et al., 1991 [171] | Randomized parallel clinical trial | Study A: Smokers: Control: BMI: 23.5 ± 1.2, age: 42.6, n = 5 Fish oil: 23.8 ± 0.8, age: 37.4, n = 6 Study B: Smokers Control: BMI: 24.5 ± 1.2, age: 31, n = 3 Fish oil: BMI: 25.0 ± 1.3, age: 29.1, n = 3 Fish oil + VitE: BMI: 23.5 ± 1.2, age: 35.2, n = 4 Study C: Non-smokers, normolipidemic: Control: BMI: 23.7, age: 36.8, n = 8 Fish oil: BMI: 24.7 ± 1.3, age: 41, n = 6 Fish oil + VitE: BMI: 24.5 ± 1.9, age: 38.8, n = 6 | Study A: Fish oil: concentrate (MaxEPA), 10 g/d for 4 weeks. Study B and C, Fish oil: (MaxEPA), 10 g/d for 4 weeks Fish oil (10 g/day) + Vit E (400 mg/d) for 4 weeks | Plasma and LDL TBARS level | 10 g/d of fish oil consumption ↑plasma LDL TBARS level in smokers and non-smokers Vitamin E counteracted the effect of fish oil more effectively in non-smokers |
| Nenseter et al., 1992 [176] | Randomized placebo-controlled parallel clinical trial | Normolipidemic subjects Treatment: women and men, BMI not reported, age: 27–63, n = 12 Control: women and men, BMI not reported, age: 23–70, n = 11 | Treatment: 6 g capsules/d of n-3 PUFA (highly concentrated ethyl esters). Control: 6 g of corn oil Duration: 4 months | Susceptibility of LDL to Lipid peroxides formation | ↔ Lipid peroxides formation |
| Frankel et al., 1994 [177] | Randomized, double-blind, clinical trial | Hypertriglycemic men and women, age, BMI, smoking status not reported. n = 9/group | Control group: fish oil absent from the diet. Supplemented group: 5.1 g of fish oil per day for 6 weeks | LDL oxidative susceptibility | ↔ LDL oxidative susceptibility |
| Brude et al., 1997 [178] | Randomized, double-blind, placebo-controlled parallel clinical trial | Male smokers, hyperlipidemia, aged 40–60, BMI not mentioned. n-3 PUFA capsule group (n = 11), antioxidant group (n = 11), n-3 PUFAS + antioxidants group (n = 11), control oil group (n = 9) | n-3 PUFAS group: 5 g DHA and EPA/d Antioxidants capsule, 75 mg Vit E, 150 mg Vit C, 15 mg β-carotene, and 30 mg coenzyme Q10 per day Control group: 8 g of oil with an FA pattern similar to an ordinary Norwegian diet Lasted for 6 weeks | LDL oxidative susceptibility, lipid peroxides | ↔ LDL oxidative susceptibility, ↔ lipid peroxides |
| Mori et al., 1999 [159] | Randomized, controlled parallel study | 49 untrained and sedentary NIDDM patients. Age: 30–65 y. BMI < 36 kg/m2 | Study 1: Group 1: Low-fat diet (30% of daily energy) (n = 14) Group 2: Low-fat diet + one daily fish meal (3.6 g n-3 PUFA/day) (n = 12) Group 3: Low-fat diet + Moderate exercise (n = 11) Group 4: Low-fat diet + Fish meal + moderate exercise (n = 12) for 8 weeks. | Urine F2- isoprostanes | Urine F2- isoprostanes |
| Higdon et al., 2000 [160] | Randomized blinded, crossover study | Post-menopausal women, aged between 50–75, BMI < 30 kg/m2, non-smokers, n = 15 | Fish oil group: 15 g/d (2.0 g EPA/d and 1.4 g DHA/d) Safflower oil group: 15g/d (10.5 g linoleate/d); Sunflower oil: 15g/d (12.3 g oleate/d) in a 3-treatment crossover trial (5 weeks with a 7-wk washout interval) | Plasma F2-isoprostanes, MDA, and TBARS | In fish oil group: ↓plasma F2-isoprostanes ↓MDA ↑TBARS |
| Wander and Du, 2000 [172] | Randomized crossover study | Post-menopausal women, aged 45–75, BMI < 30 kg/m2, smoking status not reported. n = 46 | Group 1: fish oil (2.5 g EPA and 1.8 g DHA) Group 2: fish oil (2.5 g EPA and 1.8 g DHA) + 100 mg α-tocopheryl acetate Group 3: fish oil (2.5 g EPA and 1.8 g DHA) + 200 mg α-tocopheryl acetate Group 4: fish oil (2.5 g EPA and 1.8 g DHA) + 400 mg α-tocopheryl acetate for 5 weeks (4-period crossover design) | TBARS, protein oxidation | ↑TBARS. Protein oxidation not changed |
| Mori et al., 2000 [162] | Randomized, placebo-controlled parallel study | Overweight, mildly hyperlipidemic men, age: 20–65 y, BMI: 25–30 kg/m2 | Group 1: 4 g/d of purified EPA (n = 19) Group 2: 4 g/d of purified DHA (n = 17) Group 3: 4 g/d of olive oil (n = 20) for 6 weeks | Urine F2- isoprostanes | ↓Urine F2- isoprostanes in the EPA, DHA treatment groups |
| Wu et al., 2006 [179] | Randomized, single-blind, placebo-controlled parallel clinical trial | Post-menopausal vegetarian women, aged <60. Corn oil: n = 13 DHA: n = 14 | Corn oil group: 6 g corn oil/day DHA-rich algae oil group: 2.14 g of DHA/day for 6 weeks | Plasma α-tocopherol, urine F2-isoprostanes | ↔Plasma α-tocopherol, urine F2-isoprostanes |
| Egert et al., 2007 [173] | Randomized parallel controlled study | Healthy men and women, aged: 25.9 ± 6.82; BMI: 22.2 ± 2.95, non-smokers. n = 48 | ALA group: Rapeseed oil +1% of energy of ALA (n = 15) EPA group: Rapeseed oil + 1% of energy of EPA (n = 17) DHA group: Rapeseed oil + 1% of energy of DHA (n = 16) | Ex vivo LDL oxidative susceptibility | EPA and DHA group: ↑ ex vivo LDL oxidative susceptibility |
| Cazzola et al., 2007 [163] | Randomized parallel placebo-controlled intervention | Healthy young men (age: 14–42 y, BMI: 24.1 ± 0.3). n = 93 Healthy old men (age: 53-70 y. BMI: 27.6 ± 0.0). n = 62 | 4 young and 4 older groups: 1: 1.35 g EPA + 0.27 g DHA per day; 2: 2.7 g EPA + 0.54 g DHA per day; 3: 4.05 g EPA + 0.81 g DHA per day; 4: corn oil group Lasted for 12 weeks | Plasma lipid hydroperoxides Lag time of lipoprotein peroxidation | ↓Plasma lipid hydroperoxides ↓Lag time of lipoprotein peroxidation and ↓GSH/Gluthatione in olders |
| Hanwell et al., 2009 [180] | Randomized, double-blind, placebo-controlled crossover clinical trial | Hyper-triglyceridemic, overweight, and obese men; aged > 45, smoking status not reported. n = 10 in total | High-fat, high-fructose meal in all groups: Fish oil group: 7 g of fish oil concentrate (2.8 g EPA and 1.4 g DHA) Isoflavone group: 336 mg NovaSoy (150 mg glycoside isoflavones). Fish oil + isoflavone: 7 g fish oil + 336 mg NovaSoy Placebo group: 7 g corn oil Consumed 4 days separated by 1week wash out. | Lipid peroxides, oxidized LDL, total antioxidant status | ↔ Lipid peroxides, ↔ oxidized LDL, ↔ total antioxidant status |
| Bloomer et al., 2009 [174] | Randomized, double-blind crossover study | Subjects are exercise trained man, non-smokers, no history of cardiometabolic diseases. Age: 25.5 ± 4.8 y. BMI: 24.1 ± 1.6 n = 14 | Intervention group: 2.224 g EPA and 2.208 g DHA per day Control group: same quantity of soybean oil Duration: 6 weeks (with 8-week washout) Supplementation were prior to performing a 60 min treadmill climb using a weighted pack | Blood was collected pre and post exercise and analyzed for a variety of oxidative stress (Protein carbonyls, IgG-autoantibodies, low-density lipoprotein, Malondialdehyde, Hydrogen peroxide and xanthine oxidase activity, Nitric oxide, Whole blood lactate and inflammatory biomarkers | Resting levels: ↓ CRP, ↓TNF- α, ↔ MDA, ↔ Nitric oxide. Exercise: ↑ oxidative biomarkers (mild) |
| Mas et al., 2010 [164] | Randomized, Placebo-controlled intervention | Study A: placebo-controlled intervention (BMI: 25–30), dyslipidemic men, age: 20–54 y, n = 17–20 per group. Study B: hypertensive type 2 diabetic and post-menopausal women, age: 40–75 y, n = 16–18 | In both studies, n-3 PUFA group: 4 g/day of EPA or DHA Control group: Olive oil placebo lasted for 6 weeks | Plasma F2-isoprostanes | ↓ Plasma F2-isoprostanes with n-3 PUFAS supplementation |
| Petersson et al., 2010 [181] | Randomized parallel study | Participants with metabolic syndrome, age: 35–70 y, BMI: 20–40 kg/m2, smokers or non-smokers. Saturated high-fat diet: n = 100 Monosaturated high-fat diet: n = 111 Low-fat diets with n-3 PUFA: n = 100 Low-fat diets with sunflower oil: n = 106 | Saturated high-fat diet (38% E fat): (HSFA: 16% SFA, 12% MUFA and 6% PUFA), Monosaturated high-fat diet (38% E fat): (HMUFA: 8% SFA, 20% MUFA and 6% PUFA) Low-fat (28% E) high-complex carbohydrate diets (LFHCC: 8% SFA, 11% MUFA and 6% PUFA) with 1.24 g/d n-3 PUFA Low-fat (28% E)-high-complex carbohydrate diets (LFHCC: 8% SFA, 11% MUFA and 6% PUFA) with 1g/d high-oleic acid sunflower oil For 12 weeks | Urinary levels of 8-iso-PGF2α and 15-keto-dihydro-PGF2α Serum CRP | ↔ 8-iso-PGF2α ↔ 15-keto-dihydro-PGF2α ↔ Serum CRP |
| Ulven et al., 2011 [182] | Randomized parallel study | Participants with normal or slightly elevated total blood cholesterol and/or triglyceride levels, age: 30–50 y, BMI > 30 kg/m2 | Krill oil group: 3 g/day (EPA + DHA= 543 mg/day) in 6 capsules (n = 36) Fish oil group: 1.8 g/day (EPA + DHA= 864 mg/day) in 3 capsules (n = 40) Control group: no supplementation (n = 37) Duration: 7 weeks | Urine F2-isoprostanes, plasma α-tocopherol | ↔ Urine F2-isoprostanes, ↔ plasma α-tocopherol |
| Egert et al., 2012 [184] | Randomized single-blind parallel | Men and premenopausal women; Age: 19–43 y; BMI < 28 kg/m2, non-smokers | Margarines fortified with 10% weight of EPA, DHA, or ALA EPA group: 2.2 g/day (n = 25). DHA group: 2.3 g/day (n = 25) ALA group: 4.4 g/day (n = 24) For 6 weeks | Antioxidant capacity, plasma MDA, RBC-MDA, linoleic acid hydroperoxides (LA-OOH) in RBC | ↔ Antioxidant capacity ↑Plasma MDA in EPA and DHA groups. ↔ RBC-MDA ↓ RBC-LA-OOH |
| Kirkhus et al., 2012 [185] | Open, randomized parallel study | 159 healthy men and women. Age: 18–70 y, BMI < 30 kg/m2, moderate smokers | 1g/day of EPA + DHA as: - fish pâté (34 g). n = 44 - n-3 PUFA-enriched fruit juice (500 mL). n = 38 - 3 capsuled of fish oil. n = 40 - Control: non-supplemented. n = 37 Duration: 7weeks | Urine F2-isoprostanes and plasma α-tocopherol | ↓Plasma α-tocopherol in fish pâté group when calculated in relation to the level of serum TG ↔ F2-isoprostanes |
| Ottestad et al., 2012 [183] | Randomized, double-blind, placebo-controlled parallel study | 54 Healthy men and women, age: 18–50 y, BMI < 30 kg/m2, non-smokers | Group 1: 8 g/d of fish oil (EPA/DHA) n = 17 Group 2: 8 g/d of oxidized fish oil (EPA/DHA) n = 18 Group 3: 8 g/d of high-oleic sunflower oil n = 19 For 7 weeks | Urine F2-isoprostanes and plasma oxidation products from n-3 PUFA and n-6 PUFA oxidation 4-HHE and 4-HNE; plasma α-tocopherol, enzymatic activity of GR, GPx, and CAT | ↔ Urine F2-isoprostanes and ↔ plasma oxidation products from n-3 PUFA and n-6 PUFA oxidation ↔ 4-HHE and ↔ 4-HNE; ↔ plasma α-tocopherol, ↔ enzymatic activity of GR, GPx, and CAT |
| Schimidt et al., 2012 [165] | Randomized, controlled, parallel intervention | 10 normo and 10 dyslipidemic men; Age: 29–51, BMI: 35 kg/m2. n = 20 | 6 Fish oil capsules, providing 1.14 g DHA and 1.56 g EPA per day, for 12 weeks | GST, GR, and antioxidative enzymes SOD3, CAT, and HMOX2 expression in whole blood cells, GPx, MMPs, cyrochrome P450 (CYP) enzymes expression in whole blood | ↑GST, ↑GR and antioxidative enzymes ↑SOD3, ↑CAT and HMOX2 expression, ↓GPx, ↓MMPs, ↓cytochrome P450 (CYP) enzymes expression |
| Kiecolt-Glaser et al., 2013 [166] | Randomized, double-blind, controlled parallel trial | Healthy sedentary overweight middle-aged and older adults Age: 48–85 y, BMI: 22.5–40 kg/m2. Non-smokers | Group 1: 2.5 g/day n-3 PUFA (n = 35), Group 2: l.25 g/day n-3 PUFA (n = 40) Group 3: placebo capsules that mirrored the proportions of fatty acids in the typical American diet (n = 31) Duration: 4 months | Plasma F2-isoprostanes | ↓Plasma F2-isoprostanes with n-3 PUFAS supplementation |
| Haijianfar et al., 2013 [167] | Randomized double-blind placebo-controlled clinical trial | Type 2 diabetic women. Age: 45–65 y BMI: 27.7 ± 3.4 (n-3 PUFA group). BMI: 28 ± 3.8. (Control group) | n-3 PUFA group: 2000 mg/d in 2 capsules: each contained 1,000 mg n-3PUFA (65% EPA, 360 mg and 35% DHA, 240 mg) (n = 37) Control group: 2 placebo capsules, each contains 1 g of cornstarch (n = 34) Duration: 8 weeks | Serum antioxidant capacity | ↑Antioxidant capacity in the n-3 PUFA supplemented group |
| Véricel et al. 2015 [168] | Randomized, double-blind, placebo-controlled, two-period crossover trial | Post-menopausal women with type 2 diabetes, age: 59.8 ± 4.7 y, BMI: 34.1 ± 5 kg/m2. n = 11 | Intervention: 400 mg/day of DHA (in 2 capsules/d) Control: 2 placebo (same amount of sunflower oil) Duration: 2 weeks | Plasma and platelet vitamin E, alpha- and gamma-tocopherol concentrations, plasma MDA, 8-iso-PGF2α | ↑ Platelet alpha-tocopherol, gamma-tocopherol tend to increase. ↓MDA, ↓8-iso-PGF2α. n-3 PUFAS supplementation ↓ oxidative stress associated with diabetes |
| Alves Luzia et al. (2015) [175] | Randomized, double-blind, placebo-controlled trial | Women (40 to 70 years) with low habitual fatty fish and seafood intake, who met at least two of the following criteria: total cholesterol > 200 mg/dL, LDL-C > 140 mg/dL, HDL-C < 50 mg/dL, and triglycerides >150 mg/dL | The fish oil group: daily consumption of 1 g n-3 PUFA (540 mg EPA + 360 mg DHA) and 1 capsule of placebo (n = 22) Fish+VitE group: 1 g n-3 PUFA, 400 mg vitamin E/ alpha-tocopherol (n = 19). Placebo group: 2 capsules/d mineral oil (n = 18) Duration: 3 months | Biomarkers of oxidative stress at baseline, 45 and 90 days | ↑ TBARS in the group supplemented with fish oil alone, but not in the fish oil + vitamin E group |
| Berge et al., (2015) [169] | Randomized, clinical interventional pilot study | Healthy female and male, mean age: 23 ± 4 y. BMI: 20.9 kg/m2, n=17 | 17 subjects received dietary supplementation with krill oil (832.5 mg EPA and DHA per day) for 28 days | Plasma total antioxidant capacity (AOC) | ↑AOC after krill oil intake. AOC positively correlated with plasma EPA concentration and RBC EPA concentration |
| Fayh et al., 2018 [161] | Randomized, double-blind, placebo-controlled trial | Male and female with T2DM, Age: 50–57 y. Mean BMI: 28.2 kg/m2 in n-3 PUFAS group and 28.8 kg/m2 in control group. n = 15/group | Control group: 3 capsules/day that contains 500 mg gelatin Intervention group: 3 capsules/d (each capsule contains 180 mg EPA, 120 mg DHA, 2 mg Vit E) For 8 weeks At the beginning and at the end of protocol, an acute exercise was performed (treadmill) | TBARS; Plasma F2-isoprostanes, TRAP, SOD activity, hs-CRP | n-3 PUFA supplementation: ↓ TG, ↓TRAP levels after exercise, without a significant effect on inflammatory and oxidative-stress markers |
| Reference | Study Design | Population | Intervention | Outcome Measurements | Results | Comments |
|---|---|---|---|---|---|---|
| Capanni et al., 2006. [200] | Open-label trial | Patients with NAFLD proven by US; Age range: 31–77 y. Mean BMI: 28.5 kg/m². n = 56 | Oral intake of n-3 PUFA (EPA and DHA in a 0.9/1.5 ratio), 1 g capsule a day for 12 months. Intervention group (n = 42) vs control group (n = 14) | Hematochemical tests; Liver fat changes detected by US and liver eco-texture measured by Duplex Doppler US and DPI follow up | ↓ AST, ALT, GGT; ↓ fasting TG and glucose ↓arachidonate ↓n-6/n-3 ratio Significant beneficial effects on liver US pattern and ↑ DPI | Long-term n-3 PUFAS supplementation ameliorates hepatic steatosis in NAFLD patients |
| Spadaro et al., 2008. [111] | Randomized open-label trial | Patients with NAFLD proven by US; Mean age: 51 y Mean BMI: 30.5 kg/m² | AHA diet + 2 g/d n-3 PUFA (group DP, n = 20) AHA diet (Group D, n = 20) for 6 months | Changes on liver fat via US; ALT, AST, GGT, lipid profile, TNF-α serum levels, fasting glucose, and IR by HOMA-IR | Group DP: ↓ ALT, GGT ↓ TG, TNF-α ↓ HOMA-IR ↑ HDL cholesterol Complete steatosis regression in 33.4 % of patients and an overall reduction of 50%. | n-3 PUFA have a major improvement on fatty liver in patients with NAFLD |
| Zhu et al., 2008. [201] | Randomized controlled trial | Patients with US proven NAFLD associated with hyperlipidemia; Age: 18–65 y | Oral supplementation of n-3 PUFA for 24 weeks. AHA-based diet with a caloric restriction of 25-30 kcal/kg per day Group A (n = 66): 2 g n-3 PUFA from seal oils, 3 times/day. Group B (n = 68) 2 g placebo, three times/day | Primary endpoints: fatty liver assessed by symptom scores, ALT and serum lipid levels at 8, 12, 16, and 24 weeks. Secondary endpoints: liver fat changes by US at weeks 12 and 24 | After 24 wk of treatment: ↔ body weight, ↔ FBG; ↓ Total symptom scores, ↓ALT and TG Complete fatty liver regression was observed in 19.7% of the patients, and an overall reduction was found in 53.0% (35/66) of the patients in group A | n-3 PUFA from seal oils is safe and efficacious for patients with NAFLD associated with hyperlipidemia and can improve their total symptom scores, ALT, serum lipid levels, and normalization of ultrasonographic evidence |
| Tanaka et al., 2008. [202] | Pilot Trial | 23 biopsy-proven NASH patients | Highly purified EPA (2700 mg/d) was administered for 12 months | Biochemical parameters of glucose and lipid metabolism, inflammatory and iron metabolism oxidative-stress markers Ultrasonography Histologic evaluation of liver biopsies | ↓ ALT, AST ↓Total cholesterol ↓ sTNFR1,2 ↓Ferritin ↓ Thioredoxin ↓ hepatic steatosis and fibrosis, hepatocyte ballooning, and lobular inflammation | EPA treatment seems to be safe and efficacious for patients with NASH |
| Sofi et al., 2010. [203] | Randomized | Patients with NAFLD proven by US. Age: 30–70 y, mean BMI: 29.3 kg/m² | Food consumption enriched with n-3 PUFA (0.47 g EPA + 0.24 g DHA) for 12 months. Group 1: (n = 6) 6.5ml/d enriched with olive oil + recommended diet Group 2: (n = 5) control (recommended diet + not enriched olive oil) | Liver eco-texture measured by Duplex Doppler US and DPI. Liver enzymes, TG and adiponectin levels | ↓ ALT, AST, and GGT ↓TG level ↑HDL cholesterol ↑adiponectin ↑ DPI level | Persistent consumption of food enriched with n-3 PUFA has favorable effects in patients with NAFLD |
| Scorletti et al., 2014. [204] | WELCOME study: double-blind, randomized, placebo-controlled trial | Patients with histological confirmation of NAFLD. Mean age: 50 years old. Mean BMI: 32.5 kg/m² | Intervention group (n = 51): oral supplementation of purified long-chain n-3 PUFA ethyl esters (1 g contains 460 mg of EPA and 380 mg of DHA), 4 g/day. Placebo group (n = 52): 4 g/day of olive oil. 15 to 18 months of treatment | Liver fat percentage assessed by MRS and biomarker scores for liver fibrosis, erythrocyte enrichment quantification with DHA+EPA via gas chromatography | Trend to improve liver fat% with DHA+EPA No improvement in liver fibrosis scores | Association between erythrocyte DHA enrichment with DHA+EPA treatment and a decrease of liver fat percentage |
| Sanyal et al., 2014. [205] | Double-blind, randomized, placebo-controlled trial | Patients with NASH, NAFLD activity scores ≥ 4, with minimum scores of 1 for steatosis and inflammation, along with either ballooning or at least stage 1a fibrosis. n = 243 | Subjects were randomly assigned to groups given placebo (n = 75), low- dosage EPA-E (1800 mg/d; n = 82), or high-dosage EPA-E (2700 mg/d; n = 86) for 12 months | The primary end point: NAFLD activity score ≤3, without worsening of fibrosis; or a decrease in NAFLD activity score by ≥2 with contribution from >1 parameter, without worsening of fibrosis. Liver enzymes, IR, adiponectin, keratin 18, hs-CRP, or hyaluronic acid were measured as well | No effects of EPA-E on steatosis, inflammation, ballooning, or fibrosis scores. No effects on levels of liver enzymes, IR, adiponectin, keratin 18, hs-CRP, or hyaluronic acid. High-dosage EPA-E: ↓ levels of TG | In a phase 2 trial, EPA-E had no significant effect on the histologic features of NASH. EPA-E reduced subjects’ levels of triglyceride compared with placebo, without any increase in serious adverse events |
| Li et al., 2015. [206] | Randomized placebo-controlled trial | Patients diagnosed with NASH Mean age: 51 years old. Mean BMI: 27.9 kg/m² | Intervention group (n = 39): 50 mL of PUFA with 1:1 Ratio of EPA and DHA added into daily diet placebo: saline (n = 39). Duration of treatment: 6 months | Liver enzymes, lipid profile, markers of inflammation and oxidation, and histological changes by biopsy | Liver function was significantly improved: ↓ ALT / AST ↓ TG ↓Total Cholesterol ↓ CRP (inflammation) ↓ MDA (oxidation) ↓ fibrotic parameters | 6 months of n-3 PUFA therapy is beneficial for improving NASH |
| Argo et al., 2015. [207] | Double-blind, randomized, placebo-controlled trial | Patients 34 subjects with biopsy-proven NASH; Mean age: 47 y Mean BMI: 32.5 kg/m2 | Oral supplementation of n-3 PUFA 3000 mg/d (each 1000 mg capsule contains 35% EPA, 25% DHA and 10% other n-3 PUFA), vs placebo (soybean oil). n = 17 per group 1 year of treatment | Liver biopsy, Abdominal MRI for quantitative assessment of hepatic fat, AST, ALT, total cholesterol, LDL and HDL cholesterol, and TGs. FFAs, insulin, and glucose levels | No differences for the primary end point of NASH activity score (NAS) reduction. In n-3 PUFA-treated subjects: ↓in liver fat content by MRI (among subjects with increased or stable weight) | Treatment did not exert beneficial effects towards hepatic histological improvement in NASH patients |
| Qin et al., 2015. [208] | A double-blind randomized Placebo-controlled clinical trial | Patients with NAFLD associated with hyperlipidemia, Mean age: 44.3 ± 10.9 and 46.0 ± 10.6 y for placebo or treated group Mean BMI: 26.0 ± 2.8 and 26.4±3.9 kg/m², respectively. n = 70 | Randomly assigned to consume fish oil (n = 36, 4 g/d) or corn oil capsules (n = 34, 4 g/d) for 3 months | Blood levels of lipids, glucose and insulin, liver enzymes, and cytokines at baseline and the end of the study were measured | Fish oil group: ↓ total cholesterol, ↓ TG, ↓apolipoprotein B ↓ glucose, ↓ ALT ↓ GGT ↑ Adiponectin ↓ TNF-α ↓ LTB4, ↓ FGF21, ↓ CK-18/M30 ↓PGE2 | These findings suggest that fish oil can benefit metabolic abnormalities associated with NAFLD |
| Dasarathy et al., 2015. [209] | Double-blind, randomized, placebo-controlled trial | Patients with NAFLD and NASH diagnosed by liver biopsy, Mean age: 50 y. Mean BMI: 35 kg/m². n = 37 | n-3 PUFA group: oral supplementation of 2160 mg of EPA and 1440 mg of DHA. (n = 19) and Placebo group (n = 18) using corn oil supplementation Duration: 48 months | Primary endpoints: assess the improvement of 2 points in the NAFLD activity score by liver biopsy. Secondary endpoints: changes in liver enzymes, IR, fasting glucose, and HbA1C | No differences between groups in BMI, serum transaminases, diabetes control, histological evaluation of NAFLD activity score and individual components | N-3 PUFA supplementation showed no beneficial effects in NASH patients with diabetes |
| Nogueira et al., 2016 [210] | Double-blind, randomized, placebo-controlled trial | Men and women with a proven histological diagnosis of NASH. Mean age: 53.9 ± 1.8 and 52.5 ± 7.2 y for placebo group and n-3 PUFA group. Mean BMI: 30.3 ± 4.4 and 31.1 ± 4.6, respectively. n = 50 | n-3 PUFA group: 3 capsules (0.945 g in total per day, 64% ALA, 16% EPA, and 21% DHA). (n = 27) Placebo group: 3 capsules of mineral oil (n = 23). Duration: 6 months | Primary endpoints: Plasma fatty acids (ALA, EPA, DHA and AA), NAS. Secondary endpoints: serum TG, AST, ALT, GGT, fasting lipid profile, fasting glucose, anthropometric parameters, or plasma levels of IL-6 at baseline and at endpoint, | n-3 PUFA group: ↑plasma ALA and EPA. NAS correlated with↑plasma ALA. ↓TG Control group: ↑plasma DHA and EPA, NAS correlated with↑plasma DHA and EPA | No significant changes were observed on liver histology in the n-3 PUFA or placebo group |
| Tobin et al., 2018 [211] | Double-blind, randomized, placebo-controlled trial | Patients with previously diagnosed NAFLD (hepatic steatosis stage). Mean age; 55.1 ± 10.9 and 55.3 ± 13.3 y for placebo and n-3 PUFA MF4637 group Mean BMI; 32.4 ± 5.0 and 32.1±4.8. respectively. n = 176 | n-3 PUFA group: oral supplementation of 3g capsule (1380 g of EPA and 1140 g of DHA) (n = 87) Placebo group: oral supplementation of 3 g olive oil capsule (n = 89) Duration = 24 weeks | n-3 PUFA index, n-6 PUFA: n-3 PUFA ratio, quantitative measurements of RBC EPA and DHA at the baseline and the endpoint, liver fat content measured by MRI | n-3 PUFA group: ↑n-3 PUFA index and ↑absolute values of RBC EPA and DHA, ↓RBC n-6: n-3 ratio ↓liver fat content in both groups | No significant differences in fat liver were found between n-3 PUFA and placebo group |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Fernández-Galilea, M.; Martínez-Fernández, L.; González-Muniesa, P.; Pérez-Chávez, A.; Martínez, J.A.; Moreno-Aliaga, M.J. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients 2019, 11, 872. https://doi.org/10.3390/nu11040872
Yang J, Fernández-Galilea M, Martínez-Fernández L, González-Muniesa P, Pérez-Chávez A, Martínez JA, Moreno-Aliaga MJ. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients. 2019; 11(4):872. https://doi.org/10.3390/nu11040872
Chicago/Turabian StyleYang, Jinchunzi, Marta Fernández-Galilea, Leyre Martínez-Fernández, Pedro González-Muniesa, Adriana Pérez-Chávez, J. Alfredo Martínez, and Maria J. Moreno-Aliaga. 2019. "Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation" Nutrients 11, no. 4: 872. https://doi.org/10.3390/nu11040872
APA StyleYang, J., Fernández-Galilea, M., Martínez-Fernández, L., González-Muniesa, P., Pérez-Chávez, A., Martínez, J. A., & Moreno-Aliaga, M. J. (2019). Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients, 11(4), 872. https://doi.org/10.3390/nu11040872