Choline Supplementation in Cystic Fibrosis—The Metabolic and Clinical Impact

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
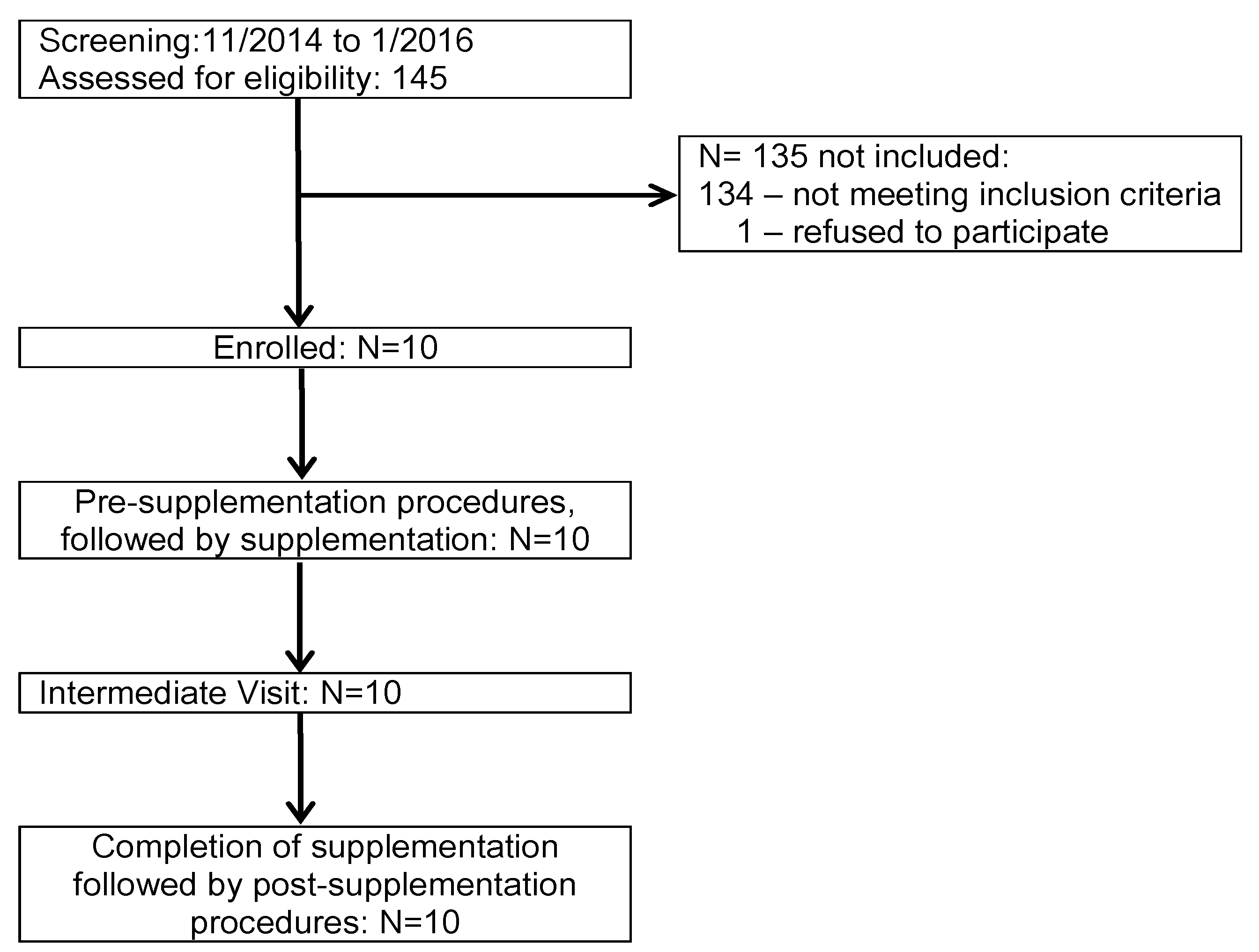
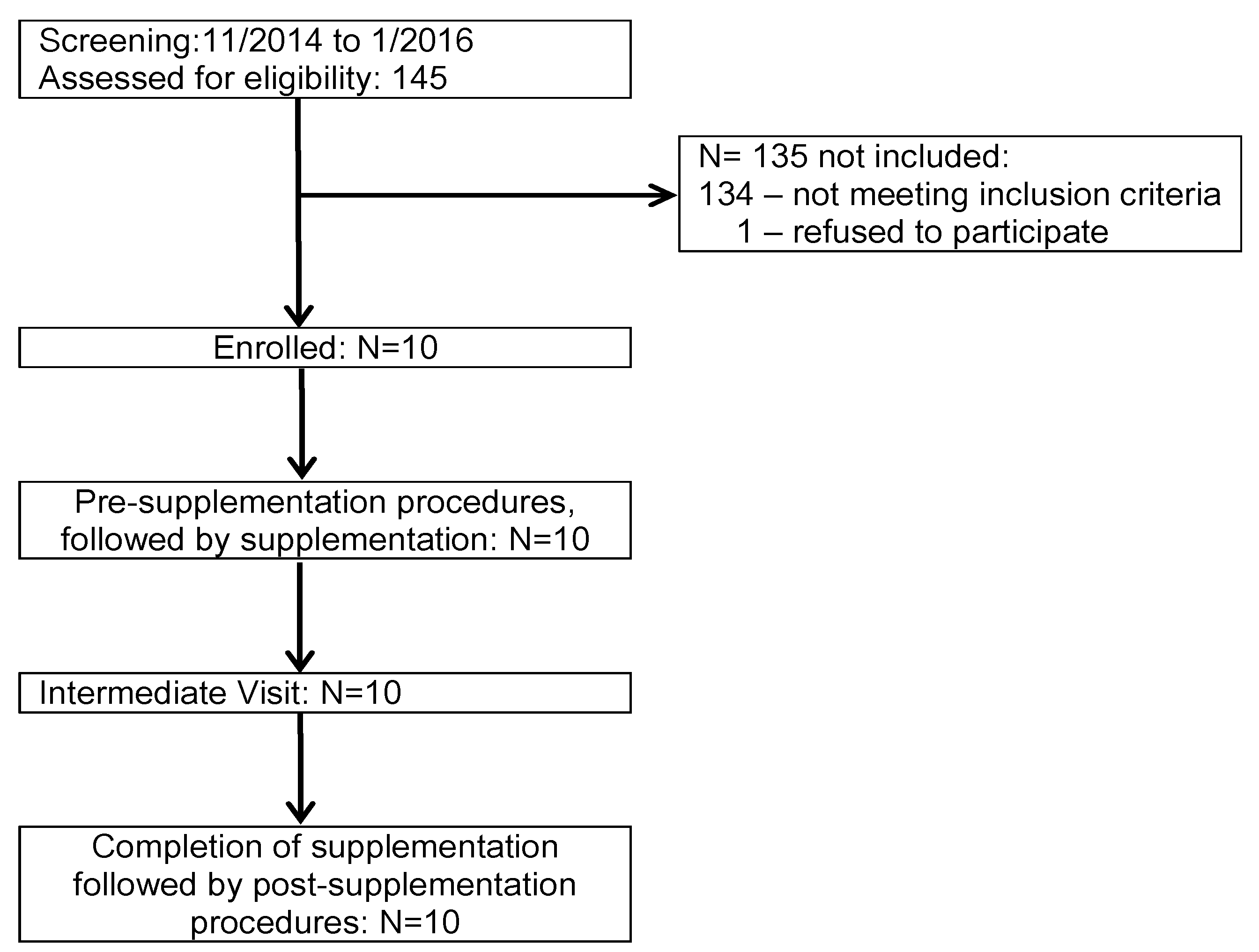
2.1. Study Population
2.2. Intervention
2.3. Outcome and Safety Parameters
2.4. Sample Size Calculation
2.5. Investigation of Choline and Phosphatidylcholine (PC) Metabolism
2.6. Blood Collection and Analytical Procedures
2.7. Analysis of Choline, Choline Metabolites, and Phospholipids
2.8. Clinical Parameters
2.9. Statistics
3. Results
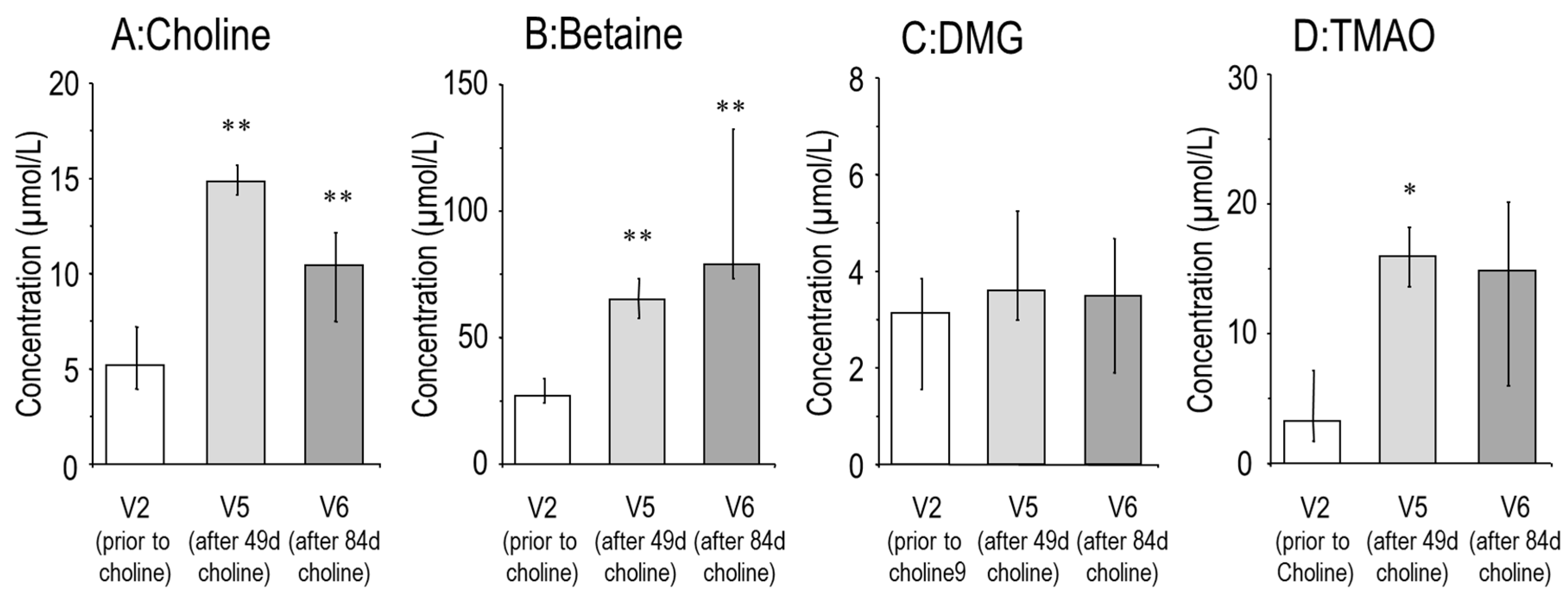
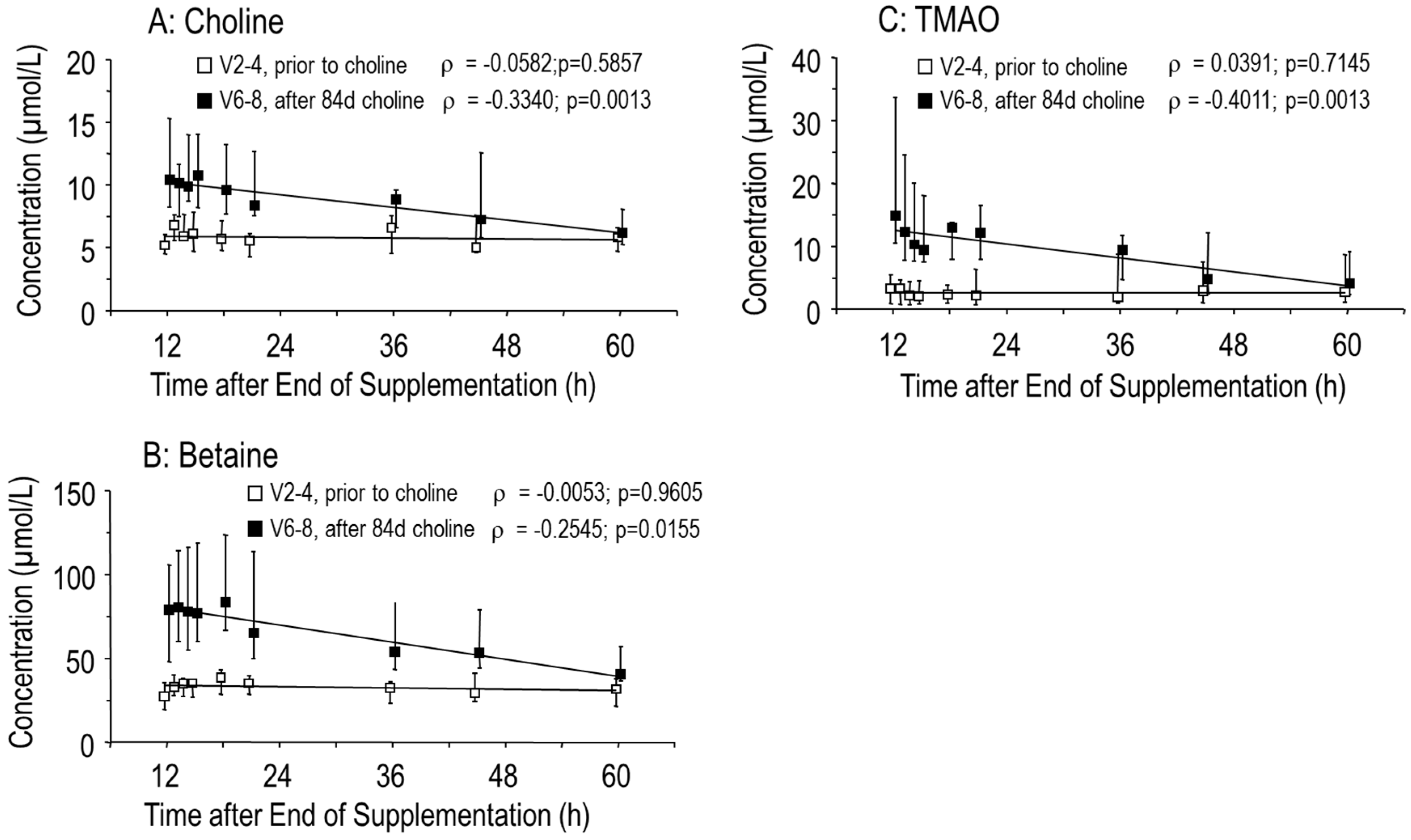
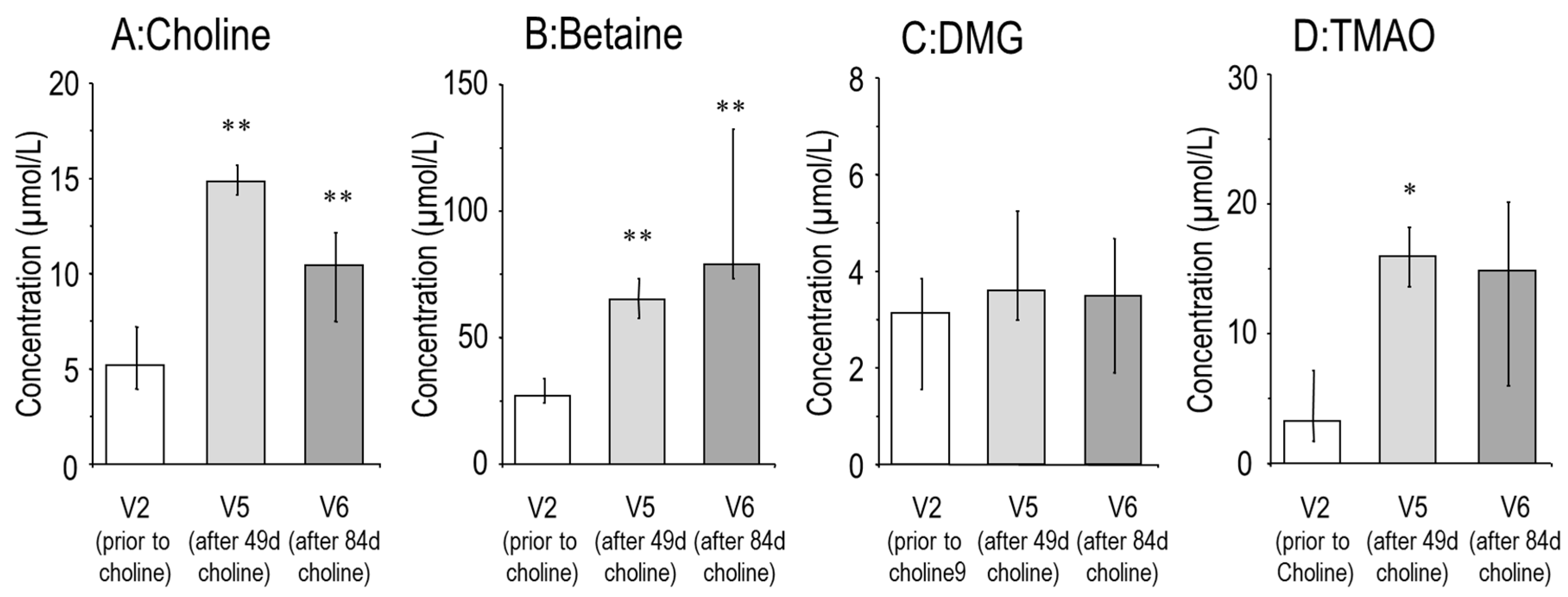
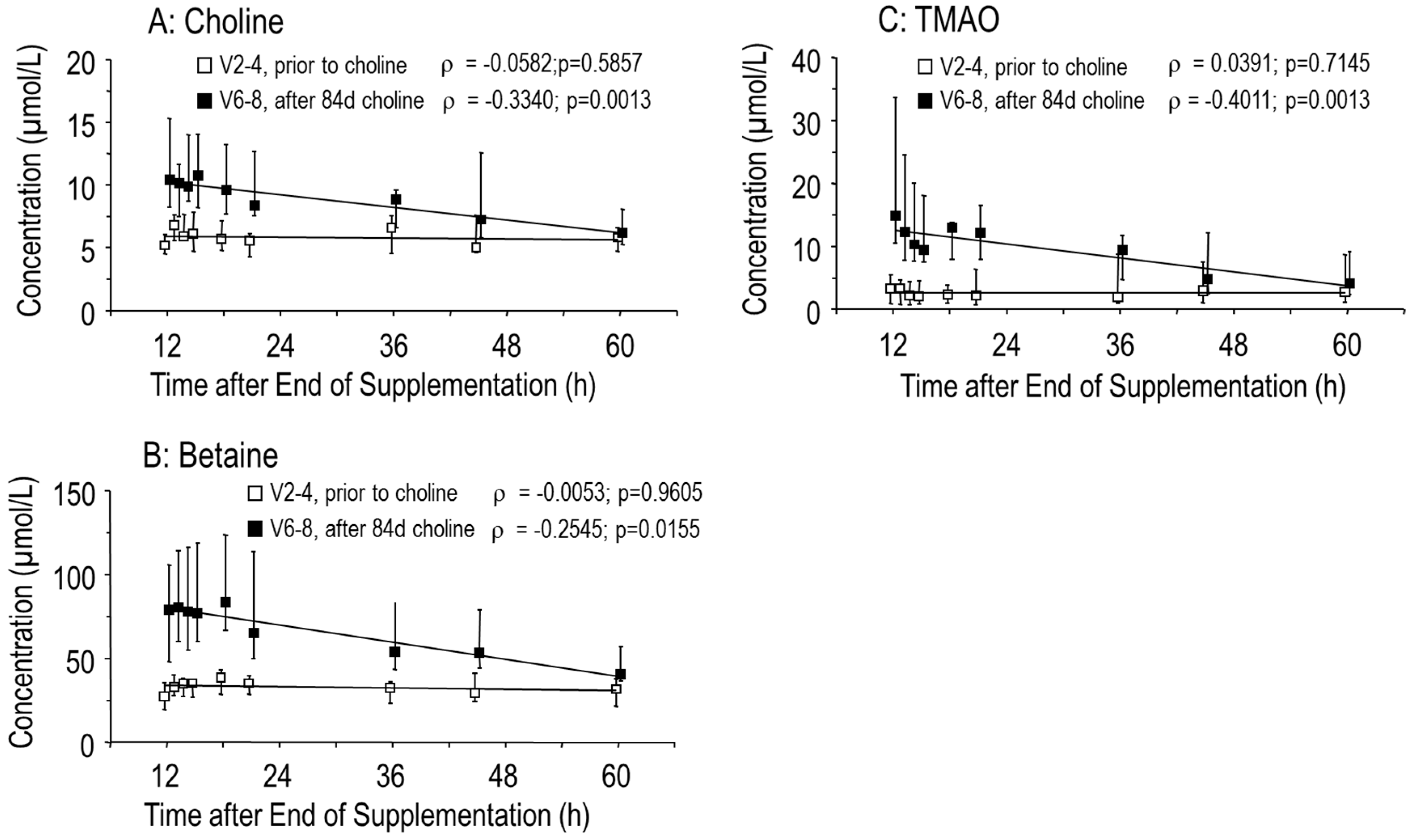
3.1. Effects of Choline Supplementation on Plasma Choline and Its Water-Soluble Metabolites
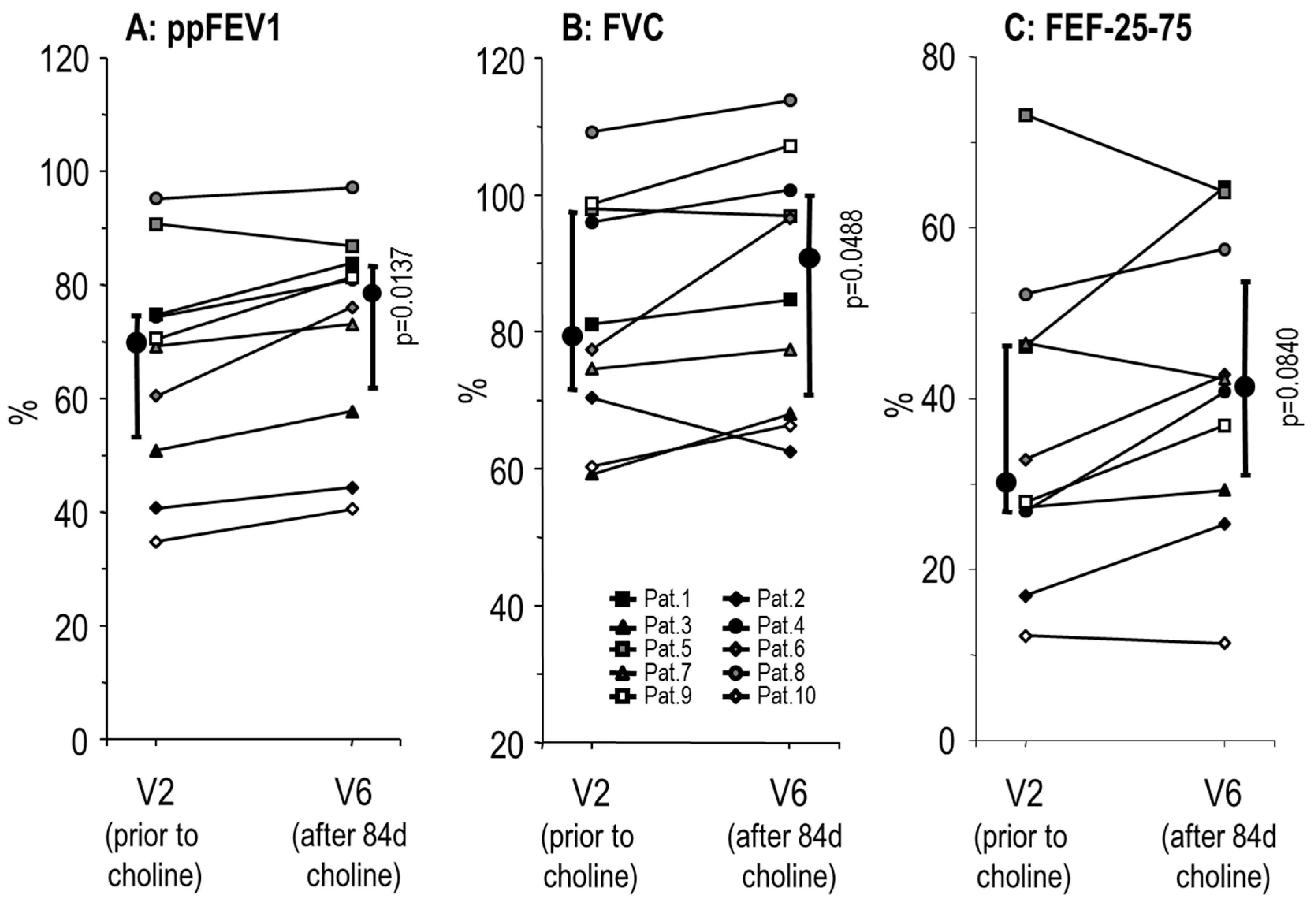
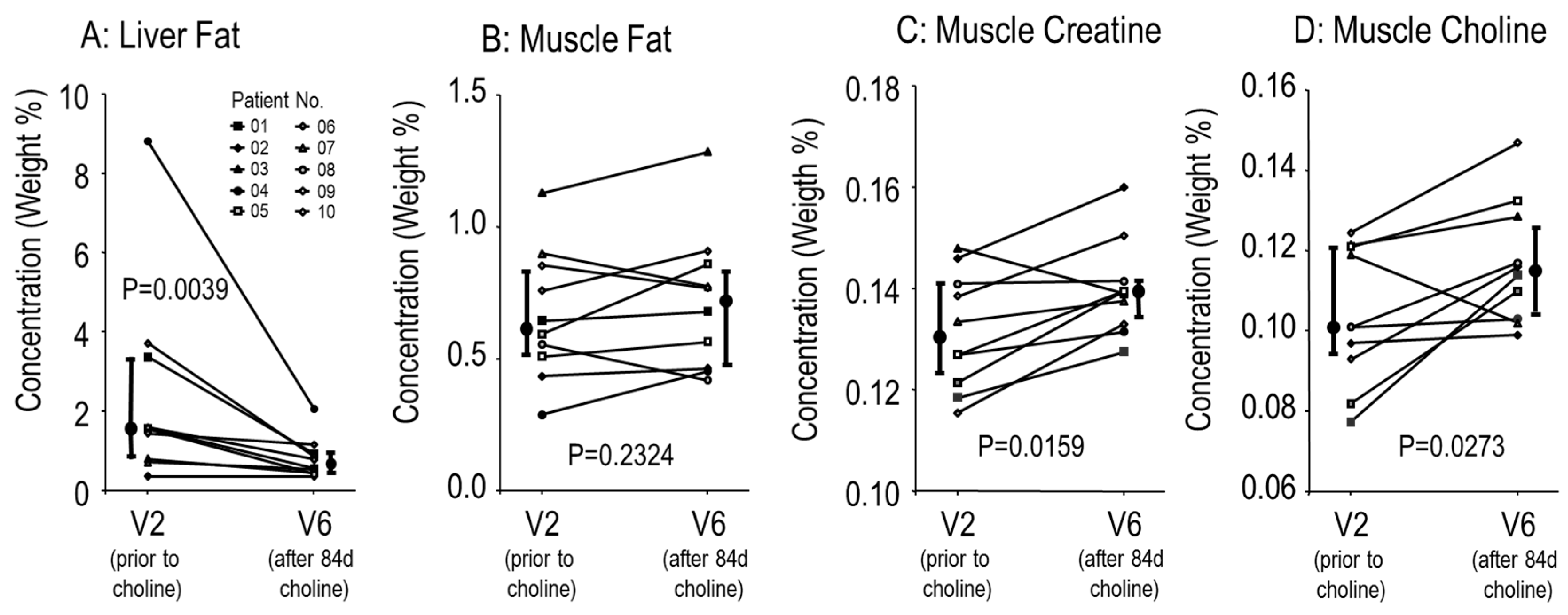
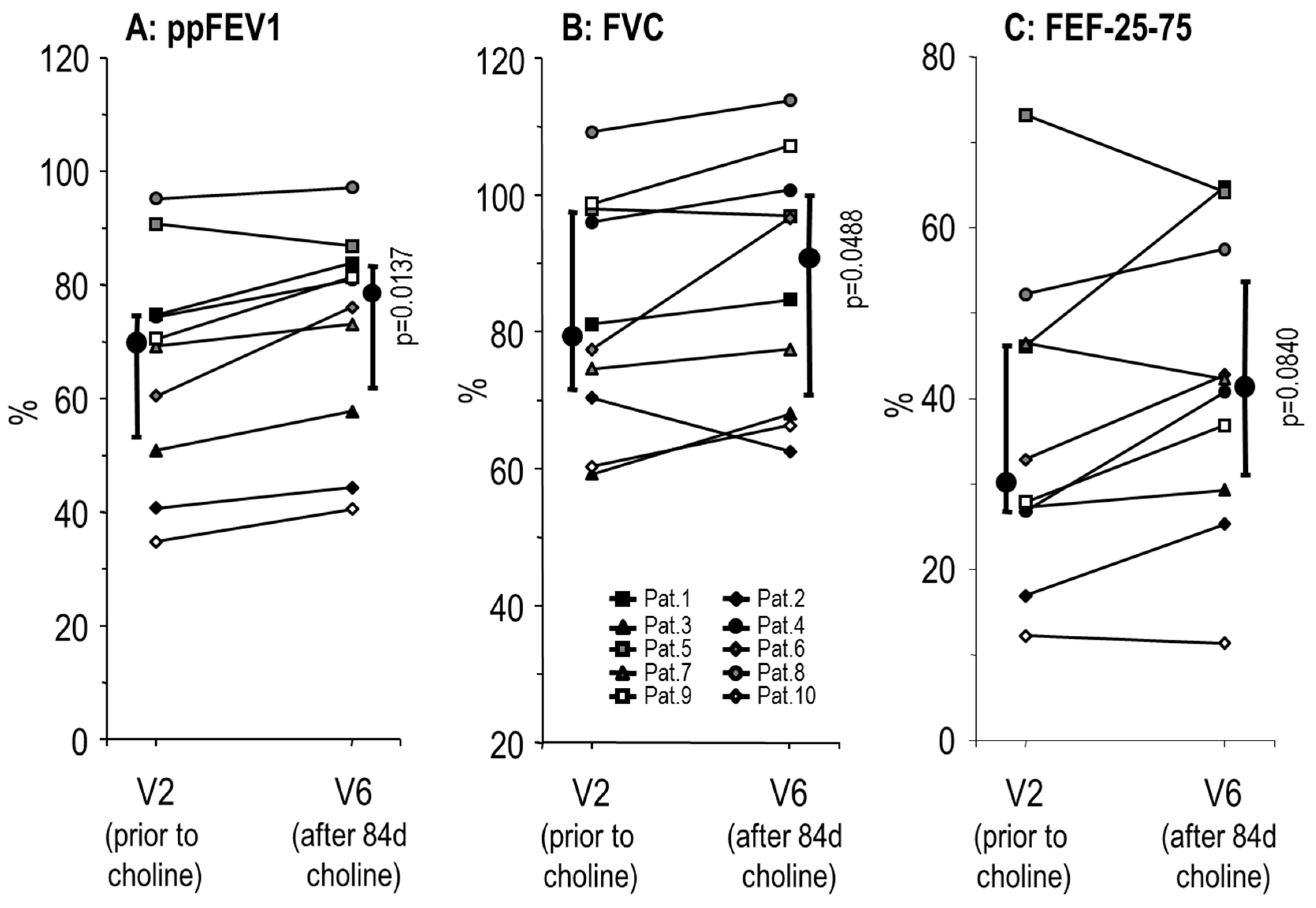
3.2. Effects of Choline Supplementation on Lung Function and Liver Triglycerides
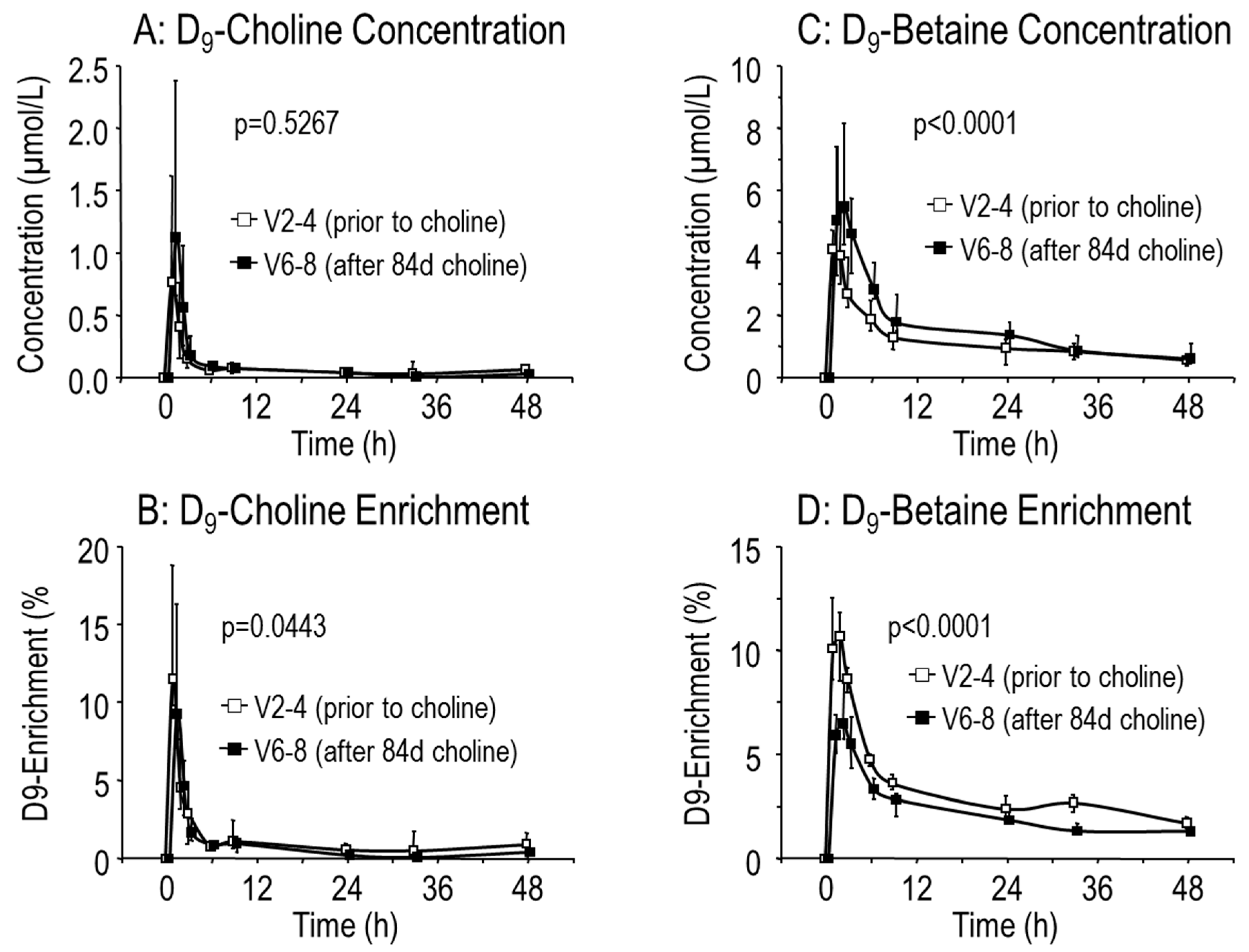
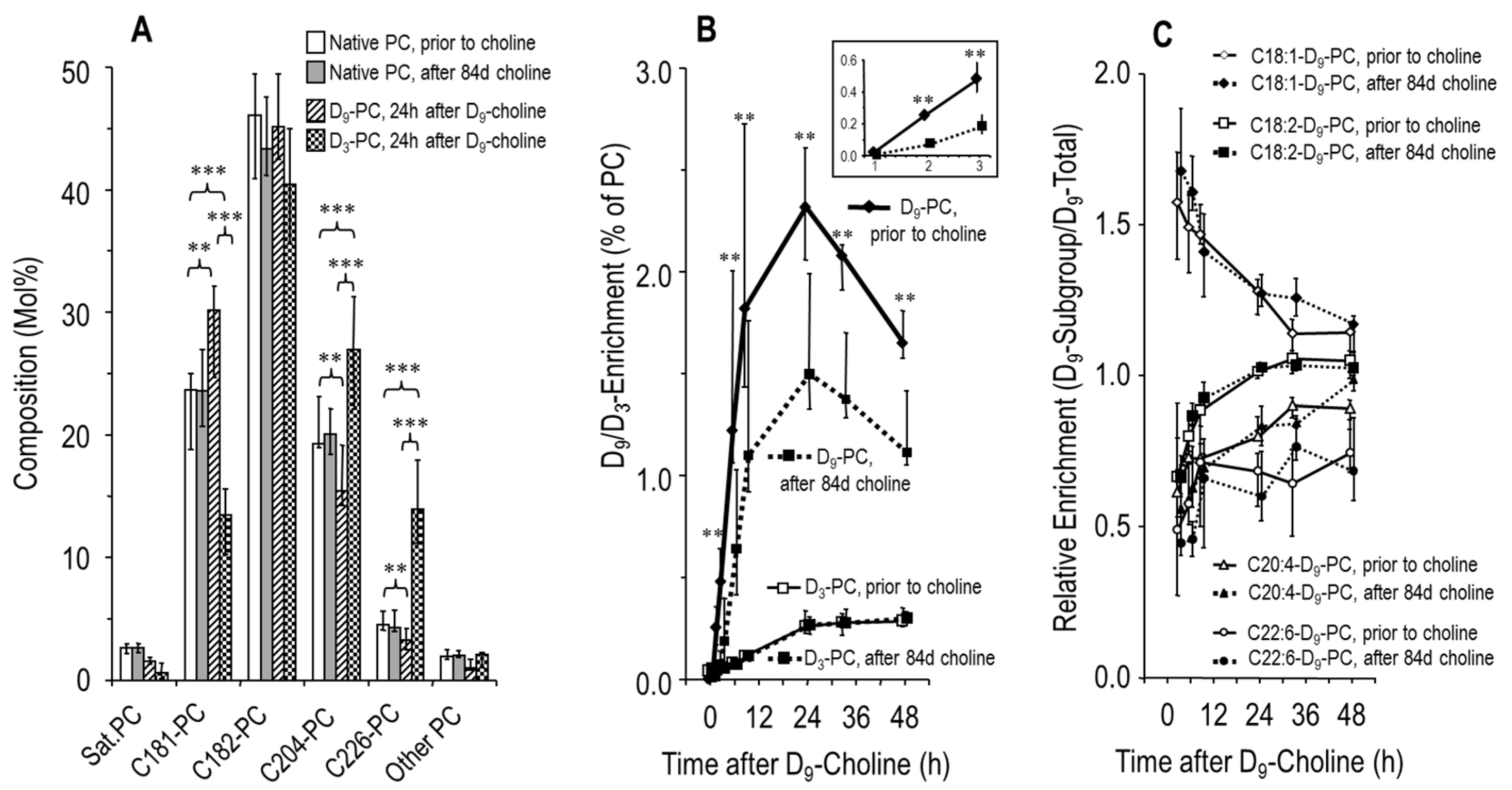
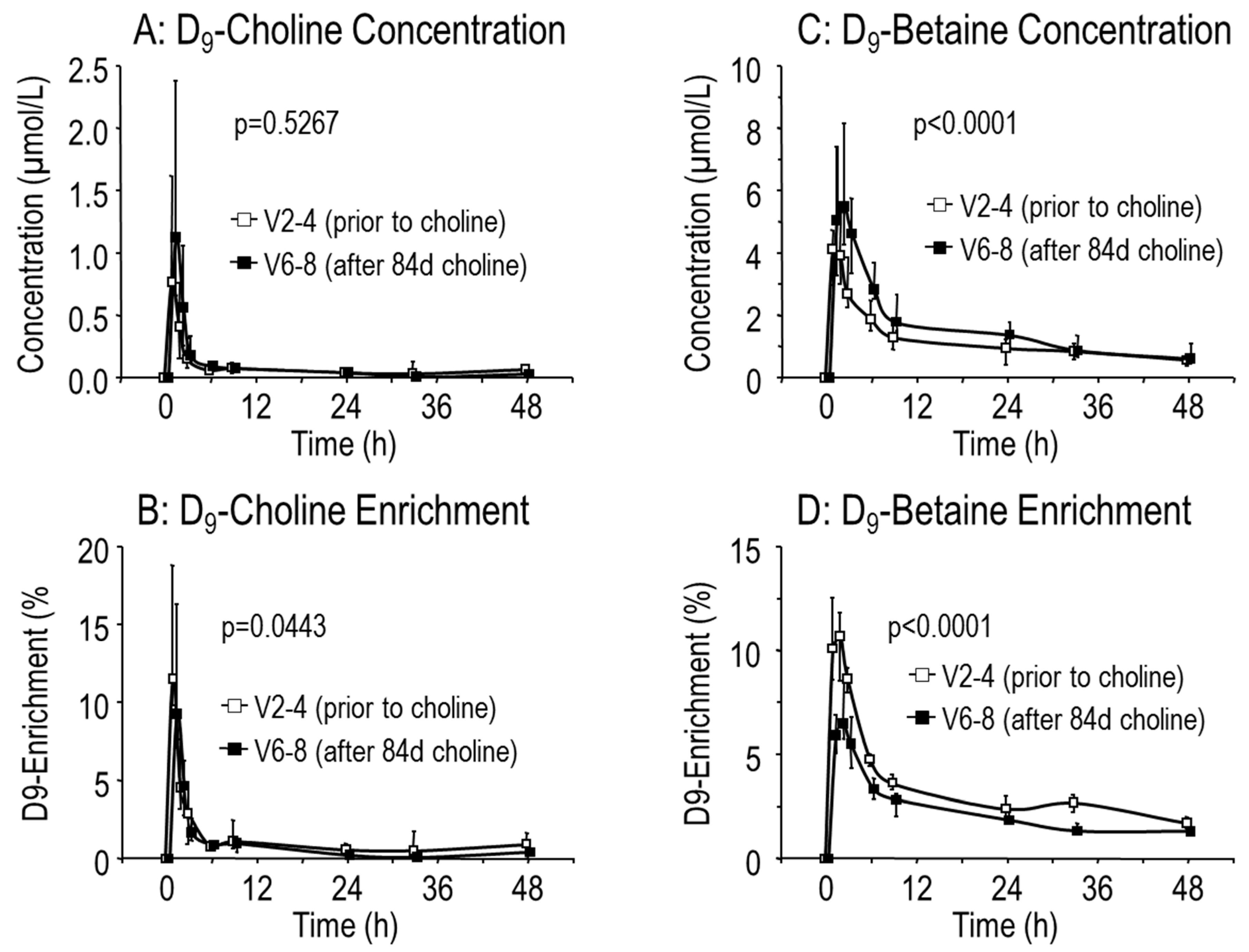
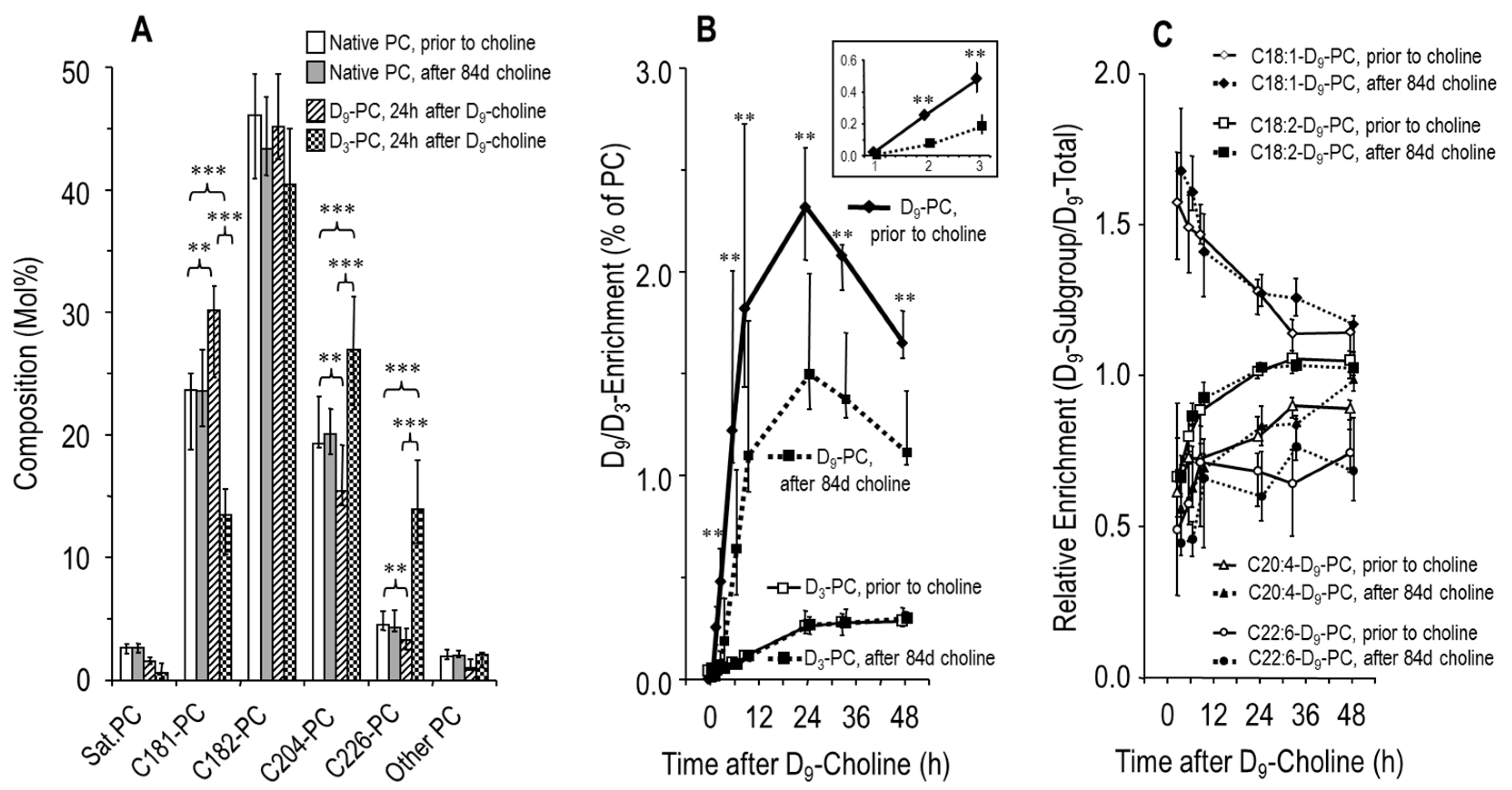
3.3. Effects of Choline Supplementation on Plasma Phosphatidylcholine Metabolism
4. Discussion
Limitations
5. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Parameters | Exclusion Parameters |
|---|---|
|
|
| Days (d) Relative to Start of Supplementation | Visit 2–4 (−2 to 0) | Visit 5 (d 49) | Visit 6–8 (d 84–86) |
|---|---|---|---|
| Primary Endpoint | |||
| Plasma D9-PC Concentration 33 h after D9-choline | x | x | |
| Predefined Secondary Endpoints | |||
| Lung Function (FEV1, FVC, MEF-25, MEF 25-75) | x | x | x |
| Liver Fat; Muscle Creatine, Choline, and Fat | x | x | |
| Plasma Choline, Betaine, Dimethylglycine, Methionine, Homocysteine, Trimethylamine Oxide | x | x | x |
| Plasma Phosphatidylcholine (PC), Sphingomyelin (SPH), Phosphatidylethanolamine (PE) | x | x | x |
| Plasma Triglycerides, HDL-Cholesterol, LDL-Cholesterol, | x | x | |
| Additional Secondary Outcome Parameters | |||
| Kinetics of D9-Choline, D9-Betaine, D9-PC, D3-PC; Molecular Composition of Plasma PC | x x | x x |
| Visit | V1 | V2–4 | V5 | V6–8 |
|---|---|---|---|---|
| Days (d) relative to | d −14 | d ‒2 to 0 | d 49 | d 84 to 86 |
| start of supplementation | ||||
| Age (years) | 24 (20–34); range 18–46 | |||
| Choline Intake (days) | – | – | 49 (45–51) | V6: 84 (84–91) |
| Body Weight (kg) | 65 (61–71) | 63 (61–70) | 62 (60–70) | |
| Length (cm) | 173 (171–175) | |||
| BMI (kg/m2) | 22.7 (21.3–23.6) | 21.6 (21.2–23.3) | 21.7 (21.0–23.3) | |
| Heart Rate (bpm) | 72 (72–81) | 77 (73–88) | 78 (72–81) | 78 (73–85) |
| Systolic BP (mmHg) | 120 (112–125) | 117 (73–88) | 115 (114–124) | 114 (110–119) |
| Diastolic BP (mmHg) | 75 (65–75) | 68 (66–76) | 70 (67–76) | 67 (65–73) |
| HbSO2 (%) | 97 (95–97) | 96 (95–97) | 96 (96–97) | 97 (95–98) |
| Sodium (mmol/L) | 141 (139–141) | 141 (140–142) | 140 (137–142) | 140 (139–141) |
| Potassium (mmol/L) | 4.0 (3.9–4.4) | 3.9 (3.7–4.2) | 4.0 (3.9–4.4) | 3.9 (3.7–4.1) |
| Creatinine (mmol/L) | 0.9 (0.7–0.9) | 0.8 (0.7–1.0) | 0.9 (0.7–0.9) | 0.8 (0.7–1.0) |
| Urea (mmol/L) | 33 (26–36) | 27 (25–33) | 31 (26–37) | 29 (23–34) |
| AST (U/L) | 41 (27–74) | 34 (27–50) | 30 (25–41) | 33 (23–37) |
| ALT (U/L) | 48 (27–77) | 35 (27–68) | 36 (27–48) | 34 (27–42) |
| GGT(U/L) | 21 (14–64) | 17 (16–56) | 22 (17–69) | 17 (14–22) |
| AP (U/L) | 116 (103–138) | 125 (102–163) | 97 (95–134) | 117 (105–121) |
| Bilirubin (mg/dL) | 0.70 (0.48–0.95) | 0.65 (0.43–0.90) | 0.60 (0.50–0.70) | 0.75 (0.50–1.08) |
| Cholesterol (mg/dL) | 124 (98–144) | 132 (106–155) | 142 (106–154) | 125 (102–142) |
| HDL-Chol. (mg/dL) | 35 (27–49) | 41 (33–46) | 38 (32–39) | 40 (34–44) |
| LDL-Chol. (mg/dL) | 58 (52–90) | 64 (59–112) | 61 (49–102) | 68 (49–93) |
| Apo-B (mg/dL) | 61 (50–71) | 60 (52–79) | 62 (49–85 | 62 (49–75) |
| Triglycerides (mg/dL) | 65 (62–90) | 64 (56–99) | 84 (65–137) | 87 (64–110) |
| Hemoglobin (g/dL) | 15.5 (15.0–15.9) | 14.7 (14.5–16.4) | 15.0 (14.0–15.2) | 14.7 (14.4–15.6) |
| Leucocytes (µL-1) | 8170 (7103–9413) | 8575 (7033–9610) | 9040 (6840–9580) | 8830 (7265–9890) |
| Thromboc. (x103/mL) | 236 (193–262) | 264 (240–281) | 252 (222–300) | 254 (210–288) |
| CRP (mg/dL) | 0.35 (0.16–0.7) | 0.33 (0.18–0.50) | 0.17 (0.10–0.60) | 0.70 (0.23–0.89) |
| Interleukin 6 (ng/L) | 2.0 (2.0–7.0) | 4.0 (2.5–6.1) | 2.5 (2.0–4.9) | 3.5 (2.5–6.4) |
References
- Uhlig, S.; Gulbins, E. Sphingolipids in the lungs. Am. J. Respir. Crit. Care Med. 2008, 178, 1100–1114. [Google Scholar] [CrossRef]
- Becker, K.A.; Riethmüller, J.; Lüth, A.; Döring, G.; Kleuser, B.; Gulbins, E. Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 42, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Tafesse, F.G.; Ternes, P.; Holthuis, J.C. The multigenic sphingomyelin synthase family. J. Biol. Chem. 2006, 281, 29421–29425. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.H.; Innis, S.M.; Davidson, A.G.; James, S.J. Phosphatidylcholine and lysophosphatidylcholine excretion is increased in children with cystic fibrosis and is associated with plasma homocysteine, S-adenosylhomocysteine, and S-adenosylmethionine. Am. J. Clin. Nutr. 2005, 81, 686–691. [Google Scholar] [CrossRef]
- Bradbury, N.A. Intracellular CFTR: Localization and function. Physiol. Rev. 2011, 79, S175–S191. [Google Scholar] [CrossRef]
- Colombo, C.; Battezzati, P.M.; Crosignani, A.; Morabito, A.; Costantini, D.; Padoan, R.; Giunta, A. Liver disease in cystic fibrosis: A prospective study on incidence, risk factors, and outcome. Hepatology 2002, 36, 1374–1382. [Google Scholar] [CrossRef]
- Williams, S.M.; Goodman, R.; Thomson, R.; Mchugh, K.; Lindsell, D.R.M. Ultrasound evaluation of liver disease in cystic fibrosis as part of an annual assessment clinic: A 9-year review. Clin. Radiol. 2002, 57, 365–370. [Google Scholar] [CrossRef]
- Herrmann, U.; Dockter, G.; Lammert, F. Cystic fibrosis-associated liver disease. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 585–592. [Google Scholar] [CrossRef]
- Freudenberg, F.; Broderick, A.L.; Yu, B.B.; Leonard, M.R.; Glickman, J.N.; Carey, M.C. Pathophysiological basis of liver disease in cystic fibrosis employing a DeltaF508 mouse model. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1411–G1420. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation Patient Registry. Annual Data Report. Bethesda, Maryland. 2017. Available online: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Patient-Registry-Annual-Data-Report.pdf (accessed on 28 January 2019).
- Canadian Cystic Fibrosis Foundation Registry. Annual Data Report. Toronto, ON, Canada, 2017. Available online: https://www.cysticfibrosis.ca/uploads/Registry%20Report%202017/2017%20Registry%20Annual%20Data%20Report.pdf (accessed on 28 January 2019).
- Lindblad, A.; Glaumann, H.; Strandvik, B. Natural history of liver disease in cystic fibrosis. Hepatology 1999, 30, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Grothe, J.; Riethmüller, J.; Tschürtz, S.M.; Raith, M.; Pynn, C.J.; Stoll, D.; Bernhard, W. Plasma phosphatidylcholine alterations in cystic fibrosis patients: Impaired metabolism and correlation with lung function and inflammation. Cell. Physiol. Biochem. 2015, 35, 1437–1453. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.P.; Nissim, I.; Brosnan, M.E.; Brosnan, J.T. Creatine synthesis: Hepatic metabolism of guanidinoacetate and creatine in the rat in vitro and in vivo. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E256–E261. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W.; Poets, C.F.; Franz, A.R. Choline and choline-related nutrients in regular and preterm infant growth. Eur. J. Nutr. 2018. [Google Scholar] [CrossRef] [PubMed]
- National Academy of Sciences USA, Institute of Medicine, Food and Nutrition Board Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline and Subcommittee on Upper Reference Levels of Nutrients; National Academies Press: Washington, DC, USA, 1998; Volume 1, pp. 390–422. Available online: https://www.nap.edu/read/6015/chapter/14 (accessed on 28 January 2019).
- Da Costa, K.A.; Kozyreva, O.G.; Song, J.; Galanko, J.A.; Fischer, L.M.; Zeisel, S.H. Common genetic polymorphisms affect the human requirement for the nutrient choline. FASEB J. 2006, 20, 1336–1344. [Google Scholar] [CrossRef]
- Zeisel, S.H. Dietary choline: Biochemistry, physiology, and pharmacology. Ann. Rev. Nutr. 1981, 1, 95–121. [Google Scholar] [CrossRef]
- Li, Z.; Agellon, L.B.; Vance, D.E. Phosphatidylcholine homeostasis and liver failure. J. Biol. Chem. 2005, 280, 37798–37802. [Google Scholar] [CrossRef]
- Scheele, G.A.; Fukuoka, S.I.; Kern, H.F.; Freedman, S.D. Pancreatic dysfunction in cystic fibrosis occurs as a result of impairments in luminal pH, apical trafficking of zymogen granule membranes, and solubilization of secretory enzymes. Pancreas 1996, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W.; Gesche, J.; Raith, M.; Poets, C.F. Phosphatidylcholine kinetics in neonatal rat lungs and the effects of rhuKGF and betamethasone. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L955–L963. [Google Scholar] [CrossRef]
- Bates, S.R.; Tao, J.Q.; Yu, K.J.; Borok, Z.; Crandall, E.D.; Collins, H.L.; Rothblat, G.H. Expression and biological activity of ABCA1 in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2008, 38, 283–292. [Google Scholar] [CrossRef]
- Zhou, J.; You, Y.; Ryan, A.J.; Mallampalli, R.K. Upregulation of surfactant synthesis triggers ABCA1-mediated basolateral phospholipid efflux. J. Lipid Res. 2004, 45, 1758–1767. [Google Scholar] [CrossRef]
- Agassandian, M.; Miakotina, O.L.; Andrews, M.; Mathur, S.N.; Mallampalli, R.K. Pseudomonas aeruginosa and sPLA2 IB stimulate ABCA1-mediated phospholipid efflux via ERK-activation of PPARalpha-RXR. Biochem. J. 2007, 403, 409–420. [Google Scholar] [CrossRef]
- Li, Z.; Agellon, L.B.; Vance, D.E. Choline redistribution during adaptation to choline deprivation. J. Biol. Chem. 2007, 282, 10283–10289. [Google Scholar] [CrossRef]
- Bernhard, W.; Pynn, C.J.; Jaworski, A.; Rau, G.A.; Hohlfeld, J.M.; Freihorst, J.; Poets, C.F.; Stoll, D.; Postle, A.D. Mass spectrometric analysis of surfactant metabolism in human volunteers using deuterated choline. Am. J. Respir. Crit. Care Med. 2004, 170, 54–58. [Google Scholar] [CrossRef]
- Pynn, C.J.; Henderson, N.G.; Clark, H.; Koster, G.; Bernhard, W.; Postle, A.D. Specificity and rate of human and mouse liver and plasma phosphatidylcholine synthesis analyzed in vivo. J. Lipid Res. 2011, 52, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M.; Davidson, A.G.; Melynk, S.; James, S.J. Choline-related supplements improve abnormal plasma methionine-homocysteine metabolites and glutathione status in children with cystic fibrosis. Am. J. Clin. Nutr. 2007, 85, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W.; Raith, M.; Kunze, R.; Koch, V.; Heni, M.; Maas, C.; Abele, H.; Poets, C.F.; Franz, A.R. Choline concentrations are lower in postnatal plasma of preterm infants than in cord plasma. Eur. J. Nutr. 2015, 54, 733–741. [Google Scholar] [CrossRef]
- Bernhard, W.; Raith, M.; Koch, V.; Kunze, R.; Maas, C.; Abele, H.; Poets, C.F.; Franz, A.R. Plasma phospholipids indicate impaired fatty acid homeostasis in preterm infants. Eur. J. Nutr. 2014, 53, 1533–1547. [Google Scholar] [CrossRef]
- Bernhard, W.; Raith, M.; Koch, V.; Maas, C.; Abele, H.; Poets, C.F.; Franz, A.R. Developmental changes in polyunsaturated fetal plasma phospholipids and feto-maternal plasma phospholipid ratios and their association with bronchopulmonary dysplasia. Eur. J. Nutr. 2016, 55, 2265–2274. [Google Scholar] [CrossRef] [PubMed]
- Machann, J.; Thamer, C.; Schnoedt, B.; Stefan, N.; Haring, H.U.; Claussen, C.D.; Fritsche, A.; Schick, F. Hepatic lipid accumulation in healthy subjects: A comparative study using spectral fat-selective MRI and volume-localized 1H-MR spectroscopy. Magn. Reson. Med. 2006, 55, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, N.J.; Isselbacher, K.J. Diseases of the gallbladder and bile ducts. In Harrison’s Principles of Internal Medicine; Fauci, A., Braunwald, E., Isselbacher, K.J., Wilson, J.D., Martin, J.B., Eds.; McGraw-Hill: New York, NY, USA, 1998; pp. 1725–1726. [Google Scholar]
- Hollenbeck, C.B. An introduction to the nutrition and metabolism of choline. Cent. Nerv. Syst. Agents Med. Chem. 2012, 12, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Lockman, P.R.; Allen, D.D. The transport of choline. Drug Dev. Ind. Pharm. 2002, 28, 749–771. [Google Scholar] [CrossRef]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [PubMed]
- Bernhard, W. Regulation of Surfactant-Associated Phospholipid Synthesis and Secretion. In Fetal and Neonatal Physiology, 5th ed.; Polin, R.A., Abman, S.H., Rowitch, D., Benitz, W.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 813–824. [Google Scholar]
- Sun, X.; Sui, H.; Fisher, J.T.; Yan, Z.; Liu, X.; Cho, H.J.; Joo, N.S.; Zhang, Y.; Zhou, W.; Yi, Y.; et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J. Clin. Investig. 2010, 120, 3149–3160. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, D.A.; Meyerholz, D.K.; Pezzulo, A.A.; Ramachandran, S.; Rogan, M.P.; Davis, G.J.; Hanfland, R.A.; Wohlford-Lenane, C.; Dohrn, C.L.; Bartlett, J.A.; et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci. Transl. Med. 2010, 2, 29ra31. [Google Scholar] [CrossRef] [PubMed]
- Vance, D.E. Role of phosphatidylcholine biosynthesis in the regulation of lipoprotein homeostasis. Curr. Opin. Lipidol. 2008, 19, 229–234. [Google Scholar] [CrossRef]
- Ji, R.R.; Xu, Z.Z.; Strichartz, G.; Serhan, C.N. Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. 2011, 34, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Leggieri, E.; De Biase, R.V.; Savi, D.; Zullo, S.; Halili, I.; Quattrucci, S. Clinical effects of diet supplementation with DHA in pediatric patients suffering from cystic fibrosis. Minerva Pediatr. 2013, 65, 389–398. [Google Scholar]
- Bernhard, W.; Maas, C.; Shunova, A.; Mathes, M.; Böckmann, K.; Bleeker, C.; Vek, J.; Poets, C.F.; Schleicher, E.; Franz, A.R. Transport of long-chain polyunsaturated fatty acids in preterm infant plasma is dominated by phosphatidylcholine. Eur. J. Nutr. 2018, 57, 2105–2112. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Growdon, J.H.; Wurtman, R.J.; Magil, S.G.; Logue, M. Normal plasma choline responses to ingested lecithin. Neurology 1980, 30, 1226–1229. [Google Scholar] [CrossRef]
- Holmes-McNary, M.Q.; Cheng, W.L.; Mar, M.H.; Fussell, S.; Zeisel, S.H. Choline and choline esters in human and rat milk and in infant formulas. Am. J. Clin. Nutr. 1996, 64, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Resseguie, M.E.; da Costa, K.A.; Galanko, J.A.; Patel, M.; Davis, I.J.; Zeisel, S.H. Aberrant estrogen regulation of PEMT results in choline deficiency-associated liver dysfunction. J. Biol. Chem. 2011, 286, 1649–1658. [Google Scholar] [CrossRef]
- Fischer, L.M.; da Costa, K.A.; Kwock, L.; Galanko, J.; Zeisel, S.H. Dietary choline requirements of women: Effects of estrogen and genetic variation. Am. J. Clin. Nutr. 2010, 92, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M.; Hasman, D. Evidence of choline depletion and reduced betaine and dimethylglycine with increased homocysteine in plasma of children with cystic fibrosis. J. Nutr. 2006, 136, 2226–2231. [Google Scholar] [CrossRef]
- Milla, C.E.; Ratjen, F.; Marigowda, G.; Liu, F.; Waltz, D.; Rosenfeld, M.; VX13-809-011 Part B Investigator Grou. Lumacaftor/ivacaftor in patients aged 6-11 years with Cystic Fibrosis and homozygous for F508del-CFTR. Am. J. Respir. Crit. Care Med. 2017, 195, 912–920. [Google Scholar] [CrossRef]
- Ratjen, F.; Hug, C.; Marigowda, G.; Tian, S.; Huang, X.; Stanojevic, S.; Milla, C.E.; Robinson, P.D.; Waltz, D.; Davies, J.C.; et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: A randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 2017, 5, 557–567. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernhard, W.; Lange, R.; Graepler-Mainka, U.; Engel, C.; Machann, J.; Hund, V.; Shunova, A.; Hector, A.; Riethmüller, J. Choline Supplementation in Cystic Fibrosis—The Metabolic and Clinical Impact. Nutrients 2019, 11, 656. https://doi.org/10.3390/nu11030656
Bernhard W, Lange R, Graepler-Mainka U, Engel C, Machann J, Hund V, Shunova A, Hector A, Riethmüller J. Choline Supplementation in Cystic Fibrosis—The Metabolic and Clinical Impact. Nutrients. 2019; 11(3):656. https://doi.org/10.3390/nu11030656
Chicago/Turabian StyleBernhard, Wolfgang, Robert Lange, Ute Graepler-Mainka, Corinna Engel, Jürgen Machann, Verena Hund, Anna Shunova, Andreas Hector, and Joachim Riethmüller. 2019. "Choline Supplementation in Cystic Fibrosis—The Metabolic and Clinical Impact" Nutrients 11, no. 3: 656. https://doi.org/10.3390/nu11030656
APA StyleBernhard, W., Lange, R., Graepler-Mainka, U., Engel, C., Machann, J., Hund, V., Shunova, A., Hector, A., & Riethmüller, J. (2019). Choline Supplementation in Cystic Fibrosis—The Metabolic and Clinical Impact. Nutrients, 11(3), 656. https://doi.org/10.3390/nu11030656