Potential Protective Mechanisms of Ketone Bodies in Migraine Prevention


 , ,
, , 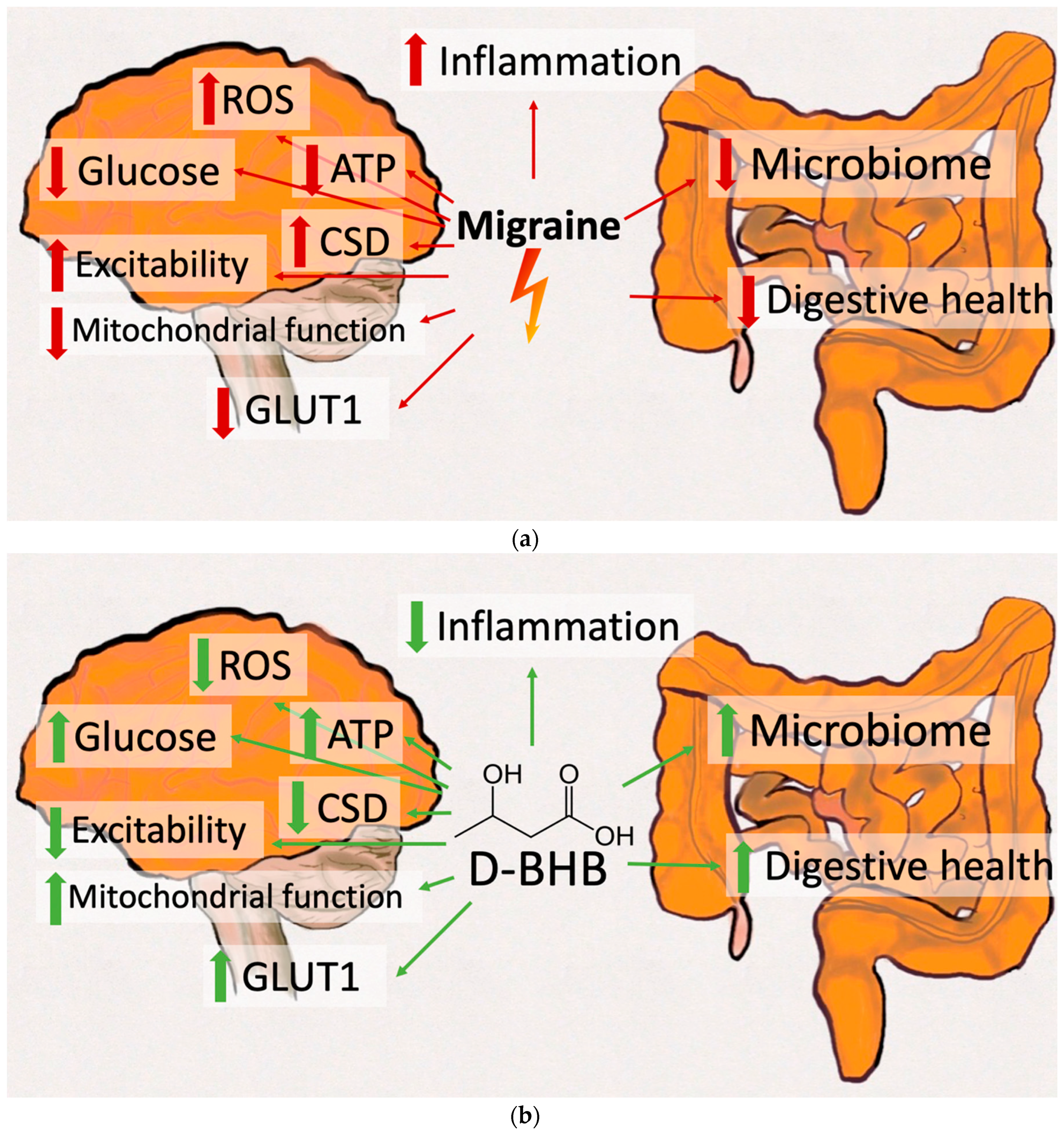{kind=link}
Abstract
1. Introduction
2. Potentially Migraine Relevant Mechanisms of Ketosis
2.1. Hypoglycemia/Hypometabolism
2.2. Glucose Transport
2.3. Mitochondrial Functoning
2.4. Oxidative Stress
2.5. Cerebral Excitability
2.6. Cortical Spreading Depression
2.7. Inflammation
2.8. Gut Microbiome
3. Discussion and Conclusions
Patents
Author Contributions
Funding
Conflicts of Interest
References
- Stovner, L.J.; Hoff, J.M.; Svalheim, S.; Gilhus, N.E. Neurological disorders in the Global Burden of Disease 2010 study. Acta Neurol. Scand. 2014, 129, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Stovner, L.J.; Hagen, K. Prevalence, burden, and cost of headache disorders. Curr. Opin. Neurol. 2006, 19, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Gustavsson, A.; Svensson, M.; Wittchen, H.-U.; Jönsson, B. The economic cost of brain disorders in Europe. Eur. J. Neurol. 2012, 19, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Buse, D.C.; Lipton, R.B. Global perspectives on the burden of episodic and chronic migraine. Cephalalgia Int. J. Headache 2013, 33, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, T.; Goadsby, P.J. Migraine pathogenesis and state of pharmacological treatment options. BMC Med. 2009, 7, 71. [Google Scholar] [CrossRef]
- Leonardi, M. Burden of migraine: What should we say more? Neurol. Sci. 2015, 36 (Suppl. 1), 1–3. [Google Scholar] [CrossRef]
- Lipton, R.B.; Buse, D.C.; Serrano, D.; Holland, S.; Reed, M.L. Examination of unmet treatment needs among persons with episodic migraine: Results of the American Migraine Prevalence and Prevention (AMPP) Study. Headache 2013, 53, 1300–1311. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, D.; Moskowitz, M.A. Pathophysiology of migraine. Annu. Rev. Physiol. 2013, 75, 365–391. [Google Scholar] [CrossRef]
- Gross, E.C.; Lisicki, M.; Fischer, D.; Sandor, P.S.; Schoenen, J. The metabilic face of migraine. Nat. Neurosci. 2019, 7, 50708–50718. [Google Scholar]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; de Vellis, J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [CrossRef]
- Bailey, E.E.; Pfeifer, H.H.; Thiele, E.A. The use of diet in the treatment of epilepsy. Epilepsy Behav. E&B 2005, 6, 4–8. [Google Scholar]
- Danial, N.N.; Hartman, A.L.; Stafstrom, C.E.; Thio, L.L. How does the ketogenic diet work? Four potential mechanisms. J. Child Neurol. 2013, 28, 1027–1033. [Google Scholar] [CrossRef]
- Stafstrom, C.E.; Rho, J.M. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharmacol. 2012, 3, 59. [Google Scholar] [CrossRef]
- Barañano, K.W.; Hartman, A.L. The ketogenic diet: Uses in epilepsy and other neurologic illnesses. Curr. Treat. Opt. Neurol. 2008, 10, 410–419. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Blatter, L.A. Role of β-hydroxybutyrate, its polymer poly-β-hydroxybutyrate and inorganic polyphosphate in mammalian health and disease. Front. Physiol. 2014, 5, 260. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essential Fatty Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
- Owen, O.E.; Felig, P.; Morgan, A.P.; Wahren, J.; Cahill, G.F. Liver and kidney metabolism during prolonged starvation. J. Clin. Investig. 1969, 48, 574–583. [Google Scholar] [CrossRef]
- Nei, M.; Ngo, L.; Sirven, J.I.; Sperling, M.R. Ketogenic diet in adolescents and adults with epilepsy. Seizure 2014, 23, 439–442. [Google Scholar] [CrossRef]
- Reid, C.A.; Mullen, S.; Kim, T.H.; Petrou, S. Epilepsy, energy deficiency and new therapeutic approaches including diet. Pharmacol. Ther. 2014, 144, 192–201. [Google Scholar] [CrossRef]
- De Almeida Rabello Oliveira, M.; da Rocha Ataíde, T.; de Oliveira, S.L.; de Melo Lucena, A.L.; de Lira, C.E.P.R.; Soares, A.A.; De Almeida, C.B.S.; Ximenes-da-Silva, A. Effects of short-term and long-term treatment with medium- and long-chain triglycerides ketogenic diet on cortical spreading depression in young rats. Neurosci. Lett. 2008, 434, 66–70. [Google Scholar] [CrossRef] [PubMed]
- SCHNABEL, T.G. An Experience with a Ketogenic Dietary in Migraine. Ann. Intern. Med. 1928, 2, 341. [Google Scholar]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Klepper, J.; Leiendecker, B.; Riemann, E.; Baumeister, F.A. [The ketogenic diet in German-speaking countries: Update 2003]. Klin. Pädiatrie 2004, 216, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Bianco, A.; Damiani, E.; Bosco, G. Ketogenic diet in neuromuscular and neurodegenerative diseases. BioMed Res. Int. 2014, 2014, 474296. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.M.; Kossoff, E.H. Ketosis and the ketogenic diet, 2010: Advances in treating epilepsy and other disorders. Adv. Pediatrics 2010, 57, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Wang, H.-S. Medium-chain triglyceride ketogenic diet, an effective treatment for drug-resistant epilepsy and a comparison with other ketogenic diets. Biomed. J. 2013, 36, 9–15. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Bajolle, F.; Arnoux, J.-B.; Dubois, S.; Sannier, N.; Baussan, C.; Petit, F.; Labrune, P.; Rabier, D.; Ottolenghi, C.; et al. Successful treatment of severe cardiomyopathy in glycogen storage disease type III With D,L-3-hydroxybutyrate, ketogenic and high-protein diet. Pediatric Res. 2011, 70, 638–641. [Google Scholar] [CrossRef]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; Todd King, M.; Musa-Veloso, K.; Ho, M.; Roberts, A.; Robertson, J.; Vanitallie, T.B.; et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul. Toxicol. Pharmacol. 2012, 63, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Cervenka, M.C.; Henry, B.J.; Haney, C.A.; Turner, Z. A decade of the modified Atkins diet (2003–2013): Results, insights, and future directions. Epilepsy Behav. E&B 2013, 29, 437–442. [Google Scholar]
- Newport, M.T.; VanItallie, T.B.; Kashiwaya, Y.; King, M.T.; Veech, R.L. A new way to produce hyperketonemia: Use of ketone ester in a case of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2015, 11, 99–103. [Google Scholar] [CrossRef]
- Douris, N.; Melman, T.; Pecherer, J.M.; Pissios, P.; Flier, J.S.; Cantley, L.C.; Locasale, J.W.; Maratos-Flier, E. Adaptive changes in amino acid metabolism permit normal longevity in mice consuming a low-carbohydrate ketogenic diet. Biochim. Biophys. Acta 2015, 1852, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Strahlman, R.S. Can ketosis help migraine sufferers? A case report. Headache 2006, 46, 182. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, C.; Currà, A.; Sirianni, G.; Coppola, G.; Bracaglia, M.; Cardillo, A.; De Nardis, L.; Pierelli, F. Diet transiently improves migraine in two twin sisters: Possible role of ketogenesis? Funct. Neurol. 2013, 28, 305–308. [Google Scholar]
- Maggioni, F.; Margoni, M.; Zanchin, G. Ketogenic diet in migraine treatment: A brief but ancient history. Cephalalgia Int. J. Headache 2011, 31, 1150–1151. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, C.; Coppola, G.; Bracaglia, M.; Di Lenola, D.; Evangelista, M.; Sirianni, G.; Rossi, P.; Di Lorenzo, G.; Serrao, M.; Parisi, V.; et al. Cortical functional correlates of responsiveness to short-lasting preventive intervention with ketogenic diet in migraine: A multimodal evoked potentials study. J. Headache Pain 2016, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, C.; Coppola, G.; Sirianni, G.; Di Lorenzo, G.; Bracaglia, M.; Di Lenola, D.; Siracusano, A.; Rossi, P.; Pierelli, F. Migraine improvement during short lasting ketogenesis: A proof-of-concept study. Eur. J. Neurol. 2015, 22, 170–177. [Google Scholar] [CrossRef]
- Lutas, A.; Yellen, G. The ketogenic diet: Metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013, 36, 32–40. [Google Scholar] [CrossRef]
- Blau, J.N.; Cumings, J.N. Method of precipitating and preventing some migraine attacks. Br. Med. J. 1966, 2, 1242–1243. [Google Scholar] [CrossRef][Green Version]
- Gray, P.A.; Burtness, H.I. HYPOGLYCEMIC HEADACHE*. Endocrinology 1935, 19, 549–560. [Google Scholar] [CrossRef]
- Roberts, H.J. Migraine and related vascular headaches due to diabetogenic hyperinsulinism. Observations on pathogenesis and rational treatment in 421 patients. Headache 1967, 7, 41–62. [Google Scholar] [CrossRef]
- Pavlovic, J.M.; Buse, D.C.; Sollars, C.M.; Haut, S.; Lipton, R.B. Trigger Factors and Premonitory Features of Migraine Attacks: Summary of Studies. Headache J. Head Face Pain 2014, 54, 1670–1679. [Google Scholar] [CrossRef]
- Peroutka, S.J. What turns on a migraine? A systematic review of migraine precipitating factors. Curr. Pain Headache Rep. 2014, 18, 454. [Google Scholar] [CrossRef]
- Yadav, R.K.; Kalita, J.; Misra, U.K. A Study of Triggers of Migraine in India. Pain Med. 2010, 11, 44–47. [Google Scholar] [CrossRef]
- Binder, C.; Bendtson, I. Endocrine emergencies. Hypoglycaemia. Bailliere’s Clinical Endocrinol. Metab. 1992, 6, 23–39. [Google Scholar] [CrossRef]
- Abu-Salameh, I.; Plakht, Y.; Ifergane, G. Migraine exacerbation during Ramadan fasting. J. Headache Pain 2010, 11, 513–517. [Google Scholar] [CrossRef]
- Haghighi, F.S.; Rahmanian, M.; Namiranian, N.; Arzaghi, S.M.; Dehghan, F.; Chavoshzade, F.; Sepehri, F. Migraine and type 2 diabetes; is there any association? J. Diabetes Metab. Disord. 2015, 15, 37. [Google Scholar] [CrossRef]
- Welch, K.M.; Levine, S.R.; D’Andrea, G.; Schultz, L.R.; Helpern, J.A. Preliminary observations on brain energy metabolism in migraine studied by in vivo phosphorus 31 NMR spectroscopy. Neurology 1989, 39, 538–541. [Google Scholar] [CrossRef]
- Barbiroli, B.; Montagna, P.; Cortelli, P.; Funicello, R.; Iotti, S.; Monari, L.; Pierangeli, G.; Zaniol, P.; Lugaresi, E. Abnormal brain and muscle energy metabolism shown by 31P magnetic resonance spectroscopy in patients affected by migraine with aura. Neurology 1992, 42, 1209–1214. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, S.; Suh, S.I.; Koh, S.B.; Park, K.W.; Oh, K. Interictal metabolic changes in episodic migraine: A voxel-based FDG-PET study. Cephalalgia 2010, 30, 53–61. [Google Scholar] [CrossRef]
- Lodi, R.; Montagna, P.; Soriani, S.; Iotti, S.; Arnaldi, C.; Cortelli, P.; Pierangeli, G.; Patuelli, A.; Zaniol, P.; Barbiroli, B. Deficit of Brain and Skeletal Muscle Bioenergetics and Low Brain Magnesium in Juvenile Migraine: An in Vivo 31P Magnetic Resonance Spectroscopy Interictal Study. Pediatric Res. 1997, 42, 866–871. [Google Scholar] [CrossRef]
- Lodi, R.; Iotti, S.; Cortelli, P.; Pierangeli, G.; Cevoli, S.; Clementi, V.; Soriani, S.; Montagna, P.; Barbiroli, B. Deficient energy metabolism is associated with low free magnesium in the brains of patients with migraine and cluster headache. Brain Res. Bull. 2001, 54, 437–441. [Google Scholar] [CrossRef]
- Montagna, P.; Cortelli, P.; Monari, L.; Pierangeli, G.; Parchi, P.; Lodi, R.; Iotti, S.; Frassineti, C.; Zaniol, P.; Lugaresi, E. 31P-magnetic resonance spectroscopy in migraine without aura. Neurology 1994, 44, 666–669. [Google Scholar] [CrossRef]
- Reyngoudt, H.; Achten, E.; Paemeleire, K. Magnetic resonance spectroscopy in migraine: What have we learned so far? Cephalalgia Int. J. Headache 2012, 32, 845–859. [Google Scholar] [CrossRef]
- Schulz, U.G.; Blamire, A.M.; Corkill, R.G.; Davies, P.; Styles, P.; Rothwell, P.M. Association between cortical metabolite levels and clinical manifestations of migrainous aura: An MR-spectroscopy study. Brain 2007, 130, 3102–3110. [Google Scholar] [CrossRef][Green Version]
- Lodi, R.; Kemp, G.J.; Pierangeli, G.; Cortelli, P.; Iotti, S.; Radda, G.K.; Barbiroli, B. Quantitative analysis of skeletal muscle bioenergetics and proton efflux in migraine and cluster headache. J. Neurol. Sci. 1997, 146, 73–80. [Google Scholar] [CrossRef]
- Barbiroli, B.; Montagna, P.; Cortelli, P.; Martinelli, P.; Sacquegna, T.; Zaniol, P.; Lugaresi, E. Complicated migraine studied by phosphorus magnetic resonance spectroscopy. Cephalalgia 1990, 10, 263–272. [Google Scholar] [CrossRef]
- Reyngoudt, H.; Paemeleire, K.; Descamps, B.; De Deene, Y.; Achten, E. 31P-MRS demonstrates a reduction in high-energy phosphates in the occipital lobe of migraine without aura patients. Cephalalgia Int. J. Headache 2011, 31, 1243–1253. [Google Scholar] [CrossRef]
- Lisicki, M.; D’Ostilio, K.; Coppola, G.; Scholtes, F.; Maertens de Noordhout, A.; Parisi, V.; Schoenen, J.; Magis, D. Evidence of an increased neuronal activation-to-resting glucose uptake ratio in the visual cortex of migraine patients: A study comparing 18FDG-PET and visual evoked potentials. J. Headache Pain 2018, 19, 49. [Google Scholar] [CrossRef]
- Julio-Amilpas, A.; Montiel, T.; Soto-Tinoco, E.; Gerónimo-Olvera, C.; Massieu, L. Protection of hypoglycemia-induced neuronal death by β-hydroxybutyrate involves the preservation of energy levels and decreased production of reactive oxygen species. J. Cereb. Blood Flow Metab. 2015, 35, 851–860. [Google Scholar] [CrossRef]
- Courchesne-Loyer, A.; Croteau, E.; Castellano, C.-A.; St-Pierre, V.; Hennebelle, M.; Cunnane, S.C. Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: A dual tracer quantitative positron emission tomography study. J. Cereb. Blood Flow Metab. 2017, 37, 2485–2493. [Google Scholar] [CrossRef]
- Zhang, Y.; Kuang, Y.; Xu, K.; Harris, D.; Lee, Z.; LaManna, J.; Puchowicz, M.A. Ketosis proportionately spares glucose utilization in brain. J. Cereb. Blood Flow Metab. 2013, 33, 1307–1311. [Google Scholar] [CrossRef]
- Chowdhury, G.M.I.; Jiang, L.; Rothman, D.L.; Behar, K.L. The contribution of ketone bodies to basal and activity-dependent neuronal oxidation in vivo. J. Cereb. Blood Flow Metab. 2014, 34, 1233–1242. [Google Scholar] [CrossRef]
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 1995, 9, 651–658. [Google Scholar] [CrossRef]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef]
- DeVivo, D.C.; Leckie, M.P.; Ferrendelli, J.S.; McDougal, D.B. Chronic ketosis and cerebral metabolism. Ann. Neurol. 1978, 3, 331–337. [Google Scholar] [CrossRef]
- Pan, J.W.; Bebin, E.M.; Chu, W.J.; Hetherington, H.P. Ketosis and epilepsy: 31P spectroscopic imaging at 4.1 T. Epilepsia 1999, 40, 703–707. [Google Scholar] [CrossRef]
- Hockaday, J.; Williamson, D.H.; Whitty, C.W.M. Blood-glucose levels and fatty-acid metabolism in migraine related to fasting. Lancet 1971, 297, 1153–1156. [Google Scholar] [CrossRef]
- Shaw, S.W.; Johnson, R.H.; Keogh, H.J. Metabolic changes during glucose tolerance tests in migraine attacks. J. Neurol. Sci. 1977, 33, 51–59. [Google Scholar] [CrossRef]
- Mohammad, S.S.; Coman, D.; Calvert, S. Glucose transporter 1 deficiency syndrome and hemiplegic migraines as a dominant presenting clinical feature. J. Paediatr. Child Health 2014, 50, 1025–1026. [Google Scholar]
- Uemura, E.; Greenlee, H.W. Insulin regulates neuronal glucose uptake by promoting translocation of glucose transporter GLUT3. Exp. Neurol. 2006, 198, 48–53. [Google Scholar] [CrossRef]
- Dexter, J.D.; Roberts, J.; Byer, J.A. The Five Hour Glucose Tolerance Test and Effect of Low Sucrose Diet in Migraine. Headache J. Head Face Pain 1978, 18, 91–94. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Diao, Y.; Meng, S.; Xing, Y.; Zhou, H.; Yang, D.; Sun, J.; Chen, H.; Zhao, Y. Are Glucose and Insulin Metabolism and Diabetes Associated with Migraine? A Community-Based, Case-Control Study. J. Oral Facial Pain Headache 2017, 31, 240–250. [Google Scholar] [CrossRef]
- Rainero, I.; Limone, P.; Ferrero, M.; Valfrè, W.; Pelissetto, C.; Rubino, E.; Gentile, S.; Lo Giudice, R.; Pinessi, L. Insulin sensitivity is impaired in patients with migraine. Cephalalgia 2005, 25, 593–597. [Google Scholar] [CrossRef]
- Fava, A.; Pirritano, D.; Consoli, D.; Plastino, M.; Casalinuovo, F.; Cristofaro, S.; Colica, C.; Ermio, C.; De Bartolo, M.; Opipari, C.; et al. Chronic migraine in women is associated with insulin resistance: A cross-sectional study. Eur. J. Neurol. 2014, 21, 267–272. [Google Scholar] [CrossRef]
- Cavestro, C.; Rosatello, A.; Micca, G.; Ravotto, M.; Marino, M.P.; Asteggiano, G.; Beghi, E. Insulin Metabolism is Altered in Migraineurs: A New Pathogenic Mechanism for Migraine? Headache J. Head Face Pain 2007, 47, 1436–1442. [Google Scholar] [CrossRef]
- Sacco, S.; Altobelli, E.; Ornello, R.; Ripa, P.; Pistoia, F.; Carolei, A. Insulin resistance in migraineurs: Results from a case-control study. Cephalalgia 2014, 34, 349–356. [Google Scholar] [CrossRef]
- Rainero, I.; Govone, F.; Gai, A.; Vacca, A.; Rubino, E. Is Migraine Primarily a Metaboloendocrine Disorder? Curr. Pain Headache Rep. 2018, 22, 36. [Google Scholar] [CrossRef]
- Curtain, R.; Tajouri, L.; Lea, R.; MacMillan, J.; Griffiths, L. No mutations detected in the INSR gene in a chromosome 19p13 linked migraine pedigree. Eur. J. Med. Genet. 2006, 49, 57–62. [Google Scholar] [CrossRef]
- Kaunisto, M.A.; Tikka, P.J.; Kallela, M.; Leal, S.M.; Papp, J.C.; Korhonen, A.; Hämäläinen, E.; Harno, H.; Havanka, H.; Nissilä, M.; et al. Chromosome 19p13 loci in Finnish migraine with aura families. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 132B, 85–89. [Google Scholar] [CrossRef]
- McCarthy, L.C.; Hosford, D.A.; Riley, J.H.; Bird, M.I.; White, N.J.; Hewett, D.R.; Peroutka, S.J.; Griffiths, L.R.; Boyd, P.R.; Lea, R.A.; et al. Single-nucleotide polymorphism alleles in the insulin receptor gene are associated with typical migraine. Genomics 2001, 78, 135–149. [Google Scholar] [CrossRef]
- Netzer, C.; Freudenberg, J.; Heinze, A.; Heinze-Kuhn, K.; Goebel, I.; McCarthy, L.C.; Roses, A.D.; Göbel, H.; Todt, U.; Kubisch, C. Replication study of the insulin receptor gene in migraine with aura. Genomics 2008, 91, 503–507. [Google Scholar] [CrossRef]
- Guzmán, M.; Blázquez, C. Ketone body synthesis in the brain: Possible neuroprotective effects. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Iizumi, T.; Mashima, K.; Abe, T.; Suzuki, N. Roles and regulation of ketogenesis in cultured astroglia and neurons under hypoxia and hypoglycemia. ASN Neuro 2014, 6, 1759091414550997. [Google Scholar] [CrossRef] [PubMed]
- Veggiotti, P.; De Giorgis, V. Dietary Treatments and New Therapeutic Perspective in GLUT1 Deficiency Syndrome. Curr. Treat. Opt. Neurol. 2014, 16, 291. [Google Scholar] [CrossRef]
- Valdebenito, R.; Ruminot, I.; Garrido-Gerter, P.; Fernández-Moncada, I.; Forero-Quintero, L.; Alegría, K.; Becker, H.M.; Deitmer, J.W.; Barros, L.F. Targeting of astrocytic glucose metabolism by beta-hydroxybutyrate. J. Cereb. Blood Flow Metab. 2016, 36, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Kraya, T.; Deschauer, M.; Joshi, P.R.; Zierz, S.; Gaul, C. Prevalence of Headache in Patients With Mitochondrial Disease: A Cross-Sectional Study. Headache 2018, 58, 45–52. [Google Scholar] [CrossRef]
- Vollono, C.; Primiano, G.; Della Marca, G.; Losurdo, A.; Servidei, S. Migraine in mitochondrial disorders: Prevalence and characteristics. Cephalalgia 2018, 38, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Montagna, P.; Gallassi, R.; Medori, R.; Govoni, E.; Zeviani, M.; Di Mauro, S.; Lugaresi, E.; Andermann, F. MELAS syndrome: Characteristic migrainous and epileptic features and maternal transmission. Neurology 1988, 38, 751–754. [Google Scholar] [CrossRef]
- Lemos, C.; Alonso, I.; Barros, J.; Sequeiros, J.; Pereira-Monteiro, J.; Mendonça, D.; Sousa, A. Assessing risk factors for migraine: Differences in gender transmission. PLoS ONE 2012, 7, e50626. [Google Scholar] [CrossRef]
- Eising, E.; Huisman, S.M.H.; Mahfouz, A.; Vijfhuizen, L.S.; Anttila, V.; Winsvold, B.S.; Kurth, T.; Ikram, M.A.; Freilinger, T.; Kaprio, J.; et al. Gene co-expression analysis identifies brain regions and cell types involved in migraine pathophysiology: A GWAS-based study using the Allen Human Brain Atlas. Hum. Genet. 2016, 135, 425–439. [Google Scholar] [CrossRef]
- Littlewood, J.; Glover, V.; Sandler, M.; Peatfield, R.; Petty, R.; Clifford Rose, F. Low platelet monoamine oxidase activity in headache: No correlation with phenolsulphotransferase, succinate dehydrogenase, platelet preparation method or smoking. J. Neurol. Neurosurg. Psychiatry 1984, 47, 338–343. [Google Scholar] [CrossRef]
- Sangiorgi, S.; Mochi, M.; Riva, R.; Cortelli, P.; Monari, L.; Pierangeli, G.; Montagna, P. Abnormal platelet mitochondrial function in patients affected by migraine with and without aura. Cephalalgia Int. J. Headache 1994, 14, 21–23. [Google Scholar] [CrossRef]
- Van Houten, B.; Hunter, S.E.; Meyer, J.N. Mitochondrial DNA damage induced autophagy, cell death, and disease. Front. Biosci. (Landmark Ed) 2016, 21, 42–54. [Google Scholar] [CrossRef]
- Yang, J.-L.; Weissman, L.; Bohr, V.A.; Mattson, M.P. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair 2008, 7, 1110–1120. [Google Scholar] [CrossRef]
- Boehnke, C.; Reuter, U.; Flach, U.; Schuh-Hofer, S.; Einhäupl, K.M.; Arnold, G. High-dose riboflavin treatment is efficacious in migraine prophylaxis: An open study in a tertiary care centre. Eur. J. Neurol. 2004, 11, 475–477. [Google Scholar] [CrossRef]
- Condò, M.; Posar, A.; Arbizzani, A.; Parmeggiani, A. Riboflavin prophylaxis in pediatric and adolescent migraine. J. Headache Pain 2009, 10, 361–365. [Google Scholar] [CrossRef]
- Gaul, C.; Diener, H.-C.; Danesch, U.; Migravent® Study Group. Improvement of migraine symptoms with a proprietary supplement containing riboflavin, magnesium and Q10: A randomized, placebo-controlled, double-blind, multicenter trial. J. Headache Pain 2015, 16, 516. [Google Scholar] [CrossRef]
- Schoenen, J.; Jacquy, J.; Lenaerts, M. Effectiveness of high-dose riboflavin in migraine prophylaxis. A randomized controlled trial. Neurology 1998, 50, 466–470. [Google Scholar] [CrossRef]
- Rahimdel, A.; Mellat, A.; Zeinali, A.; Jafari, E.; Ayatollahi, P. Comparison between Intravenous Sodium Valproate and Subcutaneous Sumatriptan for Treatment of Acute Migraine Attacks; Double-Blind Randomized Clinical Trial. Iran. J. Med. Sci. 2014, 39, 171–177. [Google Scholar]
- Dahri, M.; Hashemilar, M.; Asghari-Jafarabadi, M.; Tarighat-Esfanjani, A. Efficacy of coenzyme Q10 for the prevention of migraine in women: A randomized, double-blind, placebo-controlled study. Eur. J. Integr. Med. 2017, 16, 8–14. [Google Scholar] [CrossRef]
- Dahri, M.; Tarighat-Esfanjani, A.; Asghari-Jafarabadi, M.; Hashemilar, M. Oral coenzyme Q10 supplementation in patients with migraine: Effects on clinical features and inflammatory markers. Nutr. Neurosci. 2018, 0, 1–9. [Google Scholar] [CrossRef]
- Sándor, P.S.; Di Clemente, L.; Coppola, G.; Saenger, U.; Fumal, A.; Magis, D.; Seidel, L.; Agosti, R.M.; Schoenen, J. Efficacy of coenzyme Q10 in migraine prophylaxis: A randomized controlled trial. Neurology 2005, 64, 713–715. [Google Scholar] [CrossRef]
- Hajihashemi, P.; Askari, G.; Khorvash, F.; Reza Maracy, M.; Nourian, M. The effects of concurrent Coenzyme Q10, L-carnitine supplementation in migraine prophylaxis: A randomized, placebo-controlled, double-blind trial. Cephalalgia 2019, 6, 0333102418821661. [Google Scholar] [CrossRef]
- Shoeibi, A.; Olfati, N.; Soltani Sabi, M.; Salehi, M.; Mali, S.; Akbari Oryani, M. Effectiveness of coenzyme Q10 in prophylactic treatment of migraine headache: An open-label, add-on, controlled trial. Acta Neurol. Belg. 2017, 117, 103–109. [Google Scholar] [CrossRef]
- Rozen, T.; Oshinsky, M.; Gebeline, C.; Bradley, K.; Young, W.; Shechter, A.; Silberstein, S. Open label trial of coenzyme Q10 as a migraine preventive. Cephalalgia 2002, 22, 137–141. [Google Scholar] [CrossRef]
- Magis, D.; Ambrosini, A.; Sándor, P.; Jacquy, J.; Laloux, P.; Schoenen, J. A randomized double-blind placebo-controlled trial of thioctic acid in migraine prophylaxis. Headache 2007, 47, 52–57. [Google Scholar] [CrossRef]
- Cavestro, C.; Bedogni, G.; Molinari, F.; Mandrino, S.; Rota, E.; Frigeri, M.C. Alpha-Lipoic Acid Shows Promise to Improve Migraine in Patients with Insulin Resistance: A 6-Month Exploratory Study. J. Med. Food 2018, 21, 269–273. [Google Scholar] [CrossRef]
- Ali, A.M.; Awad, T.G.; Al-Adl, N.M. Efficacy of combined topiramate/thioctic acid therapy in migraine prophylaxis. Saudi Pharm. J. 2010, 18, 239–243. [Google Scholar] [CrossRef]
- Lea, R.; Colson, N.; Quinlan, S.; Macmillan, J.; Griffiths, L. The effects of vitamin supplementation and MTHFR (C677T) genotype on homocysteine-lowering and migraine disability. Pharmacogenet. Genom. 2009, 19, 422–428. [Google Scholar] [CrossRef]
- Menon, S.; Lea, R.A.; Roy, B.; Hanna, M.; Wee, S.; Haupt, L.M.; Oliver, C.; Griffiths, L.R. Genotypes of the MTHFR C677T and MTRR A66G genes act independently to reduce migraine disability in response to vitamin supplementation. Pharmacogenet. Genom. 2012, 22, 741–749. [Google Scholar] [CrossRef]
- Prousky, J.; Seely, D. The treatment of migraines and tension-type headaches with intravenous and oral niacin (nicotinic acid): Systematic review of the literature. Nutr. J. 2005, 4, 3. [Google Scholar] [CrossRef]
- Chiu, H.-Y.; Yeh, T.-H.; Huang, Y.-C.; Chen, P.-Y. Effects of Intravenous and Oral Magnesium on Reducing Migraine: A Meta-analysis of Randomized Controlled Trials. Pain Physician 2016, 19, E97–E112. [Google Scholar]
- Kudin, A.P.; Debska-Vielhaber, G.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. The mechanism of neuroprotection by topiramate in an animal model of epilepsy. Epilepsia 2004, 45, 1478–1487. [Google Scholar] [CrossRef]
- Motaghinejad, M.; Motevalian, M.; Shabab, B. Neuroprotective effects of various doses of topiramate against methylphenidate induced oxidative stress and inflammation in rat isolated hippocampus. Clin. Exp. Pharmacol. Physiol. 2016, 43, 360–371. [Google Scholar] [CrossRef]
- Wilkes, J.J.; Nelson, E.; Osborne, M.; Demarest, K.T.; Olefsky, J.M. Topiramate is an insulin-sensitizing compound in vivo with direct effects on adipocytes in female ZDF rats. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E617–E624. [Google Scholar] [CrossRef]
- Tripathi, G.M.; Kalita, J.; Misra, U.K. A study of oxidative stress in migraine with special reference to prophylactic therapy. Int. J. Neurosci. 2018, 128, 318–324. [Google Scholar] [CrossRef]
- Li, R.; Liu, Y.; Chen, N.; Zhang, Y.; Song, G.; Zhang, Z. Valproate Attenuates Nitroglycerin-Induced Trigeminovascular Activation by Preserving Mitochondrial Function in a Rat Model of Migraine. Med. Sci. Monit. 2016, 22, 3229–3237. [Google Scholar] [CrossRef]
- Sitarz, K.S.; Elliott, H.R.; Karaman, B.S.; Relton, C.; Chinnery, P.F.; Horvath, R. Valproic acid triggers increased mitochondrial biogenesis in POLG-deficient fibroblasts. Mol. Genet. Metab. 2014, 112, 57–63. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef]
- Milder, J.; Patel, M. Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. 2012, 100, 295–303. [Google Scholar] [CrossRef]
- Prins, M.L.; Lee, S.M.; Fujima, L.S.; Hovda, D.A. Increased cerebral uptake and oxidation of exogenous betaHB improves ATP following traumatic brain injury in adult rats. J. Neurochem. 2004, 90, 666–672. [Google Scholar] [CrossRef]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.-C.; Yan, S.-D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef]
- Bough, K. Energy metabolism as part of the anticonvulsant mechanism of the ketogenic diet. Epilepsia 2008, 49 (Suppl. 8), 91–93. [Google Scholar] [CrossRef]
- Srivastava, S.; Kashiwaya, Y.; King, M.T.; Baxa, U.; Tam, J.; Niu, G.; Chen, X.; Clarke, K.; Veech, R.L. Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB J. 2012, 26, 2351–2362. [Google Scholar] [CrossRef]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar]
- Kelman, L. The Triggers or Precipitants of the Acute Migraine Attack. Cephalalgia 2007, 27, 394–402. [Google Scholar] [CrossRef]
- Borkum, J.M. Migraine Triggers and Oxidative Stress: A Narrative Review and Synthesis. Headache 2015. [Google Scholar] [CrossRef]
- Welch, K.M.; Nagesh, V.; Aurora, S.K.; Gelman, N. Periaqueductal gray matter dysfunction in migraine: Cause or the burden of illness? Headache 2001, 41, 629–637. [Google Scholar] [CrossRef]
- Gonullu, H.; Gonullu, E.; Karadas, S.; Arslan, M.; Kalemci, O.; Aycan, A.; Sayin, R.; Demir, H. The levels of trace elements and heavy metals in patients with acute migraine headache. J. Pak. Med. Assoc. 2015, 65, 694–697. [Google Scholar]
- Alp, R.; Selek, S.; Alp, S.I.; Taşkin, A.; Koçyiğit, A. Oxidative and antioxidative balance in patients of migraine. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 877–882. [Google Scholar]
- Aytaç, B.; Coşkun, Ö.; Alioğlu, B.; Durak, Z.E.; Büber, S.; Tapçi, E.; Öcal, R.; İnan, L.E.; Durak, İ.; Yoldaş, T.K. Decreased antioxidant status in migraine patients with brain white matter hyperintensities. Neurol. Sci. 2014, 35, 1925–1929. [Google Scholar] [CrossRef]
- Bernecker, C.; Ragginer, C.; Fauler, G.; Horejsi, R.; Möller, R.; Zelzer, S.; Lechner, A.; Wallner-Blazek, M.; Weiss, S.; Fazekas, F.; et al. Oxidative stress is associated with migraine and migraine-related metabolic risk in females. Eur. J. Neurol. 2011, 18, 1233–1239. [Google Scholar] [CrossRef]
- Bolayir, E.; Celik, K.; Kugu, N.; Yilmaz, A.; Topaktas, S.; Bakir, S. Intraerythrocyte antioxidant enzyme activities in migraine and tension-type headaches. J. Chin. Med. Assoc. 2004, 67, 263–267. [Google Scholar]
- Ciancarelli, I.; Tozzi-Ciancarelli, M.; Massimo, C.D.; Marini, C.; Carolei, A. Urinary Nitric Oxide Metabolites and Lipid Peroxidation By-Products in Migraine. Cephalalgia 2003, 23, 39–42. [Google Scholar] [CrossRef]
- Ciancarelli, I.; Tozzi-Ciancarelli, M.; Spacca, G.; Massimo, C.D.; Carolei, A. Relationship Between Biofeedback and Oxidative Stress in Patients With Chronic Migraine. Cephalalgia 2007, 27, 1136–1141. [Google Scholar] [CrossRef]
- Eren, Y.; Dirik, E.; Neşelioğlu, S.; Erel, Ö. Oxidative stress and decreased thiol level in patients with migraine: Cross-sectional study. Acta Neurol. Belg. 2015, 115, 643–649. [Google Scholar] [CrossRef]
- Geyik, S.; Altunısık, E.; Neyal, A.M.; Taysi, S. Oxidative stress and DNA damage in patients with migraine. J. Headache Pain 2016, 17, 10. [Google Scholar] [CrossRef]
- Gumusyayla, S.; Vural, G.; Bektas, H.; Neselioglu, S.; Deniz, O.; Erel, O. A novel oxidative stress marker in migraine patients: Dynamic thiol-disulphide homeostasis. Neurol. Sci. 2016, 37, 1311–1317. [Google Scholar] [CrossRef]
- Shimomura, T.; Kowa, H.; Nakano, T.; Kitano, A.; Marukawa, H.; Urakami, K.; Takahashi, K. Platelet Superoxide Dismutase in Migraine and Tension-Type Headache. Cephalalgia 1994, 14, 215–218. [Google Scholar] [CrossRef]
- Tozzi-Ciancarelli, M.; De Matteis, G.; Di Massimo, C.; Marini, C.; Ciancarelli, I.; Carolei, A. Oxidative Stress and Platelet Responsiveness in Migraine. Cephalalgia 1997, 17, 580–584. [Google Scholar] [CrossRef]
- Tuncel, D.; Tolun, F.I.; Gokce, M.; İmrek, S.; Ekerbiçer, H. Oxidative Stress in Migraine with and Without Aura. Biol. Trace Elem. Res. 2008, 126, 92–97. [Google Scholar] [CrossRef]
- Yilmaz, G.; Sürer, H.; Inan, L.E.; Coskun, O.; Yücel, D. Increased nitrosative and oxidative stress in platelets of migraine patients. Tohoku J. Exp. Med. 2007, 211, 23–30. [Google Scholar] [CrossRef]
- Neri, M.; Frustaci, A.; Milic, M.; Valdiglesias, V.; Fini, M.; Bonassi, S.; Barbanti, P. A meta-analysis of biomarkers related to oxidative stress and nitric oxide pathway in migraine. Cephalalgia 2015, 35, 931–937. [Google Scholar] [CrossRef]
- Palmirotta, R.; Barbanti, P.; De Marchis, M.L.; Egeo, G.; Aurilia, C.; Fofi, L.; Ialongo, C.; Valente, M.G.; Ferroni, P.; Della-Morte, D.; et al. Is SOD2 Ala16Val polymorphism associated with migraine with aura phenotype? Antioxid. Redox Signal. 2015, 22, 275–279. [Google Scholar] [CrossRef]
- Saygi, S.; Erol, İ.; Alehan, F.; Yalçın, Y.Y.; Kubat, G.; Ataç, F.B. Superoxide Dismutase and Catalase Genotypes in Pediatric Migraine Patients. J. Child Neurol. 2015, 30, 1586–1590. [Google Scholar] [CrossRef]
- Haces, M.L.; Hernández-Fonseca, K.; Medina-Campos, O.N.; Montiel, T.; Pedraza-Chaverri, J.; Massieu, L. Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp. Neurol. 2008, 211, 85–96. [Google Scholar] [CrossRef]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef]
- Kong, G.; Huang, Z.; Ji, W.; Wang, X.; Liu, J.; Wu, X.; Huang, Z.; Li, R.; Zhu, Q. The Ketone Metabolite β-Hydroxybutyrate Attenuates Oxidative Stress in Spinal Cord Injury by Suppression of Class I Histone Deacetylases. J. Neurotrauma 2017, 34, 2645–2655. [Google Scholar] [CrossRef]
- Nagao, M.; Toh, R.; Irino, Y.; Mori, T.; Nakajima, H.; Hara, T.; Honjo, T.; Satomi-Kobayashi, S.; Shinke, T.; Tanaka, H.; et al. β-Hydroxybutyrate elevation as a compensatory response against oxidative stress in cardiomyocytes. Biochem. Biophys. Res. Commun. 2016, 475, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Wang, X.; Wu, X.; Liu, Q.; Kong, G.; Zhou, J.; Jiang, J.; Wu, X.; Huang, Z.; Su, W.; Zhu, Q. Ketogenic Metabolism Inhibits Histone Deacetylase (HDAC) and Reduces Oxidative Stress After Spinal Cord Injury in Rats. Neuroscience 2017, 366, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.R.; Kim, D.H.; Park, M.H.; Lee, B.; Kim, M.J.; Lee, E.K.; Chung, K.W.; Kim, S.M.; Im, D.S.; Chung, H.Y. β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 2016, 7, 66444–66454. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Milder, J.B.; Liang, L.-P.; Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 2008, 106, 1044–1051. [Google Scholar] [CrossRef]
- Winesett, S.P.; Bessone, S.K.; Kossoff, E.H.W. The ketogenic diet in pharmacoresistant childhood epilepsy. Expert Rev. Neurother 2015, 15, 621–628. [Google Scholar] [CrossRef]
- Winawer, M.R.; Connors, R. Evidence for a shared genetic susceptibility to migraine and epilepsy. Epilepsia 2013, 54, 288–295. [Google Scholar] [CrossRef]
- Coppola, G.; Pierelli, F.; Schoenen, J. Habituation and migraine. Neurobiol. Learn. Mem. 2009, 92, 249–259. [Google Scholar] [CrossRef]
- Aurora, S.K.; Wilkinson, F. The brain is hyperexcitable in migraine. Cephalalgia Int. J. Headache 2007, 27, 1442–1453. [Google Scholar] [CrossRef]
- Cestèle, S.; Scalmani, P.; Rusconi, R.; Terragni, B.; Franceschetti, S.; Mantegazza, M. Self-limited hyperexcitability: Functional effect of a familial hemiplegic migraine mutation of the Nav1.1 (SCN1A) Na+ channel. J. Neurosci. 2008, 28, 7273–7283. [Google Scholar] [CrossRef]
- Lang, E.; Kaltenhäuser, M.; Neundörfer, B.; Seidler, S. Hyperexcitability of the primary somatosensory cortex in migraine—A magnetoencephalographic study. Brain J. Neurol. 2004, 127, 2459–2469. [Google Scholar] [CrossRef]
- Boulloche, N.; Denuelle, M.; Payoux, P.; Fabre, N.; Trotter, Y.; Géraud, G. Photophobia in migraine: An interictal PET study of cortical hyperexcitability and its modulation by pain. J. Neurol. Neurosurg. Psychiatry 2010, 81, 978–984. [Google Scholar] [CrossRef]
- Moulton, E.A.; Becerra, L.; Maleki, N.; Pendse, G.; Tully, S.; Hargreaves, R.; Burstein, R.; Borsook, D. Painful heat reveals hyperexcitability of the temporal pole in interictal and ictal migraine States. Cerebral Cortex 2011, 21, 435–448. [Google Scholar] [CrossRef]
- Ducros, A.; Denier, C.; Joutel, A.; Cecillon, M.; Lescoat, C.; Vahedi, K.; Darcel, F.; Vicaut, E.; Bousser, M.G.; Tournier-Lasserve, E. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. New England J. Med. 2001, 345, 17–24. [Google Scholar] [CrossRef]
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.E.; Mohrenweiser, H.W.; Bulman, D.E.; Ferrari, M.; et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef]
- De Fusco, M.; Marconi, R.; Silvestri, L.; Atorino, L.; Rampoldi, L.; Morgante, L.; Ballabio, A.; Aridon, P.; Casari, G. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat. Genet. 2003, 33, 192–196. [Google Scholar] [CrossRef]
- Dichgans, M.; Freilinger, T.; Eckstein, G.; Babini, E.; Lorenz-Depiereux, B.; Biskup, S.; Ferrari, M.D.; Herzog, J.; van den Maagdenberg, A.M.J.M.; Pusch, M.; et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 2005, 366, 371–377. [Google Scholar] [CrossRef]
- Anttila, V.; Stefansson, H.; Kallela, M.; Todt, U.; Terwindt, G.M.; Calafato, M.S.; Nyholt, D.R.; Dimas, A.S.; Freilinger, T.; Müller-Myhsok, B.; et al. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat. Genet. 2010, 42, 869–873. [Google Scholar] [CrossRef]
- Chasman, D.I.; Schürks, M.; Anttila, V.; de Vries, B.; Schminke, U.; Launer, L.J.; Terwindt, G.M.; van den Maagdenberg, A.M.J.M.; Fendrich, K.; Völzke, H.; et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat. Genet. 2011, 43, 695–698. [Google Scholar] [CrossRef]
- Freilinger, T.; Anttila, V.; de Vries, B.; Malik, R.; Kallela, M.; Terwindt, G.M.; Pozo-Rosich, P.; Winsvold, B.; Nyholt, D.R.; van Oosterhout, W.P.J.; et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat. Genet. 2012, 44, 777–782. [Google Scholar] [CrossRef]
- Ferrari, M.D.; Klever, R.R.; Terwindt, G.M.; Ayata, C.; van den Maagdenberg, A.M.J.M. Migraine pathophysiology: Lessons from mouse models and human genetics. Lancet Neurol. 2015, 14, 65–80. [Google Scholar] [CrossRef]
- Bough, K.J.; Rho, J.M. Anticonvulsant Mechanisms of the Ketogenic Diet. Epilepsia 2007, 48, 43–58. [Google Scholar] [CrossRef]
- Yudkoff, M.; Daikhin, Y.; Melø, T.M.; Nissim, I.; Sonnewald, U.; Nissim, I. The ketogenic diet and brain metabolism of amino acids: Relationship to the anticonvulsant effect. Annu. Rev. Nutr. 2007, 27, 415–430. [Google Scholar] [CrossRef]
- Ma, W.; Berg, J.; Yellen, G. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J. Neurosci. 2007, 27, 3618–3625. [Google Scholar] [CrossRef]
- Juge, N.; Gray, J.A.; Omote, H.; Miyaji, T.; Inoue, T.; Hara, C.; Uneyama, H.; Edwards, R.H.; Nicoll, R.A.; Moriyama, Y. Metabolic control of vesicular glutamate transport and release. Neuron 2010, 68, 99–112. [Google Scholar] [CrossRef]
- Masino, S.A.; Li, T.; Theofilas, P.; Sandau, U.S.; Ruskin, D.N.; Fredholm, B.B.; Geiger, J.D.; Aronica, E.; Boison, D. A ketogenic diet suppresses seizures in mice through adenosine A₁ receptors. J. Clin. Investig. 2011, 121, 2679–2683. [Google Scholar] [CrossRef]
- Sada, N.; Lee, S.; Katsu, T.; Otsuki, T.; Inoue, T. Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 2015, 347, 1362–1367. [Google Scholar] [CrossRef]
- Won, Y.-J.; Lu, V.B.; Puhl, H.L.; Ikeda, S.R. β-Hydroxybutyrate Modulates N-Type Calcium Channels in Rat Sympathetic Neurons by Acting as an Agonist for the G-Protein-Coupled Receptor FFA3. J. Neurosci. 2013, 33, 19314–19325. [Google Scholar] [CrossRef]
- Tanner, G.R.; Lutas, A.; Martínez-François, J.R.; Yellen, G. Single K ATP channel opening in response to action potential firing in mouse dentate granule neurons. J. Neurosci. 2011, 31, 8689–8696. [Google Scholar] [CrossRef]
- Giménez-Cassina, A.; Martínez-François, J.R.; Fisher, J.K.; Szlyk, B.; Polak, K.; Wiwczar, J.; Tanner, G.R.; Lutas, A.; Yellen, G.; Danial, N.N. BAD-Dependent Regulation of Fuel Metabolism and KATP Channel Activity Confers Resistance to Epileptic Seizures. Neuron 2012, 74, 719–730. [Google Scholar] [CrossRef]
- Gerich, F.J.; Hepp, S.; Probst, I.; Müller, M. Mitochondrial inhibition prior to oxygen-withdrawal facilitates the occurrence of hypoxia-induced spreading depression in rat hippocampal slices. J. Neurophysiol. 2006, 96, 492–504. [Google Scholar] [CrossRef]
- Takano, T.; Tian, G.-F.; Peng, W.; Lou, N.; Lovatt, D.; Hansen, A.J.; Kasischke, K.A.; Nedergaard, M. Cortical spreading depression causes and coincides with tissue hypoxia. Nat. Neurosci. 2007, 10, 754–762. [Google Scholar] [CrossRef]
- Hoffmann, U.; Sukhotinsky, I.; Eikermann-Haerter, K.; Ayata, C. Glucose modulation of spreading depression susceptibility. J. Cereb. Blood Flow Metab. 2013, 33, 191–195. [Google Scholar] [CrossRef]
- Kilic, K.; Karatas, H.; Dönmez-Demir, B.; Eren-Kocak, E.; Gursoy-Ozdemir, Y.; Can, A.; Petit, J.-M.; Magistretti, P.J.; Dalkara, T. Inadequate brain glycogen or sleep increases spreading depression susceptibility. Ann. Neurol. 2018, 83, 61–73. [Google Scholar] [CrossRef]
- Peroutka, S.J. Neurogenic inflammation and migraine: implications for the therapeutics. Mol. Interv. 2005, 5, 304. [Google Scholar] [CrossRef]
- Lukacs, M.; Tajti, J.; Fulop, F.; Toldi, J.; Edvinsson, L.; Vecsei, L. Migraine, Neurogenic Inflammation, Drug Development—Pharmacochemical Aspects. Curr. Med. Chem. 2017, 24, 3649–3665. [Google Scholar] [CrossRef]
- Ramachandran, R. Neurogenic inflammation and its role in migraine. Semin. Immunopathol. 2018, 40, 301–314. [Google Scholar] [CrossRef]
- Diener, H.-C.; Goadsby, P.; Asghar, M.; Hansen, A.; Kapijimpanga, T.; Edvinsson, L.; Warfvinge, K.; Olesen, J.; Diener, H. CGRP as a new target in prevention and treatment of migraine. Lancet. Neurol. 2014, 13, 1065–1067. [Google Scholar] [CrossRef]
- Durham, P.L. Calcitonin Gene-Related Peptide (CGRP) and Migraine. Headache J. Head Face Pain 2006, 46, S3–S8. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Edvinsson, L.; Ekman, R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann. Neurol. 1990, 28, 183–187. [Google Scholar] [CrossRef]
- Lassen, L.H.; Haderslev, P.A.; Jacobsen, V.B.; Iversen, H.K.; Sperling, B.; Olesen, J. CGRP may play a causative role in migraine. Cephalalgia Int. J. Headache 2002, 22, 54–61. [Google Scholar] [CrossRef]
- Khan, S.; Olesen, A.; Ashina, M. CGRP, a target for preventive therapy in migraine and cluster headache: Systematic review of clinical data. Cephalalgia 2017, 39, 333102417741297. [Google Scholar] [CrossRef]
- Yuan, H.; Lauritsen, C.G.; Kaiser, E.A.; Silberstein, S.D. CGRP Monoclonal Antibodies for Migraine: Rationale and Progress. BioDrugs 2017, 31, 487–501. [Google Scholar] [CrossRef]
- Akerman, S.; Williamson, D.J.; Kaube, H.; Goadsby, P.J. The effect of anti-migraine compounds on nitric oxide-induced dilation of dural meningeal vessels. Eur. J. Pharmacol. 2002, 452, 223–228. [Google Scholar] [CrossRef]
- Ashina, M.; Bendtsen, L.; Jensen, R.; Schifter, S.; Olesen, J. Calcitonin gene-related peptide levels during nitric oxide-induced headache in patients with chronic tension-type headache. Eur. J. Neurol. 2001, 8, 173–178. [Google Scholar] [CrossRef]
- OLESEN, J. The role of nitric oxide (NO) in migraine, tension-type headache and cluster headache. Pharmacol. Ther. 2008, 120, 157–171. [Google Scholar] [CrossRef]
- Olesen, J.; Ashina, M. Can nitric oxide induce migraine in normal individuals? Cephalalgia 2015, 35, 1125–1129. [Google Scholar] [CrossRef]
- Boćkowski, L.; Smigielska-Kuzia, J.; Sobaniec, W.; Zelazowska-Rutkowska, B.; Kułak, W.; Sendrowski, K. Anti-inflammatory plasma cytokines in children and adolescents with migraine headaches. Pharmacol. Rep. 2010, 62, 287–291. [Google Scholar] [CrossRef]
- Longoni, M.; Ferrarese, C. Inflammation and excitotoxicity: Role in migraine pathogenesis. Neurol. Sci. 2006, 27, s107–s110. [Google Scholar] [CrossRef]
- Yılmaz, I.A.; Özge, A.; Erdal, M.E.; Edgünlü, T.G.; Çakmak, S.E.; Yalın, O.Ö. Cytokine Polymorphism in Patients with Migraine: Some Suggestive Clues of Migraine and Inflammation. Pain Med. 2010, 11, 492–497. [Google Scholar] [CrossRef]
- Levy, D. Migraine pain, meningeal inflammation, and mast cells. Curr. Pain Headache Rep. 2009, 13, 237–240. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Shao, B.-Z.; Xu, Z.-Q.; Han, B.-Z.; Su, D.-F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front Pharmacol 2015, 6, 262. [Google Scholar] [CrossRef]
- Masino, S.A.; Ruskin, D.N. Ketogenic diets and pain. J. Child Neurol. 2013, 28, 993–1001. [Google Scholar] [CrossRef]
- Ruskin, D.N.; Suter, T.A.C.S.; Ross, J.L.; Masino, S.A. Ketogenic diets and thermal pain: Dissociation of hypoalgesia, elevated ketones, and lowered glucose in rats. J. Pain 2013, 14, 467–474. [Google Scholar] [CrossRef]
- Ruskin, D.N.; Kawamura, M.; Masino, S.A. Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PLoS ONE 2009, 4, e8349. [Google Scholar] [CrossRef]
- Cámara-Lemarroy, C.R.; Rodriguez-Gutierrez, R.; Monreal-Robles, R.; Marfil-Rivera, A. Gastrointestinal disorders associated with migraine: A comprehensive review. World J. Gastroenterol. 2016, 22, 8149–8160. [Google Scholar] [CrossRef]
- Hindiyeh, N.; Aurora, S.K. What the Gut Can Teach Us About Migraine. Curr. Pain Headache Rep. 2015, 19, 33. [Google Scholar] [CrossRef]
- Van Hemert, S.; Breedveld, A.C.; Rovers, J.M.P.; Vermeiden, J.P.W.; Witteman, B.J.M.; Smits, M.G.; de Roos, N.M. Migraine associated with gastrointestinal disorders: Review of the literature and clinical implications. Front. Neurol. 2014, 5, 241. [Google Scholar] [CrossRef]
- De Roos, N.M.; van Hemert, S.; Rovers, J.M.P.; Smits, M.G.; Witteman, B.J.M. The effects of a multispecies probiotic on migraine and markers of intestinal permeability-results of a randomized placebo-controlled study. Eur. J. Clin. Nutr. 2017, 71, 1455–1462. [Google Scholar] [CrossRef]
- Straube, A.; Müller, H.; Stiegelbauer, V.; Frauwallner, A. [Migraine prophylaxis with a probiotic. Results of an uncontrolled observational study with 1020 patients]. MMW Fortschr. Med. 2018, 160, 16–21. [Google Scholar] [CrossRef]
- Aydinlar, E.I.; Dikmen, P.Y.; Tiftikci, A.; Saruc, M.; Aksu, M.; Gunsoy, H.G.; Tozun, N. IgG-Based Elimination Diet in Migraine Plus Irritable Bowel Syndrome. Headache J. Head Face Pain 2013, 53, 514–525. [Google Scholar] [CrossRef]
- Lindefeldt, M.; Eng, A.; Darban, H.; Bjerkner, A.; Zetterström, C.K.; Allander, T.; Andersson, B.; Borenstein, E.; Dahlin, M.; Prast-Nielsen, S. The ketogenic diet influences taxonomic and functional composition of the gut microbiota in children with severe epilepsy. NPJ Biofilms Microbiomes 2019, 5, 5. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, S.; Zhou, Y.; Yu, L.; Zhang, L.; Wang, Y. Altered gut microbiome composition in children with refractory epilepsy after ketogenic diet. Epilepsy Res. 2018, 145, 163–168. [Google Scholar] [CrossRef]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 173, 1728–1741.e13. [Google Scholar] [CrossRef]
- Xie, G.; Zhou, Q.; Qiu, C.-Z.; Dai, W.-K.; Wang, H.-P.; Li, Y.-H.; Liao, J.-X.; Lu, X.-G.; Lin, S.-F.; Ye, J.-H.; et al. Ketogenic diet poses a significant effect on imbalanced gut microbiota in infants with refractory epilepsy. World J. Gastroenterol 2017, 23, 6164–6171. [Google Scholar] [CrossRef]
- Newell, C.; Bomhof, M.R.; Reimer, R.A.; Hittel, D.S.; Rho, J.M.; Shearer, J. Ketogenic diet modifies the gut microbiota in a murine model of autism spectrum disorder. Mol. Autism 2016, 7, 37. [Google Scholar] [CrossRef]
- Swidsinski, A.; Dörffel, Y.; Loening-Baucke, V.; Gille, C.; Göktas, Ö.; Reißhauer, A.; Neuhaus, J.; Weylandt, K.-H.; Guschin, A.; Bock, M. Reduced Mass and Diversity of the Colonic Microbiome in Patients with Multiple Sclerosis and Their Improvement with Ketogenic Diet. Front. Microbiol. 2017, 8, 1141. [Google Scholar] [CrossRef]
- Tagliabue, A.; Ferraris, C.; Uggeri, F.; Trentani, C.; Bertoli, S.; de Giorgis, V.; Veggiotti, P.; Elli, M. Short-term impact of a classical ketogenic diet on gut microbiota in GLUT1 Deficiency Syndrome: A 3-month prospective observational study. Clin. Nutr. ESPEN 2017, 17, 33–37. [Google Scholar] [CrossRef]
- Klement, R.J.; Pazienza, V. Impact of Different Types of Diet on Gut Microbiota Profiles and Cancer Prevention and Treatment. Medicina (Kaunas) 2019, 55, 84. [Google Scholar] [CrossRef]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Gross, E.; Putananickal, N.; Orsini, A.-L.; Schmidt, S.; Vogt, D.R.; Cichon, S.; Sandor, P.; Fischer, D. Efficacy and safety of exogenous ketone bodies for preventive treatment of migraine: A study protocol for a single-centred, randomised, placebo-controlled, double-blind crossover trial. Trials 2019, 20, 61. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gross, E.C.; Klement, R.J.; Schoenen, J.; D’Agostino, D.P.; Fischer, D. Potential Protective Mechanisms of Ketone Bodies in Migraine Prevention. Nutrients 2019, 11, 811. https://doi.org/10.3390/nu11040811
Gross EC, Klement RJ, Schoenen J, D’Agostino DP, Fischer D. Potential Protective Mechanisms of Ketone Bodies in Migraine Prevention. Nutrients. 2019; 11(4):811. https://doi.org/10.3390/nu11040811
Chicago/Turabian StyleGross, Elena C., Rainer J. Klement, Jean Schoenen, Dominic P. D’Agostino, and Dirk Fischer. 2019. "Potential Protective Mechanisms of Ketone Bodies in Migraine Prevention" Nutrients 11, no. 4: 811. https://doi.org/10.3390/nu11040811
APA StyleGross, E. C., Klement, R. J., Schoenen, J., D’Agostino, D. P., & Fischer, D. (2019). Potential Protective Mechanisms of Ketone Bodies in Migraine Prevention. Nutrients, 11(4), 811. https://doi.org/10.3390/nu11040811