Methyl Donor Micronutrients that Modify DNA Methylation and Cancer Outcome


Abstract
1. Introduction
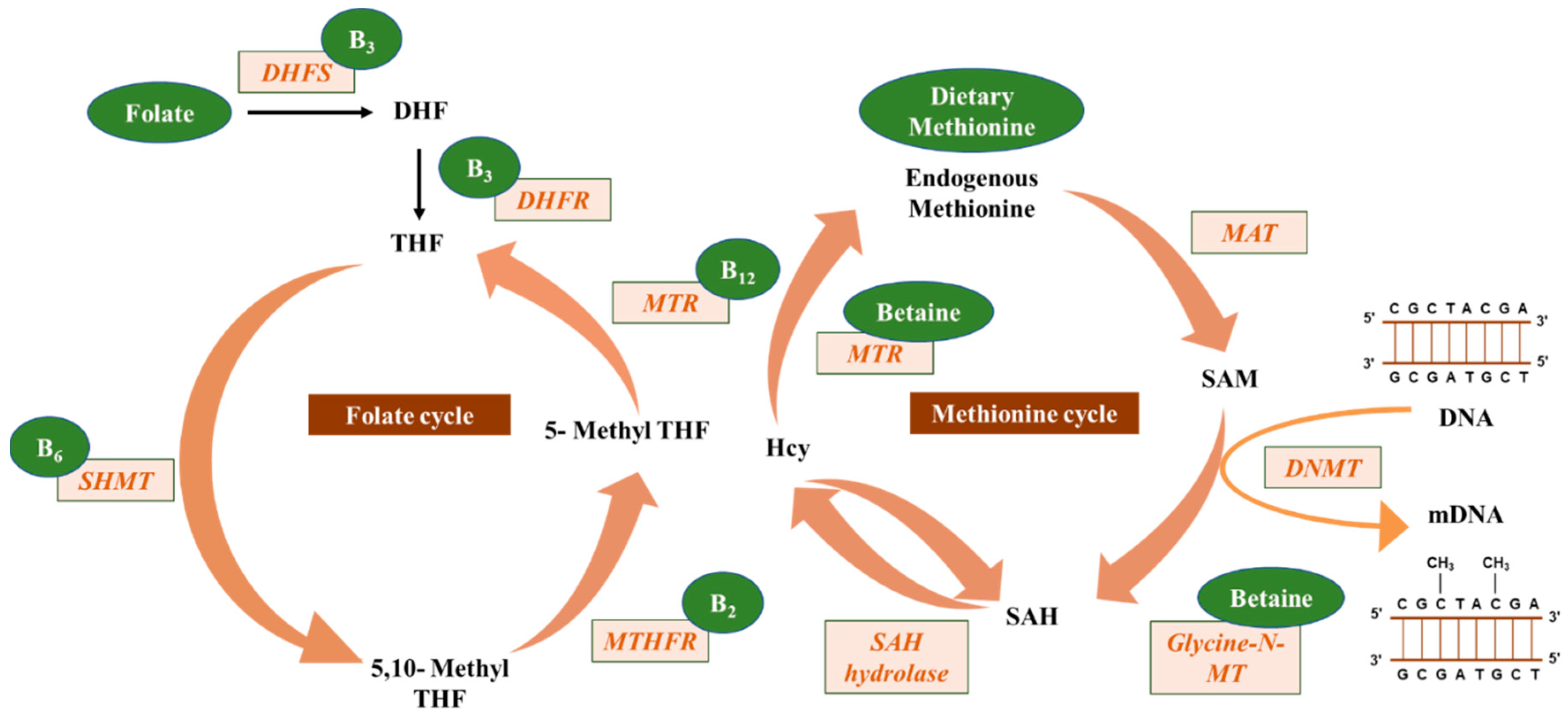
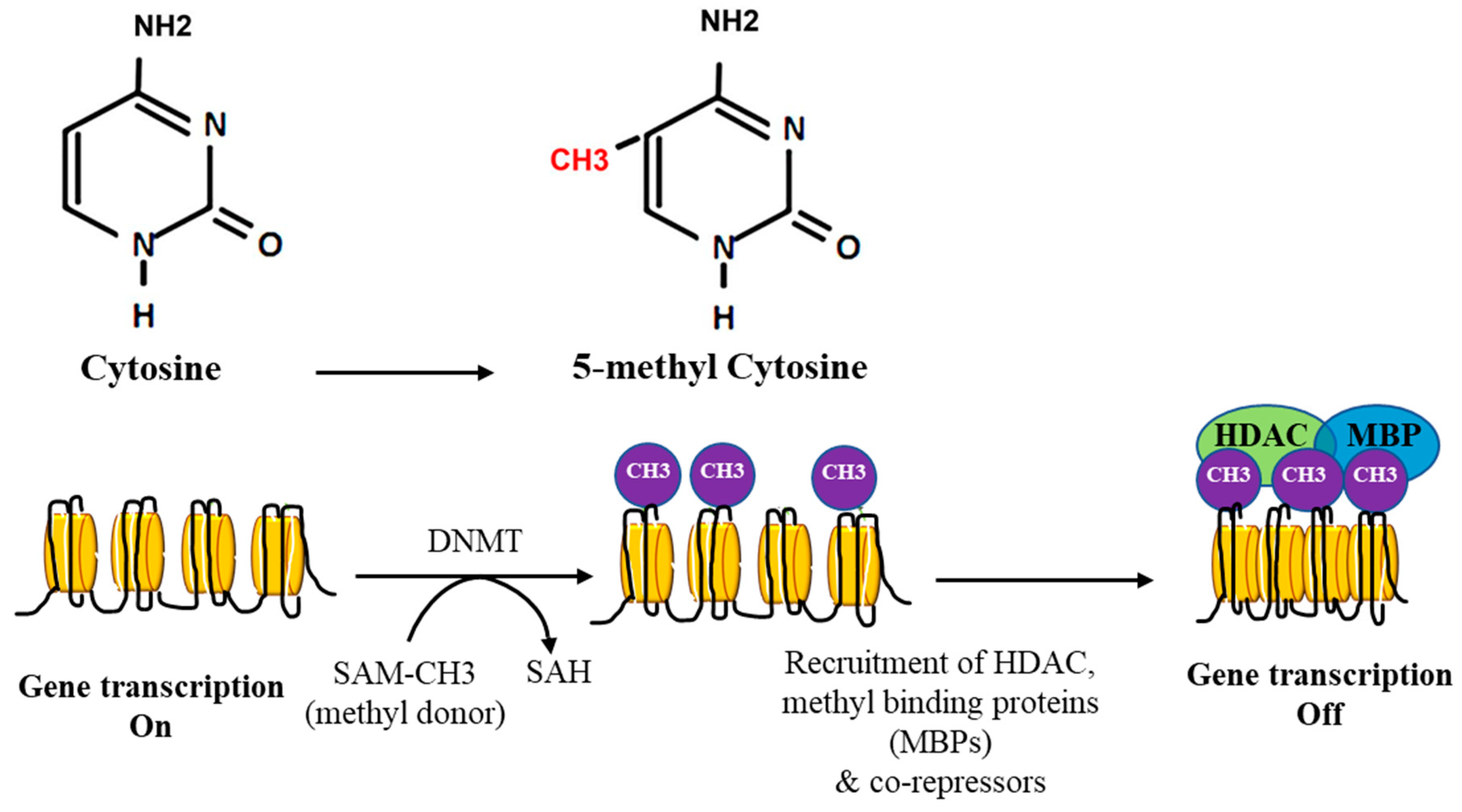
2. DNA Methylation
3. DNA Methylation in Cancer
4. Effects of Nutrients and Bioactive Food Components on DNA Methylation
5. Micronutrients and DNA Methylation and Their Impact on Cancer
5.1. Folate
5.2. Riboflavin, Pyridoxine, and Cobalamin
5.3. Choline and Betaine
5.4. Methionine
5.5. The Impact of Alcohol and Smoking on Nutrient-Mediated DNA Methylation
5.6. The Impact of Early Nutrition in Modulating DNA Methylation
6. Perspectives of Nutritional Modification of DNA Methylation
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Biswas, S.; Rao, C.M. Epigenetics in cancer: Fundamentals and beyond. Pharmacol. Ther. 2017, 173, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-carbon metabolism in health and disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S. Choline, other methyl-donors and epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Burdge, G.C. Epigenetic mechanisms linking early nutrition to long term health. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA methylation in cancer and aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef]
- Kulis, M.; Queiros, A.C.; Beekman, R.; Martin-Subero, J.I. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim. Biophys. Acta 2013, 1829, 1161–1174. [Google Scholar] [CrossRef]
- Bakshi, A.; Bretz, C.L.; Cain, T.L.; Kim, J. Intergenic and intronic DNA hypomethylated regions as putative regulators of imprinted domains. Epigenomics 2018, 10, 445–461. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Rhee, I.; Bachman, K.E.; Park, B.H.; Jair, K.W.; Yen, R.W.; Schuebel, K.E.; Cui, H.; Feinberg, A.P.; Lengauer, C.; Kinzler, K.W.; et al. Dnmt1 and dnmt3b cooperate to silence genes in human cancer cells. Nature 2002, 416, 552–556. [Google Scholar] [CrossRef]
- Leu, Y.W.; Rahmatpanah, F.; Shi, H.; Wei, S.H.; Liu, J.C.; Yan, P.S.; Huang, T.H. Double rna interference of dnmt3b and dnmt1 enhances DNA demethylation and gene reactivation. Cancer Res. 2003, 63, 6110–6115. [Google Scholar]
- Sowinska, A.; Jagodzinski, P.P. Rna interference-mediated knockdown of dnmt1 and dnmt3b induces cxcl12 expression in mcf-7 breast cancer and aspc1 pancreatic carcinoma cell lines. Cancer Lett. 2007, 255, 153–159. [Google Scholar] [CrossRef]
- Liang, G.; Chan, M.F.; Tomigahara, Y.; Tsai, Y.C.; Gonzales, F.A.; Li, E.; Laird, P.W.; Jones, P.A. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell Biol. 2002, 22, 480–491. [Google Scholar] [CrossRef]
- Fatemi, M.; Hermann, A.; Gowher, H.; Jeltsch, A. Dnmt3a and dnmt1 functionally cooperate during de novo methylation of DNA. Eur. J. Biochem. 2002, 269, 4981–4984. [Google Scholar] [CrossRef]
- Lorincz, M.C.; Schubeler, D.; Hutchinson, S.R.; Dickerson, D.R.; Groudine, M. DNA methylation density influences the stability of an epigenetic imprint and dnmt3a/b-independent de novo methylation. Mol. Cell Biol. 2002, 22, 7572–7580. [Google Scholar] [CrossRef]
- Pradhan, S.; Bacolla, A.; Wells, R.D.; Roberts, R.J. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 1999, 274, 33002–33010. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.F. Specific or not specific recruitment of dnmts for DNA methylation, an epigenetic dilemma. Clin. Epigenetics 2018, 10, 17. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. Tet enzymes, tdg and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef]
- Pidugu, L.S.; Flowers, J.W.; Coey, C.T.; Pozharski, E.; Greenberg, M.M.; Drohat, A.C. Structural basis for excision of 5-formylcytosine by thymine DNA glycosylase. Biochemistry 2016, 55, 6205–6208. [Google Scholar] [CrossRef]
- Williams, K.; Christensen, J.; Pedersen, M.T.; Johansen, J.V.; Cloos, P.A.; Rappsilber, J.; Helin, K. Tet1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 2011, 473, 343–348. [Google Scholar] [CrossRef]
- Schuermann, D.; Weber, A.R.; Schar, P. Active DNA demethylation by DNA repair: Facts and uncertainties. DNA Repair 2016, 44, 92–102. [Google Scholar] [CrossRef]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, G.; Zhou, F.; Su, B.; Li, Y. DNA methylation profiles in cancer diagnosis and therapeutics. Clin. Exp. Med. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Qi, M.; Xiong, X. Promoter hypermethylation of rarbeta2, dapk, hmlh1, p14, and p15 is associated with progression of breast cancer: A prisma-compliant meta-analysis. Medicine 2018, 97, e13666. [Google Scholar] [CrossRef]
- Yamashita, K.; Hosoda, K.; Nishizawa, N.; Katoh, H.; Watanabe, M. Epigenetic biomarkers of promoter DNA methylation in the new era of cancer treatment. Cancer Sci. 2018, 109, 3695–3706. [Google Scholar] [CrossRef]
- Rahmani, M.; Talebi, M.; Hagh, M.F.; Feizi, A.A.H.; Solali, S. Aberrant DNA methylation of key genes and acute lymphoblastic leukemia. Biomed. Pharmacother. 2018, 97, 1493–1500. [Google Scholar] [CrossRef]
- Lasseigne, B.N.; Brooks, J.D. The role of DNA methylation in renal cell carcinoma. Mol. Diagn. Ther. 2018, 22, 431–442. [Google Scholar] [CrossRef]
- Mekky, M.A.; Salama, R.H.; Abdel-Aal, M.F.; Ghaliony, M.A.; Zaky, S. Studying the frequency of aberrant DNA methylation of apc, p14, and e-cadherin genes in hcv-related hepatocarcinogenesis. Cancer Biomark. 2018, 22, 503–509. [Google Scholar] [CrossRef]
- Curtis, C.D.; Goggins, M. DNA methylation analysis in human cancer. Methods Mol. Med. 2005, 103, 123–136. [Google Scholar]
- Esteller, M. Cancer epigenetics: DNA methylation and chromatin alterations in human cancer. Adv. Exp. Med. Biol. 2003, 532, 39–49. [Google Scholar]
- Sproul, D.; Meehan, R.R. Genomic insights into cancer-associated aberrant cpg island hypermethylation. Brief. Funct. Genom. 2013, 12, 174–190. [Google Scholar] [CrossRef]
- Estecio, M.R.; Issa, J.P. Dissecting DNA hypermethylation in cancer. FEBS Lett. 2011, 585, 2078–2086. [Google Scholar] [CrossRef]
- Ashktorab, H.; Brim, H. DNA methylation and colorectal cancer. Curr. Colorectal Cancer Rep. 2014, 10, 425–430. [Google Scholar] [CrossRef]
- Funaki, S.; Nakamura, T.; Nakatani, T.; Umehara, H.; Nakashima, H.; Okumura, M.; Oboki, K.; Matsumoto, K.; Saito, H.; Nakano, T. Global DNA hypomethylation coupled to cellular transformation and metastatic ability. FEBS Lett. 2015, 589, 4053–4060. [Google Scholar] [CrossRef]
- Kisseljova, N.P.; Kisseljov, F.L. DNA demethylation and carcinogenesis. Biochemistry (Moscow) 2005, 70, 743–752. [Google Scholar] [CrossRef]
- Song, C.X.; He, C. Balance of DNA methylation and demethylation in cancer development. Genome Biol. 2012, 13, 173. [Google Scholar] [CrossRef]
- Ghobadi, A.; Choi, J.; Fiala, M.A.; Fletcher, T.; Liu, J.; Eissenberg, L.G.; Abboud, C.; Cashen, A.; Vij, R.; Schroeder, M.A.; et al. Phase i study of azacitidine following donor lymphocyte infusion for relapsed acute myeloid leukemia post allogeneic stem cell transplantation. Leuk. Res. 2016, 49, 1–6. [Google Scholar] [CrossRef]
- Harman, R.M.; Curtis, T.M.; Argyle, D.J.; Coonrod, S.A.; Van de Walle, G.R. A comparative study on the in vitro effects of the DNA methyltransferase inhibitor 5-azacytidine (5-azac) in breast/mammary cancer of different mammalian species. J. Mammary Gland Biol. Neoplasia 2016, 21, 51–66. [Google Scholar] [CrossRef]
- Wang, X.M.; Wang, X.; Li, J.; Evers, B.M. Effects of 5-azacytidine and butyrate on differentiation and apoptosis of hepatic cancer cell lines. Ann. Surg. 1998, 227, 922–931. [Google Scholar] [CrossRef]
- Kuykendall, J.R. 5-azacytidine and decitabine monotherapies of myelodysplastic disorders. Ann. Pharmacother. 2005, 39, 1700–1709. [Google Scholar] [CrossRef]
- Shin, T.H.; Paterson, A.J.; Grant, J.H., 3rd; Meluch, A.A.; Kudlow, J.E. 5-azacytidine treatment of ha-a melanoma cells induces sp1 activity and concomitant transforming growth factor alpha expression. Mol. Cell Biol. 1992, 12, 3998–4006. [Google Scholar] [CrossRef]
- Sapienza, C.; Issa, J.P. Diet, nutrition, and cancer epigenetics. Annu. Rev. Nutr. 2016, 36, 665–681. [Google Scholar] [CrossRef]
- Kadayifci, F.Z.; Zheng, S.; Pan, Y.X. Molecular mechanisms underlying the link between diet and DNA methylation. Int. J. Mol. Sci. 2018, 19, 4055. [Google Scholar] [CrossRef]
- Park, J.H.; Yoo, Y.; Park, Y.J. Epigenetics: Linking nutrition to molecular mechanisms in aging. Prev. Nutr. Food Sci. 2017, 22, 81–89. [Google Scholar]
- de Luca, A.; Hankard, R.; Borys, J.M.; Sinnett, D.; Marcil, V.; Levy, E. Nutriepigenomics and malnutrition. Epigenomics 2017, 9, 893–917. [Google Scholar] [CrossRef]
- Remely, M.; Stefanska, B.; Lovrecic, L.; Magnet, U.; Haslberger, A.G. Nutriepigenomics: The role of nutrition in epigenetic control of human diseases. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 328–333. [Google Scholar] [CrossRef]
- Friso, S.; Udali, S.; De Santis, D.; Choi, S.W. One-carbon metabolism and epigenetics. Mol. Asp. Med. 2017, 54, 28–36. [Google Scholar] [CrossRef]
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and epigenetics: An interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 2012, 23, 853–859. [Google Scholar] [CrossRef]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef]
- Kandi, V.; Vadakedath, S. Effect of DNA methylation in various diseases and the probable protective role of nutrition: A mini-review. Cureus 2015, 7, e309. [Google Scholar] [CrossRef]
- Liteplo, R.G. DNA (cytosine) methylation in murine and human tumor cell lines treated with s-adenosylhomocysteine hydrolase inhibitors. Cancer Lett. 1988, 39, 319–327. [Google Scholar] [CrossRef]
- Soda, K. Polyamine metabolism and gene methylation in conjunction with one-carbon metabolism. Int. J. Mol. Sci. 2018, 19, 3106. [Google Scholar] [CrossRef]
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246. [Google Scholar] [CrossRef]
- Tamanaha, E.; Guan, S.; Marks, K.; Saleh, L. Distributive processing by the iron(ii)/alpha-ketoglutarate-dependent catalytic domains of the tet enzymes is consistent with epigenetic roles for oxidized 5-methylcytosine bases. J. Am. Chem. Soc. 2016, 138, 9345–9348. [Google Scholar] [CrossRef]
- Wallace, K.; Grau, M.V.; Levine, A.J.; Shen, L.; Hamdan, R.; Chen, X.; Gui, J.; Haile, R.W.; Barry, E.L.; Ahnen, D.; et al. Association between folate levels and cpg island hypermethylation in normal colorectal mucosa. Cancer Prev. Res. 2010, 3, 1552–1564. [Google Scholar] [CrossRef]
- Pufulete, M.; Al-Ghnaniem, R.; Leather, A.J.; Appleby, P.; Gout, S.; Terry, C.; Emery, P.W.; Sanders, T.A. Folate status, genomic DNA hypomethylation, and risk of colorectal adenoma and cancer: A case control study. Gastroenterology 2003, 124, 1240–1248. [Google Scholar] [CrossRef]
- Piyathilake, C.J.; Azrad, M.; Jhala, D.; Macaluso, M.; Kabagambe, E.K.; Brill, I.; Niveleau, A.; Jhala, N.; Grizzle, W.E. Mandatory fortification with folic acid in the united states is not associated with changes in the degree or the pattern of global DNA methylation in cells involved in cervical carcinogenesis. Cancer Biomark. 2006, 2, 259–266. [Google Scholar] [CrossRef]
- Moore, L.E.; Pfeiffer, R.M.; Poscablo, C.; Real, F.X.; Kogevinas, M.; Silverman, D.; Garcia-Closas, R.; Chanock, S.; Tardon, A.; Serra, C.; et al. Genomic DNA hypomethylation as a biomarker for bladder cancer susceptibility in the spanish bladder cancer study: A case-control study. Lancet Oncol. 2008, 9, 359–366. [Google Scholar] [CrossRef]
- Piyathilake, C.J.; Macaluso, M.; Alvarez, R.D.; Chen, M.; Badiga, S.; Siddiqui, N.R.; Edberg, J.C.; Partridge, E.E.; Johanning, G.L. A higher degree of line-1 methylation in peripheral blood mononuclear cells, a one-carbon nutrient related epigenetic alteration, is associated with a lower risk of developing cervical intraepithelial neoplasia. Nutrition 2011, 27, 513–519. [Google Scholar] [CrossRef]
- Pufulete, M.; Al-Ghnaniem, R.; Rennie, J.A.; Appleby, P.; Harris, N.; Gout, S.; Emery, P.W.; Sanders, T.A. Influence of folate status on genomic DNA methylation in colonic mucosa of subjects without colorectal adenoma or cancer. Br. J. Cancer 2005, 92, 838–842. [Google Scholar] [CrossRef]
- O’Reilly, S.L.; McGlynn, A.P.; McNulty, H.; Reynolds, J.; Wasson, G.R.; Molloy, A.M.; Strain, J.J.; Weir, D.G.; Ward, M.; McKerr, G.; et al. Folic acid supplementation in postpolypectomy patients in a randomized controlled trial increases tissue folate concentrations and reduces aberrant DNA biomarkers in colonic tissues adjacent to the former polyp site. J. Nutr. 2016, 146, 933–939. [Google Scholar] [CrossRef]
- Cravo, M.L.; Pinto, A.G.; Chaves, P.; Cruz, J.A.; Lage, P.; Nobre Leitao, C.; Costa Mira, F. Effect of folate supplementation on DNA methylation of rectal mucosa in patients with colonic adenomas: Correlation with nutrient intake. Clin. Nutr. 1998, 17, 45–49. [Google Scholar] [CrossRef]
- Kim, Y.I.; Baik, H.W.; Fawaz, K.; Knox, T.; Lee, Y.M.; Norton, R.; Libby, E.; Mason, J.B. Effects of folate supplementation on two provisional molecular markers of colon cancer: A prospective, randomized trial. Am. J. Gastroenterol. 2001, 96, 184–195. [Google Scholar] [CrossRef]
- Coppede, F.; Migheli, F.; Lopomo, A.; Failli, A.; Legitimo, A.; Consolini, R.; Fontanini, G.; Sensi, E.; Servadio, A.; Seccia, M.; et al. Gene promoter methylation in colorectal cancer and healthy adjacent mucosa specimens: Correlation with physiological and pathological characteristics, and with biomarkers of one-carbon metabolism. Epigenetics 2014, 9, 621–633. [Google Scholar] [CrossRef]
- Christensen, B.C.; Kelsey, K.T.; Zheng, S.; Houseman, E.A.; Marsit, C.J.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Kushi, L.H.; et al. Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 2010, 6, e1001043. [Google Scholar] [CrossRef]
- Vineis, P.; Chuang, S.C.; Vaissiere, T.; Cuenin, C.; Ricceri, F.; Genair, E.C.; Johansson, M.; Ueland, P.; Brennan, P.; Herceg, Z. DNA methylation changes associated with cancer risk factors and blood levels of vitamin metabolites in a prospective study. Epigenetics 2011, 6, 195–201. [Google Scholar] [CrossRef]
- van Engeland, M.; Weijenberg, M.P.; Roemen, G.M.; Brink, M.; de Bruine, A.P.; Goldbohm, R.A.; van den Brandt, P.A.; Baylin, S.B.; de Goeij, A.F.; Herman, J.G. Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: The netherlands cohort study on diet and cancer. Cancer Res. 2003, 63, 3133–3137. [Google Scholar]
- Ba, Y.; Yu, H.; Liu, F.; Geng, X.; Zhu, C.; Zhu, Q.; Zheng, T.; Ma, S.; Wang, G.; Li, Z.; et al. Relationship of folate, vitamin b12 and methylation of insulin-like growth factor-ii in maternal and cord blood. Eur. J. Clin. Nutr. 2011, 65, 480–485. [Google Scholar] [CrossRef]
- Hoyo, C.; Murtha, A.P.; Schildkraut, J.M.; Jirtle, R.L.; Demark-Wahnefried, W.; Forman, M.R.; Iversen, E.S.; Kurtzberg, J.; Overcash, F.; Huang, Z.; et al. Methylation variation at igf2 differentially methylated regions and maternal folic acid use before and during pregnancy. Epigenetics 2011, 6, 928–936. [Google Scholar] [CrossRef]
- Shelnutt, K.P.; Kauwell, G.P.; Gregory, J.F., 3rd; Maneval, D.R.; Quinlivan, E.P.; Theriaque, D.W.; Henderson, G.N.; Bailey, L.B. Methylenetetrahydrofolate reductase 677c-->t polymorphism affects DNA methylation in response to controlled folate intake in young women. J. Nutr. Biochem. 2004, 15, 554–560. [Google Scholar] [CrossRef]
- Colacino, J.A.; Arthur, A.E.; Dolinoy, D.C.; Sartor, M.A.; Duffy, S.A.; Chepeha, D.B.; Bradford, C.R.; Walline, H.M.; McHugh, J.B.; D’Silva, N.; et al. Pretreatment dietary intake is associated with tumor suppressor DNA methylation in head and neck squamous cell carcinomas. Epigenetics 2012, 7, 883–891. [Google Scholar] [CrossRef]
- Piyathilake, C.J.; Johanning, G.L.; Macaluso, M.; Whiteside, M.; Oelschlager, D.K.; Heimburger, D.C.; Grizzle, W.E. Localized folate and vitamin b-12 deficiency in squamous cell lung cancer is associated with global DNA hypomethylation. Nutr. Cancer 2000, 37, 99–107. [Google Scholar] [CrossRef]
- Perng, W.; Rozek, L.S.; Mora-Plazas, M.; Duchin, O.; Marin, C.; Forero, Y.; Baylin, A.; Villamor, E. Micronutrient status and global DNA methylation in school-age children. Epigenetics 2012, 7, 1133–1141. [Google Scholar] [CrossRef]
- Hubner, U.; Geisel, J.; Kirsch, S.H.; Kruse, V.; Bodis, M.; Klein, C.; Herrmann, W.; Obeid, R. Effect of 1 year b and d vitamin supplementation on line-1 repetitive element methylation in older subjects. Clin. Chem. Lab. Med. 2013, 51, 649–655. [Google Scholar] [CrossRef]
- Piyathilake, C.J.; Macaluso, M.; Chambers, M.M.; Badiga, S.; Siddiqui, N.R.; Bell, W.C.; Edberg, J.C.; Partridge, E.E.; Alvarez, R.D.; Johanning, G.L. Folate and vitamin b12 may play a critical role in lowering the hpv 16 methylation-associated risk of developing higher grades of cin. Cancer Prev. Res. 2014, 7, 1128–1137. [Google Scholar] [CrossRef]
- Pauwels, S.; Duca, R.C.; Devlieger, R.; Freson, K.; Straetmans, D.; Van Herck, E.; Huybrechts, I.; Koppen, G.; Godderis, L. Maternal methyl-group donor intake and global DNA (hydroxy)methylation before and during pregnancy. Nutrients 2016, 8, 474. [Google Scholar] [CrossRef]
- Chiuve, S.E.; Giovannucci, E.L.; Hankinson, S.E.; Zeisel, S.H.; Dougherty, L.W.; Willett, W.C.; Rimm, E.B. The association between betaine and choline intakes and the plasma concentrations of homocysteine in women. Am. J. Clin. Nutr. 2007, 86, 1073–1081. [Google Scholar] [CrossRef]
- Schwab, U.; Torronen, A.; Toppinen, L.; Alfthan, G.; Saarinen, M.; Aro, A.; Uusitupa, M. Betaine supplementation decreases plasma homocysteine concentrations but does not affect body weight, body composition, or resting energy expenditure in human subjects. Am. J. Clin. Nutr. 2002, 76, 961–967. [Google Scholar] [CrossRef]
- Olthof, M.R.; Brink, E.J.; Katan, M.B.; Verhoef, P. Choline supplemented as phosphatidylcholine decreases fasting and postmethionine-loading plasma homocysteine concentrations in healthy men. Am. J. Clin. Nutr. 2005, 82, 111–117. [Google Scholar] [CrossRef]
- Perng, W.; Villamor, E.; Shroff, M.R.; Nettleton, J.A.; Pilsner, J.R.; Liu, Y.; Diez-Roux, A.V. Dietary intake, plasma homocysteine, and repetitive element DNA methylation in the multi-ethnic study of atherosclerosis (mesa). Nutr. Metab. Cardiovasc. Dis. 2014, 24, 614–622. [Google Scholar] [CrossRef]
- Tao, M.H.; Mason, J.B.; Marian, C.; McCann, S.E.; Platek, M.E.; Millen, A.; Ambrosone, C.; Edge, S.B.; Krishnan, S.S.; Trevisan, M.; et al. Promoter methylation of e-cadherin, p16, and rar-beta(2) genes in breast tumors and dietary intake of nutrients important in one-carbon metabolism. Nutr. Cancer 2011, 63, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Stampfer, M.J.; Colditz, G.A.; Rimm, E.B.; Trichopoulos, D.; Rosner, B.A.; Speizer, F.E.; Willett, W.C. Folate, methionine, and alcohol intake and risk of colorectal adenoma. J. Natl. Cancer Inst. 1993, 85, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Arab, L. Nutritional status of folate and colon cancer risk: Evidence from nhanes i epidemiologic follow-up study. Ann. Epidemiol. 2001, 11, 65–72. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Willett, W.C.; Colditz, G.A.; Hunter, D.J.; Stampfer, M.J.; Speizer, F.E.; Giovannucci, E.L. The influence of folate and multivitamin use on the familial risk of colon cancer in women. Cancer Epidemiol. Biomark. Prev. 2002, 11, 227–234. [Google Scholar]
- Stevens, V.L.; Rodriguez, C.; Pavluck, A.L.; McCullough, M.L.; Thun, M.J.; Calle, E.E. Folate nutrition and prostate cancer incidence in a large cohort of us men. Am. J. Epidemiol. 2006, 163, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Gylling, B.; Van Guelpen, B.; Schneede, J.; Hultdin, J.; Ueland, P.M.; Hallmans, G.; Johansson, I.; Palmqvist, R. Low folate levels are associated with reduced risk of colorectal cancer in a population with low folate status. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2136–2144. [Google Scholar] [CrossRef]
- Giovannucci, E.; Stampfer, M.J.; Colditz, G.A.; Hunter, D.J.; Fuchs, C.; Rosner, B.A.; Speizer, F.E.; Willett, W.C. Multivitamin use, folate, and colon cancer in women in the nurses’ health study. Ann. Intern. Med. 1998, 129, 517–524. [Google Scholar] [CrossRef]
- Konings, E.J.; Goldbohm, R.A.; Brants, H.A.; Saris, W.H.; van den Brandt, P.A. Intake of dietary folate vitamers and risk of colorectal carcinoma: Results from the netherlands cohort study. Cancer 2002, 95, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Terry, P.; Jain, M.; Miller, A.B.; Howe, G.R.; Rohan, T.E. Dietary intake of folic acid and colorectal cancer risk in a cohort of women. Int. J. Cancer 2002, 97, 864–867. [Google Scholar] [CrossRef]
- Wei, E.K.; Giovannucci, E.; Wu, K.; Rosner, B.; Fuchs, C.S.; Willett, W.C.; Colditz, G.A. Comparison of risk factors for colon and rectal cancer. Int. J. Cancer 2004, 108, 433–442. [Google Scholar] [CrossRef]
- Harnack, L.; Jacobs, D.R., Jr.; Nicodemus, K.; Lazovich, D.; Anderson, K.; Folsom, A.R. Relationship of folate, vitamin b-6, vitamin b-12, and methionine intake to incidence of colorectal cancers. Nutr. Cancer 2002, 43, 152–158. [Google Scholar] [CrossRef]
- Benito, E.; Stiggelbout, A.; Bosch, F.X.; Obrador, A.; Kaldor, J.; Mulet, M.; Munoz, N. Nutritional factors in colorectal cancer risk: A case-control study in majorca. Int. J. Cancer 1991, 49, 161–167. [Google Scholar] [CrossRef]
- Ferraroni, M.; La Vecchia, C.; D’Avanzo, B.; Negri, E.; Franceschi, S.; Decarli, A. Selected micronutrient intake and the risk of colorectal cancer. Br. J. Cancer 1994, 70, 1150–1155. [Google Scholar] [CrossRef]
- Freudenheim, J.L.; Graham, S.; Marshall, J.R.; Haughey, B.P.; Cholewinski, S.; Wilkinson, G. Folate intake and carcinogenesis of the colon and rectum. Int. J. Epidemiol. 1991, 20, 368–374. [Google Scholar] [CrossRef]
- Glynn, S.A.; Albanes, D.; Pietinen, P.; Brown, C.C.; Rautalahti, M.; Tangrea, J.A.; Gunter, E.W.; Barrett, M.J.; Virtamo, J.; Taylor, P.R. Colorectal cancer and folate status: A nested case-control study among male smokers. Cancer Epidemiol. Biomark. Prev. 1996, 5, 487–494. [Google Scholar]
- La Vecchia, C.; Braga, C.; Negri, E.; Franceschi, S.; Russo, A.; Conti, E.; Falcini, F.; Giacosa, A.; Montella, M.; Decarli, A. Intake of selected micronutrients and risk of colorectal cancer. Int. J. Cancer 1997, 73, 525–530. [Google Scholar] [CrossRef]
- Le Marchand, L.; Donlon, T.; Hankin, J.H.; Kolonel, L.N.; Wilkens, L.R.; Seifried, A. B-vitamin intake, metabolic genes, and colorectal cancer risk (united states). Cancer Causes Control. 2002, 13, 239–248. [Google Scholar] [CrossRef]
- Levi, F.; Pasche, C.; Lucchini, F.; La Vecchia, C. Selected micronutrients and colorectal cancer. A case-control study from the canton of vaud, switzerland. Eur. J. Cancer 2000, 36, 2115–2119. [Google Scholar] [CrossRef]
- Boutron-Ruault, M.C.; Senesse, P.; Faivre, J.; Couillault, C.; Belghiti, C. Folate and alcohol intakes: Related or independent roles in the adenoma-carcinoma sequence? Nutr. Cancer 1996, 26, 337–346. [Google Scholar] [CrossRef]
- Kato, I.; Dnistrian, A.M.; Schwartz, M.; Toniolo, P.; Koenig, K.; Shore, R.E.; Akhmedkhanov, A.; Zeleniuch-Jacquotte, A.; Riboli, E. Serum folate, homocysteine and colorectal cancer risk in women: A nested case-control study. Br. J. Cancer 1999, 79, 1917–1922. [Google Scholar] [CrossRef]
- Cole, B.F.; Baron, J.A.; Sandler, R.S.; Haile, R.W.; Ahnen, D.J.; Bresalier, R.S.; McKeown-Eyssen, G.; Summers, R.W.; Rothstein, R.I.; Burke, C.A.; et al. Folic acid for the prevention of colorectal adenomas: A randomized clinical trial. JAMA 2007, 297, 2351–2359. [Google Scholar] [CrossRef]
- Ebbing, M.; Bonaa, K.H.; Nygard, O.; Arnesen, E.; Ueland, P.M.; Nordrehaug, J.E.; Rasmussen, K.; Njolstad, I.; Refsum, H.; Nilsen, D.W.; et al. Cancer incidence and mortality after treatment with folic acid and vitamin b12. JAMA 2009, 302, 2119–2126. [Google Scholar] [CrossRef]
- Otani, T.; Iwasaki, M.; Hanaoka, T.; Kobayashi, M.; Ishihara, J.; Natsukawa, S.; Shaura, K.; Koizumi, Y.; Kasuga, Y.; Yoshimura, K.; et al. Folate, vitamin b6, vitamin b12, and vitamin b2 intake, genetic polymorphisms of related enzymes, and risk of colorectal cancer in a hospital-based case-control study in japan. Nutr. Cancer 2005, 53, 42–50. [Google Scholar] [CrossRef]
- Hultdin, J.; Van Guelpen, B.; Bergh, A.; Hallmans, G.; Stattin, P. Plasma folate, vitamin b12, and homocysteine and prostate cancer risk: A prospective study. Int. J. Cancer 2005, 113, 819–824. [Google Scholar] [CrossRef]
- Du, Y.F.; Lin, F.Y.; Long, W.Q.; Luo, W.P.; Yan, B.; Xu, M.; Mo, X.F.; Zhang, C.X. Serum betaine but not choline is inversely associated with breast cancer risk: A case-control study in china. Eur. J. Nutr. 2017, 56, 1329–1337. [Google Scholar] [CrossRef]
- Lu, M.S.; Fang, Y.J.; Pan, Z.Z.; Zhong, X.; Zheng, M.C.; Chen, Y.M.; Zhang, C.X. Choline and betaine intake and colorectal cancer risk in chinese population: A case-control study. PLoS ONE 2015, 10, e0118661. [Google Scholar] [CrossRef]
- Zeng, F.F.; Xu, C.H.; Liu, Y.T.; Fan, Y.Y.; Lin, X.L.; Lu, Y.K.; Zhang, C.X.; Chen, Y.M. Choline and betaine intakes are associated with reduced risk of nasopharyngeal carcinoma in adults: A case-control study. Br. J. Cancer 2014, 110, 808–816. [Google Scholar] [CrossRef]
- Zhou, R.F.; Chen, X.L.; Zhou, Z.G.; Zhang, Y.J.; Lan, Q.Y.; Liao, G.C.; Chen, Y.M.; Zhu, H.L. Higher dietary intakes of choline and betaine are associated with a lower risk of primary liver cancer: A case-control study. Sci. Rep. 2017, 7, 679. [Google Scholar] [CrossRef]
- Nitter, M.; Norgard, B.; de Vogel, S.; Eussen, S.J.; Meyer, K.; Ulvik, A.; Ueland, P.M.; Nygard, O.; Vollset, S.E.; Bjorge, T.; et al. Plasma methionine, choline, betaine, and dimethylglycine in relation to colorectal cancer risk in the european prospective investigation into cancer and nutrition (epic). Ann. Oncol. 2014, 25, 1609–1615. [Google Scholar] [CrossRef]
- Feigelson, H.S.; Jonas, C.R.; Robertson, A.S.; McCullough, M.L.; Thun, M.J.; Calle, E.E. Alcohol, folate, methionine, and risk of incident breast cancer in the american cancer society cancer prevention study ii nutrition cohort. Cancer Epidemiol. Biomark. Prev. 2003, 12, 161–164. [Google Scholar]
- Donnelly, J.G. Folic acid. Crit. Rev. Clin. Lab. Sci. 2001, 38, 183–223. [Google Scholar] [CrossRef]
- Li, W.; Jiang, M.; Xiao, Y.; Zhang, X.; Cui, S.; Huang, G. Folic acid inhibits tau phosphorylation through regulation of pp2a methylation in sh-sy5y cells. J. Nutr. Health Aging 2015, 19, 123–129. [Google Scholar] [CrossRef]
- Keyes, M.K.; Jang, H.; Mason, J.B.; Liu, Z.; Crott, J.W.; Smith, D.E.; Friso, S.; Choi, S.W. Older age and dietary folate are determinants of genomic and p16-specific DNA methylation in mouse colon. J. Nutr. 2007, 137, 1713–1717. [Google Scholar] [CrossRef]
- Cartron, P.F.; Hervouet, E.; Debien, E.; Olivier, C.; Pouliquen, D.; Menanteau, J.; Loussouarn, D.; Martin, S.A.; Campone, M.; Vallette, F.M. Folate supplementation limits the tumourigenesis in rodent models of gliomagenesis. Eur. J. Cancer 2012, 48, 2431–2441. [Google Scholar] [CrossRef]
- McKay, J.A.; Waltham, K.J.; Williams, E.A.; Mathers, J.C. Folate depletion during pregnancy and lactation reduces genomic DNA methylation in murine adult offspring. Genes Nutr. 2011, 6, 189–196. [Google Scholar] [CrossRef]
- Ly, A.; Lee, H.; Chen, J.; Sie, K.K.; Renlund, R.; Medline, A.; Sohn, K.J.; Croxford, R.; Thompson, L.U.; Kim, Y.I. Effect of maternal and postweaning folic acid supplementation on mammary tumor risk in the offspring. Cancer Res. 2011, 71, 988–997. [Google Scholar] [CrossRef]
- Sie, K.K.; Medline, A.; van Weel, J.; Sohn, K.J.; Choi, S.W.; Croxford, R.; Kim, Y.I. Effect of maternal and postweaning folic acid supplementation on colorectal cancer risk in the offspring. Gut 2011, 60, 1687–1694. [Google Scholar] [CrossRef]
- Song, J.; Sohn, K.J.; Medline, A.; Ash, C.; Gallinger, S.; Kim, Y.I. Chemopreventive effects of dietary folate on intestinal polyps in apc+/-msh2-/- mice. Cancer Res. 2000, 60, 3191–3199. [Google Scholar]
- Kotsopoulos, J.; Sohn, K.J.; Kim, Y.I. Postweaning dietary folate deficiency provided through childhood to puberty permanently increases genomic DNA methylation in adult rat liver. J. Nutr. 2008, 138, 703–709. [Google Scholar] [CrossRef]
- Pieroth, R.; Paver, S.; Day, S.; Lammersfeld, C. Folate and its impact on cancer risk. Curr. Nutr. Rep. 2018, 7, 70–84. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, C.; Hu, H.; Zheng, L.; Ma, J.; Jiang, L.; Zhao, E.; Li, H. Folate intake, serum folate levels and esophageal cancer risk: An overall and dose-response meta-analysis. Oncotarget 2017, 8, 10458–10469. [Google Scholar] [CrossRef]
- Burr, N.E.; Hull, M.A.; Subramanian, V. Folic acid supplementation may reduce colorectal cancer risk in patients with inflammatory bowel disease: A systematic review and meta-analysis. J. Clin. Gastroenterol. 2017, 51, 247–253. [Google Scholar] [CrossRef]
- Wang, R.; Zheng, Y.; Huang, J.Y.; Zhang, A.Q.; Zhou, Y.H.; Wang, J.N. Folate intake, serum folate levels, and prostate cancer risk: A meta-analysis of prospective studies. BMC Public Health 2014, 14, 1326. [Google Scholar] [CrossRef]
- Guo, S.; Jiang, X.; Chen, X.; Chen, L.; Li, X.; Jia, Y. The protective effect of methylenetetrahydrofolate reductase c677t polymorphism against prostate cancer risk: Evidence from 23 case-control studies. Gene 2015, 565, 90–95. [Google Scholar] [CrossRef]
- Yi, K.; Yang, L.; Lan, Z.; Xi, M. The association between mthfr polymorphisms and cervical cancer risk: A system review and meta analysis. Arch. Gynecol. Obstet. 2016, 294, 579–588. [Google Scholar] [CrossRef]
- Joseph, D.B.; Strand, D.W.; Vezina, C.M. DNA methylation in development and disease: An overview for prostate researchers. Am. J. Clin. Exp. Urol. 2018, 6, 197–218. [Google Scholar]
- Okugawa, Y.; Grady, W.M.; Goel, A. Epigenetic alterations in colorectal cancer: Emerging biomarkers. Gastroenterology 2015, 149, 1204–1225. [Google Scholar] [CrossRef]
- Giovannucci, E. Epidemiologic studies of folate and colorectal neoplasia: A review. J. Nutr. 2002, 132, 2350S–2355S. [Google Scholar] [CrossRef]
- Flood, A.; Velie, E.M.; Chaterjee, N.; Subar, A.F.; Thompson, F.E.; Lacey, J.V., Jr.; Schairer, C.; Troisi, R.; Schatzkin, A. Fruit and vegetable intakes and the risk of colorectal cancer in the breast cancer detection demonstration project follow-up cohort. Am. J. Clin. Nutr. 2002, 75, 936–943. [Google Scholar] [CrossRef]
- Meyer, F.; White, E. Alcohol and nutrients in relation to colon cancer in middle-aged adults. Am. J. Epidemiol. 1993, 138, 225–236. [Google Scholar] [CrossRef]
- Slattery, M.L.; Potter, J.D.; Coates, A.; Ma, K.N.; Berry, T.D.; Duncan, D.M.; Caan, B.J. Plant foods and colon cancer: An assessment of specific foods and their related nutrients (united states). Cancer Causes Control. 1997, 8, 575–590. [Google Scholar] [CrossRef]
- Sanjoaquin, M.A.; Allen, N.; Couto, E.; Roddam, A.W.; Key, T.J. Folate intake and colorectal cancer risk: A meta-analytical approach. Int. J. Cancer 2005, 113, 825–828. [Google Scholar] [CrossRef]
- Qin, X.; Cui, Y.; Shen, L.; Sun, N.; Zhang, Y.; Li, J.; Xu, X.; Wang, B.; Xu, X.; Huo, Y.; et al. Folic acid supplementation and cancer risk: A meta-analysis of randomized controlled trials. Int. J. Cancer 2013, 133, 1033–1041. [Google Scholar] [CrossRef]
- Cravo, M.; Fidalgo, P.; Pereira, A.D.; Gouveia-Oliveira, A.; Chaves, P.; Selhub, J.; Mason, J.B.; Mira, F.C.; Leitao, C.N. DNA methylation as an intermediate biomarker in colorectal cancer: Modulation by folic acid supplementation. Eur. J. Cancer Prev. 1994, 3, 473–479. [Google Scholar] [CrossRef]
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin b6, Folate, Vitamin b12, Pantothenic Acid, Biotin, and Choline; National Academies Press: Washington, DC, USA, 1998.
- Zhang, Y.F.; Shi, W.W.; Gao, H.F.; Zhou, L.; Hou, A.J.; Zhou, Y.H. Folate intake and the risk of breast cancer: A dose-response meta-analysis of prospective studies. PLoS ONE 2014, 9, e100044. [Google Scholar] [CrossRef]
- Crary-Dooley, F.K.; Tam, M.E.; Dunaway, K.W.; Hertz-Picciotto, I.; Schmidt, R.J.; LaSalle, J.M. A comparison of existing global DNA methylation assays to low-coverage whole-genome bisulfite sequencing for epidemiological studies. Epigenetics 2017, 12, 206–214. [Google Scholar] [CrossRef]
- Shiratori, H.; Feinweber, C.; Knothe, C.; Lotsch, J.; Thomas, D.; Geisslinger, G.; Parnham, M.J.; Resch, E. High-throughput analysis of global DNA methylation using methyl-sensitive digestion. PLoS ONE 2016, 11, e0163184. [Google Scholar] [CrossRef]
- Kurdyukov, S.; Bullock, M. DNA methylation analysis: Choosing the right method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Wu, H.C.; Delgado-Cruzata, L.; Flom, J.D.; Kappil, M.; Ferris, J.S.; Liao, Y.; Santella, R.M.; Terry, M.B. Global methylation profiles in DNA from different blood cell types. Epigenetics 2011, 6, 76–85. [Google Scholar] [CrossRef]
- Duman, E.A.; Kriaucionis, S.; Dunn, J.J.; Hatchwell, E. A simple modification to the luminometric methylation assay to control for the effects of DNA fragmentation. Biotechniques 2015, 58, 262–264. [Google Scholar] [CrossRef]
- Caiazza, F.; Ryan, E.J.; Doherty, G.; Winter, D.C.; Sheahan, K. Estrogen receptors and their implications in colorectal carcinogenesis. Front. Oncol. 2015, 5, 19. [Google Scholar] [CrossRef]
- Wang, Z.; Li, R.; He, Y.; Huang, S. Effects of secreted frizzled-related protein 1 on proliferation, migration, invasion, and apoptosis of colorectal cancer cells. Cancer Cell Int. 2018, 18, 48. [Google Scholar] [CrossRef]
- Bruno, E.J., Jr.; Ziegenfuss, T.N. Water-soluble vitamins: Research update. Curr. Sports Med. Rep. 2005, 4, 207–213. [Google Scholar] [CrossRef]
- Qiang, Y.; Li, Q.; Xin, Y.; Fang, X.; Tian, Y.; Ma, J.; Wang, J.; Wang, Q.; Zhang, R.; Wang, J.; et al. Intake of dietary one-carbon metabolism-related b vitamins and the risk of esophageal cancer: A dose-response meta-analysis. Nutrients 2018, 10, 835. [Google Scholar] [CrossRef]
- Kulkarni, A.; Dangat, K.; Kale, A.; Sable, P.; Chavan-Gautam, P.; Joshi, S. Effects of altered maternal folic acid, vitamin b12 and docosahexaenoic acid on placental global DNA methylation patterns in wistar rats. PLoS ONE 2011, 6, e17706. [Google Scholar] [CrossRef]
- Sinclair, K.D.; Allegrucci, C.; Singh, R.; Gardner, D.S.; Sebastian, S.; Bispham, J.; Thurston, A.; Huntley, J.F.; Rees, W.D.; Maloney, C.A.; et al. DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional b vitamin and methionine status. Proc. Natl. Acad. Sci. USA 2007, 104, 19351–19356. [Google Scholar] [CrossRef]
- Ray, J.G.; Cole, D.E.; Boss, S.C. An ontario-wide study of vitamin b12, serum folate, and red cell folate levels in relation to plasma homocysteine: Is a preventable public health issue on the rise? Clin. Biochem. 2000, 33, 337–343. [Google Scholar] [CrossRef]
- Robertson, J.; Iemolo, F.; Stabler, S.P.; Allen, R.H.; Spence, J.D. Vitamin b12, homocysteine and carotid plaque in the era of folic acid fortification of enriched cereal grain products. CMAJ 2005, 172, 1569–1573. [Google Scholar] [CrossRef]
- Azadibakhsh, N.; Hosseini, R.S.; Atabak, S.; Nateghiyan, N.; Golestan, B.; Rad, A.H. Efficacy of folate and vitamin b12 in lowering homocysteine concentrations in hemodialysis patients. Saudi J. Kidney Dis. Transplant. 2009, 20, 779–788. [Google Scholar]
- Gonin, J.M.; Nguyen, H.; Gonin, R.; Sarna, A.; Michels, A.; Masri-Imad, F.; Bommareddy, G.; Chassaing, C.; Wainer, I.; Loya, A.; et al. Controlled trials of very high dose folic acid, vitamins b12 and b6, intravenous folinic acid and serine for treatment of hyperhomocysteinemia in esrd. J. Nephrol. 2003, 16, 522–534. [Google Scholar]
- Johanning, G.L.; Heimburger, D.C.; Piyathilake, C.J. DNA methylation and diet in cancer. J. Nutr. 2002, 132, 3814S–3818S. [Google Scholar] [CrossRef]
- Hollenbeck, C.B. An introduction to the nutrition and metabolism of choline. Cent. Nerv. Syst. Agents Med. Chem. 2012, 12, 100–113. [Google Scholar] [CrossRef]
- Ueland, P.M. Choline and betaine in health and disease. J. Inherit. Metab. Dis. 2011, 34, 3–15. [Google Scholar] [CrossRef]
- Craig, S.A. Betaine in human nutrition. Am. J. Clin. Nutr. 2004, 80, 539–549. [Google Scholar] [CrossRef]
- Zeisel, S.H. Choline: An essential nutrient for humans. Nutrition 2000, 16, 669–671. [Google Scholar] [CrossRef]
- Jacob, R.A.; Jenden, D.J.; Allman-Farinelli, M.A.; Swendseid, M.E. Folate nutriture alters choline status of women and men fed low choline diets. J. Nutr. 1999, 129, 712–717. [Google Scholar] [CrossRef]
- Barak, A.J.; Beckenhauer, H.C.; Kharbanda, K.K.; Tuma, D.J. Chronic ethanol consumption increases homocysteine accumulation in hepatocytes. Alcohol 2001, 25, 77–81. [Google Scholar] [CrossRef]
- Melse-Boonstra, A.; Holm, P.I.; Ueland, P.M.; Olthof, M.; Clarke, R.; Verhoef, P. Betaine concentration as a determinant of fasting total homocysteine concentrations and the effect of folic acid supplementation on betaine concentrations. Am. J. Clin. Nutr. 2005, 81, 1378–1382. [Google Scholar] [CrossRef]
- Lee, J.E.; Jacques, P.F.; Dougherty, L.; Selhub, J.; Giovannucci, E.; Zeisel, S.H.; Cho, E. Are dietary choline and betaine intakes determinants of total homocysteine concentration? Am. J. Clin. Nutr. 2010, 91, 1303–1310. [Google Scholar] [CrossRef]
- Brouwer, I.A.; Verhoef, P.; Urgert, R. Betaine supplementation and plasma homocysteine in healthy volunteers. Arch. Intern. Med. 2000, 160, 2546–2547. [Google Scholar] [CrossRef]
- Niculescu, M.D.; Craciunescu, C.N.; Zeisel, S.H. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. FASEB J. 2006, 20, 43–49. [Google Scholar] [CrossRef]
- Mehedint, M.G.; Niculescu, M.D.; Craciunescu, C.N.; Zeisel, S.H. Choline deficiency alters global histone methylation and epigenetic marking at the re1 site of the calbindin 1 gene. FASEB J. 2010, 24, 184–195. [Google Scholar] [CrossRef]
- Mehedint, M.G.; Craciunescu, C.N.; Zeisel, S.H. Maternal dietary choline deficiency alters angiogenesis in fetal mouse hippocampus. Proc. Natl. Acad. Sci. USA 2010, 107, 12834–12839. [Google Scholar] [CrossRef]
- Kovacheva, V.P.; Mellott, T.J.; Davison, J.M.; Wagner, N.; Lopez-Coviella, I.; Schnitzler, A.C.; Blusztajn, J.K. Gestational choline deficiency causes global and igf2 gene DNA hypermethylation by up-regulation of dnmt1 expression. J. Biol. Chem. 2007, 282, 31777–31788. [Google Scholar] [CrossRef]
- Stefanska, B.; Karlic, H.; Varga, F.; Fabianowska-Majewska, K.; Haslberger, A. Epigenetic mechanisms in anti-cancer actions of bioactive food components--the implications in cancer prevention. Br. J. Pharmacol. 2012, 167, 279–297. [Google Scholar] [CrossRef]
- da Costa, K.A.; Cochary, E.F.; Blusztajn, J.K.; Garner, S.C.; Zeisel, S.H. Accumulation of 1,2-sn-diradylglycerol with increased membrane-associated protein kinase c may be the mechanism for spontaneous hepatocarcinogenesis in choline-deficient rats. J. Biol. Chem. 1993, 268, 2100–2105. [Google Scholar]
- da Costa, K.A.; Garner, S.C.; Chang, J.; Zeisel, S.H. Effects of prolonged (1 year) choline deficiency and subsequent re-feeding of choline on 1,2-sn-diradylglycerol, fatty acids and protein kinase c in rat liver. Carcinogenesis 1995, 16, 327–334. [Google Scholar] [CrossRef]
- Shivapurkar, N.; Poirier, L.A. Tissue levels of s-adenosylmethionine and s-adenosylhomocysteine in rats fed methyl-deficient, amino acid-defined diets for one to five weeks. Carcinogenesis 1983, 4, 1051–1057. [Google Scholar] [CrossRef]
- Tsujiuchi, T.; Tsutsumi, M.; Sasaki, Y.; Takahama, M.; Konishi, Y. Hypomethylation of cpg sites and c-myc gene overexpression in hepatocellular carcinomas, but not hyperplastic nodules, induced by a choline-deficient l-amino acid-defined diet in rats. Jpn. J. Cancer Res. 1999, 90, 909–913. [Google Scholar] [CrossRef]
- Tryndyak, V.P.; Han, T.; Muskhelishvili, L.; Fuscoe, J.C.; Ross, S.A.; Beland, F.A.; Pogribny, I.P. Coupling global methylation and gene expression profiles reveal key pathophysiological events in liver injury induced by a methyl-deficient diet. Mol. Nutr. Food Res. 2011, 55, 411–418. [Google Scholar] [CrossRef]
- Lupu, D.S.; Orozco, L.D.; Wang, Y.; Cullen, J.M.; Pellegrini, M.; Zeisel, S.H. Altered methylation of specific DNA loci in the liver of bhmt-null mice results in repression of iqgap2 and f2rl2 and is associated with development of preneoplastic foci. FASEB J. 2017, 31, 2090–2103. [Google Scholar] [CrossRef]
- Sun, S.; Li, X.; Ren, A.; Du, M.; Du, H.; Shu, Y.; Zhu, L.; Wang, W. Choline and betaine consumption lowers cancer risk: A meta-analysis of epidemiologic studies. Sci. Rep. 2016, 6, 35547. [Google Scholar] [CrossRef]
- Guedes, R.L.; Prosdocimi, F.; Fernandes, G.R.; Moura, L.K.; Ribeiro, H.A.; Ortega, J.M. Amino acids biosynthesis and nitrogen assimilation pathways: A great genomic deletion during eukaryotes evolution. BMC Genom. 2011, 12 (Suppl. 4), S2. [Google Scholar] [CrossRef]
- Martinez, Y.; Li, X.; Liu, G.; Bin, P.; Yan, W.; Mas, D.; Valdivie, M.; Hu, C.A.; Ren, W.; Yin, Y. The role of methionine on metabolism, oxidative stress, and diseases. Amino Acids 2017, 49, 2091–2098. [Google Scholar] [CrossRef]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Zhang, N. Role of methionine on epigenetic modification of DNA methylation and gene expression in animals. Anim. Nutr. 2018, 4, 11–16. [Google Scholar] [CrossRef]
- Finkelstein, J.D.; Martin, J.J. Methionine metabolism in mammals. Adaptation to methionine excess. J. Biol. Chem. 1986, 261, 1582–1587. [Google Scholar]
- Regina, M.; Korhonen, V.P.; Smith, T.K.; Alakuijala, L.; Eloranta, T.O. Methionine toxicity in the rat in relation to hepatic accumulation of s-adenosylmethionine: Prevention by dietary stimulation of the hepatic transsulfuration pathway. Arch. Biochem. Biophys. 1993, 300, 598–607. [Google Scholar] [CrossRef]
- Waterland, R.A. Assessing the effects of high methionine intake on DNA methylation. J. Nutr. 2006, 136, 1706S–1710S. [Google Scholar] [CrossRef]
- Rowling, M.J.; McMullen, M.H.; Chipman, D.C.; Schalinske, K.L. Hepatic glycine n-methyltransferase is up-regulated by excess dietary methionine in rats. J. Nutr. 2002, 132, 2545–2550. [Google Scholar] [CrossRef]
- Amaral, C.L.; Bueno Rde, B.; Burim, R.V.; Queiroz, R.H.; Bianchi Mde, L.; Antunes, L.M. The effects of dietary supplementation of methionine on genomic stability and p53 gene promoter methylation in rats. Mutat. Res. 2011, 722, 78–83. [Google Scholar] [CrossRef]
- Uekawa, A.; Katsushima, K.; Ogata, A.; Kawata, T.; Maeda, N.; Kobayashi, K.; Maekawa, A.; Tadokoro, T.; Yamamoto, Y. Change of epigenetic control of cystathionine beta-synthase gene expression through dietary vitamin b12 is not recovered by methionine supplementation. J. Nutr. Nutr. 2009, 2, 29–36. [Google Scholar]
- Miousse, I.R.; Pathak, R.; Garg, S.; Skinner, C.M.; Melnyk, S.; Pavliv, O.; Hendrickson, H.; Landes, R.D.; Lumen, A.; Tackett, A.J.; et al. Short-term dietary methionine supplementation affects one-carbon metabolism and DNA methylation in the mouse gut and leads to altered microbiome profiles, barrier function, gene expression and histomorphology. Genes Nutr. 2017, 12, 22. [Google Scholar] [CrossRef]
- Zhou, Z.Y.; Wan, X.Y.; Cao, J.W. Dietary methionine intake and risk of incident colorectal cancer: A meta-analysis of 8 prospective studies involving 431,029 participants. PLoS ONE 2013, 8, e83588. [Google Scholar] [CrossRef]
- Vidal, A.C.; Grant, D.J.; Williams, C.D.; Masko, E.; Allott, E.H.; Shuler, K.; McPhail, M.; Gaines, A.; Calloway, E.; Gerber, L.; et al. Associations between intake of folate, methionine, and vitamins b-12, b-6 and prostate cancer risk in american veterans. J. Cancer Epidemiol. 2012, 2012, 957467. [Google Scholar] [CrossRef]
- Durando, X.; Farges, M.C.; Buc, E.; Abrial, C.; Petorin-Lesens, C.; Gillet, B.; Vasson, M.P.; Pezet, D.; Chollet, P.; Thivat, E. Dietary methionine restriction with folfox regimen as first line therapy of metastatic colorectal cancer: A feasibility study. Oncology 2010, 78, 205–209. [Google Scholar] [CrossRef]
- Epner, D.E.; Morrow, S.; Wilcox, M.; Houghton, J.L. Nutrient intake and nutritional indexes in adults with metastatic cancer on a phase i clinical trial of dietary methionine restriction. Nutr. Cancer 2002, 42, 158–166. [Google Scholar] [CrossRef]
- Thivat, E.; Durando, X.; Demidem, A.; Farges, M.C.; Rapp, M.; Cellarier, E.; Guenin, S.; D’Incan, M.; Vasson, M.P.; Chollet, P. A methionine-free diet associated with nitrosourea treatment down-regulates methylguanine-DNA methyl transferase activity in patients with metastatic cancer. Anticancer Res. 2007, 27, 2779–2783. [Google Scholar]
- Thivat, E.; Farges, M.C.; Bacin, F.; D’Incan, M.; Mouret-Reynier, M.A.; Cellarier, E.; Madelmont, J.C.; Vasson, M.P.; Chollet, P.; Durando, X. Phase ii trial of the association of a methionine-free diet with cystemustine therapy in melanoma and glioma. Anticancer Res. 2009, 29, 5235–5240. [Google Scholar]
- Wu, W.; Kang, S.; Zhang, D. Association of vitamin b6, vitamin b12 and methionine with risk of breast cancer: A dose-response meta-analysis. Br. J. Cancer 2013, 109, 1926–1944. [Google Scholar] [CrossRef]
- Lu, S.C.; Huang, Z.Z.; Yang, H.; Mato, J.M.; Avila, M.A.; Tsukamoto, H. Changes in methionine adenosyltransferase and s-adenosylmethionine homeostasis in alcoholic rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G178–G185. [Google Scholar] [CrossRef]
- Martinez-Chantar, M.L.; Corrales, F.J.; Martinez-Cruz, L.A.; Garcia-Trevijano, E.R.; Huang, Z.Z.; Chen, L.; Kanel, G.; Avila, M.A.; Mato, J.M.; Lu, S.C. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1a. FASEB J. 2002, 16, 1292–1294. [Google Scholar] [CrossRef]
- Medici, V.; Halsted, C.H. Folate, alcohol, and liver disease. Mol. Nutr. Food Res. 2013, 57, 596–606. [Google Scholar] [CrossRef]
- Richmond, R.C.; Joubert, B.R. Contrasting the effects of intra-uterine smoking and one-carbon micronutrient exposures on offspring DNA methylation. Epigenomics 2017, 9, 351–367. [Google Scholar] [CrossRef]
- Markunas, C.A.; Xu, Z.; Harlid, S.; Wade, P.A.; Lie, R.T.; Taylor, J.A.; Wilcox, A.J. Identification of DNA methylation changes in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2014, 122, 1147–1153. [Google Scholar] [CrossRef]
- Tuenter, A.; Bautista Nino, P.K.; Vitezova, A.; Pantavos, A.; Bramer, W.M.; Franco, O.H.; Felix, J.F. Folate, vitamin b12, and homocysteine in smoking-exposed pregnant women: A systematic review. Matern. Child. Nutr. 2018, 15, e12675. [Google Scholar] [CrossRef]
- Piyathilake, C.J.; Macaluso, M.; Hine, R.J.; Richards, E.W.; Krumdieck, C.L. Local and systemic effects of cigarette smoking on folate and vitamin b-12. Am. J. Clin. Nutr. 1994, 60, 559–566. [Google Scholar] [CrossRef]
- Mozhui, K.; Smith, A.K.; Tylavsky, F.A. Ancestry dependent DNA methylation and influence of maternal nutrition. PLoS ONE 2015, 10, e0118466. [Google Scholar] [CrossRef]
- Altmann, S.; Murani, E.; Schwerin, M.; Metges, C.C.; Wimmers, K.; Ponsuksili, S. Dietary protein restriction and excess of pregnant german landrace sows induce changes in hepatic gene expression and promoter methylation of key metabolic genes in the offspring. J. Nutr. Biochem. 2013, 24, 484–495. [Google Scholar] [CrossRef]
- Sandovici, I.; Smith, N.H.; Nitert, M.D.; Ackers-Johnson, M.; Uribe-Lewis, S.; Ito, Y.; Jones, R.H.; Marquez, V.E.; Cairns, W.; Tadayyon, M.; et al. Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the hnf4a gene in rat pancreatic islets. Proc. Natl. Acad. Sci. USA 2011, 108, 5449–5454. [Google Scholar] [CrossRef]
- Tosh, D.N.; Fu, Q.; Callaway, C.W.; McKnight, R.A.; McMillen, I.C.; Ross, M.G.; Lane, R.H.; Desai, M. Epigenetics of programmed obesity: Alteration in iugr rat hepatic igf1 mrna expression and histone structure in rapid vs. Delayed postnatal catch-up growth. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G1023–G1029. [Google Scholar] [CrossRef]
- Marco, A.; Kisliouk, T.; Tabachnik, T.; Meiri, N.; Weller, A. Overweight and cpg methylation of the pomc promoter in offspring of high-fat-diet-fed dams are not “reprogrammed” by regular chow diet in rats. FASEB J. 2014, 28, 4148–4157. [Google Scholar] [CrossRef]
- Ge, Z.J.; Luo, S.M.; Lin, F.; Liang, Q.X.; Huang, L.; Wei, Y.C.; Hou, Y.; Han, Z.M.; Schatten, H.; Sun, Q.Y. DNA methylation in oocytes and liver of female mice and their offspring: Effects of high-fat-diet-induced obesity. Environ. Health Perspect. 2014, 122, 159–164. [Google Scholar] [CrossRef]
- Medici, V.; Kieffer, D.A.; Shibata, N.M.; Chima, H.; Kim, K.; Canovas, A.; Medrano, J.F.; Islas-Trejo, A.D.; Kharbanda, K.K.; Olson, K.; et al. Wilson disease: Epigenetic effects of choline supplementation on phenotype and clinical course in a mouse model. Epigenetics 2016, 11, 804–818. [Google Scholar] [CrossRef]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef]
- Steegers-Theunissen, R.P.; Obermann-Borst, S.A.; Kremer, D.; Lindemans, J.; Siebel, C.; Steegers, E.A.; Slagboom, P.E.; Heijmans, B.T. Periconceptional maternal folic acid use of 400 microg per day is related to increased methylation of the igf2 gene in the very young child. PLoS ONE 2009, 4, e7845. [Google Scholar] [CrossRef]
- Pauwels, S.; Ghosh, M.; Duca, R.C.; Bekaert, B.; Freson, K.; Huybrechts, I.; Langie, S.A.S.; Koppen, G.; Devlieger, R.; Godderis, L. Maternal intake of methyl-group donors affects DNA methylation of metabolic genes in infants. Clin. Epigenet. 2017, 9, 16. [Google Scholar] [CrossRef]
- Fryer, A.A.; Nafee, T.M.; Ismail, K.M.; Carroll, W.D.; Emes, R.D.; Farrell, W.E. Line-1 DNA methylation is inversely correlated with cord plasma homocysteine in man: A preliminary study. Epigenetics 2009, 4, 394–398. [Google Scholar] [CrossRef]
- Fryer, A.A.; Emes, R.D.; Ismail, K.M.; Haworth, K.E.; Mein, C.; Carroll, W.D.; Farrell, W.E. Quantitative, high-resolution epigenetic profiling of cpg loci identifies associations with cord blood plasma homocysteine and birth weight in humans. Epigenetics 2011, 6, 86–94. [Google Scholar] [CrossRef]
- Wang, M.; Li, K.; Zhao, D.; Li, L. The association between maternal use of folic acid supplements during pregnancy and risk of autism spectrum disorders in children: A meta-analysis. Mol. Autism 2017, 8, 51. [Google Scholar] [CrossRef]
- Dessypris, N.; Karalexi, M.A.; Ntouvelis, E.; Diamantaras, A.A.; Papadakis, V.; Baka, M.; Hatzipantelis, E.; Kourti, M.; Moschovi, M.; Polychronopoulou, S.; et al. Association of maternal and index child’s diet with subsequent leukemia risk: A systematic review and meta analysis. Cancer Epidemiol. 2017, 47, 64–75. [Google Scholar] [CrossRef]
- Xu, A.; Cao, X.; Lu, Y.; Li, H.; Zhu, Q.; Chen, X.; Jiang, H.; Li, X. A meta-analysis of the relationship between maternal folic acid supplementation and the risk of congenital heart defects. Int. Heart J. 2016, 57, 725–728. [Google Scholar] [CrossRef]
- Blanco, R.; Colombo, A.; Pardo, R.; Suazo, J. Maternal biomarkers of methylation status and non-syndromic orofacial cleft risk: A meta-analysis. Int. J. Oral. Maxillofac. Surg. 2016, 45, 1323–1332. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Authors | Population/Tissue | Study Design | Methylation Assay | Conclusion/Outcome |
|---|---|---|---|---|
| Folate | ||||
| Wallace et al. [56] | Adults with history of colorectal adenoma Colorectal tissues | Randomized, double-blind controlled trial 1 mg/day for 3 years | Gene-specific quantitative bisulfite pyrosequencing ERα and SFRP1 genes | Higher folate levels were associated with higher levels of ERα (estrogen receptor alpha) and SFRP1 (Secreted Frizzled Related Protein 1) methylation |
| Pufulete et al. [57] | Colorectal adenoma and cancer patients and heathy controls Colonic tissues | Case-control study Estimates of dietary intake and serum and erythrocyte folate | Global DNA methylation via [(3)H] methyl incorporation | High folate status was associated with decreased plasma homocysteine and increased colonic DNA methylation. Low folate intake and colonic DNA hypomethylation were associated with increased risk for adenoma and cancer |
| Piyathilake et al. [58] | Patients with cervical intraepithelial neoplasia Cervical tissues | Cross-sectional study Dietary intake pre and post folic acid fortification | Global DNA methylation via Immunohistochemical staining for 5-methyl cytosine | Folic acid fortification did not change global DNA methylation in cells involved in cervical carcinogenesis |
| Moore et al. [59] | Patients with bladder cancer and controls Blood | Case-control study Dietary intake via food frequency questionnaire (FFQ) | Global DNA methylation via 5-methyl cytosine antibody | Global DNA methylation was significantly lower in cases than control. No significant differences in folate intake between cases and control |
| Piyathilake et al. [60] | Patients with cervical intraepithelial neoplasia and controls Blood and exfoliated cervical cells | Case-control study Serum levels measured | Global DNA methylation via bisulfite pyrosequencing LINE-1 (Long Interspersed Nucleotide Element 1) analysis | Blood cell (but not cervical cell) DNA was hypomethylated in cases compared to controls and hypermethylated in the highest folate compared to the lowest folate tertile. |
| Pufulete et al. [61] | Healthy adults Colonic tissues | Cross-sectional study Serum and erythrocyte levels measured | Global DNA methylation via [(3)H] methyl incorporation | Observed weak inverted associations between serum and erythrocyte folate and colonic DNA hypomethylation |
| O’Reilly et al. [62] | Patients with colorectal adenoma Colonic tissues | Randomized, double-blind controlled trial 600 μg folic acid/day for 6 months | Global DNA methylation via methylation-sensitive restriction enzymes | Folate treatment significantly reversed global DNA hypomethylation in colonic tissues |
| Cravo et al. [63] | Patients with colorectal adenoma Colonic tissues | Randomized, controlled, cross-over study 5 mg/day for 3 months then switched to placebo for additional 3 months | Global DNA methylation via [(3)H] methyl incorporation | Folate supplementation reversed DNA Hypomethylation, which returned to baseline values after switching to placebo treatment |
| Kim et al. [64] | Patients with colorectal adenoma Colonic tissues | Randomized, double-blind controlled trial 5 mg/day for 1 year | Global DNA methylation | Folate supplementation increased genomic DNA methylation at 6 months and 1 year |
| Coppedè et al. [65] | Patients with colorectal cancer Colonic tissues (cancer and adjacent healthy) | Cross-sectional analysis Serum levels were measured | Gene-specific quantitative bisulfite pyrosequencing APC (adenomatous polyposis coli), MGMT (Methylguanine-DNA Methyltransferase), hMLH1 (MutL homolog 1), RASSF1A and CDKN2A (cyclin-dependent kinase 2A) genes | Low folate levels were associated with hMLH1 hypermethylation |
| Christensen et al. [66] | Breast cancer patients Breast cancer tissues | The Pathways Study: a prospective cohort study Estimates of dietary intake | Genome-wide methylation analysis via Illumina GoldenGate methylation bead-array platform | Higher folate intake was associated with a trend toward increased CpG methylation in several genes |
| Vineis et al. [67] | Patients with lung cancer and healthy controls Blood | Nested case-control study in The European Prospective Investigation into Cancer and Nutrition (EPIC) Serum levels were measured | Genome-wide quantitative bisulfite pyrosequencing | Folate was associated with increased methylation levels of RASSF1A (Ras association domain family member 1) and MTHFR (methylenetetrahydrofolate reductase) |
| van Engeland et al. [68] | Patients with colorectal cancer Colorectal biopsies | Netherland Cohort Study (NLCS) Estimated dietary intake via FFQ | Methylation-specific PCR (polymerase chain reaction) for APC-1A (adenomatous polyposis coli-1A), p14(ARF) (alternate reading frame protein of cyclin-dependent kinase 2A), p16(INK4A) (cyclin-dependent kinase inhibitor 4A), hMLH1, O(6)-MGMT (O-6-methylguanine-DNA methyltransferase), and RASSF1A genes | Gene promoters were hypermethylated in patients with low folate intake compared with high folate intake; differences were not statistically significant |
| Ba et al. [69] | Pregnant women Maternal and cord blood | Cross-sectional study Serum levels were measured | Methylation-specific PCR for IGF2 gene | IGF2 promoter methylation was not associated with serum folate levels in either cord or maternal blood |
| Hoyo et al. [70] | Pregnant women Cord blood | Cross-sectional study Estimated dietary intake via FFQ | Gene-specific (IGF-2) quantitative bisulfite pyrosequencing | IGF-2 methylation decreased with increasing folate intake |
| Shelnutt et al. [71] | Healthy non-pregnant women Blood | Folate depletion-repletion clinical trial 115 μg/day for 7 weeks followed by 400 μg/day for additional 7 weeks | Global DNA methylation via [(3)H] methyl incorporation | Observed global DNA hypomethylation during depletion and increases in DNA methylation during repletion |
| Vitamin B | ||||
| Colacino et al. [72] | Patients with head and neck cancer | Cross-sectional study Estimated dietary intake via FFQ | Gene-specific methylation analysis via Illumina Goldengate Methylation Cancer Panel | Patients with the highest quartile of vitamin B12 intake showed significantly less tumor suppressor gene methylation compared with those in the lowest quartile |
| Piyathilake et al. [73] | Patients with lung cancer Cancer tissue and adjacent normal bronchial tissue | Cross-sectional study Tissue levels were measured | Global DNA methylation via [(3)H] methyl incorporation | A direct association was reported between vitamin B-12 and global DNA methylation in cancer tissues but not in normal tissues |
| Perng et al. [74] | School-age children Blood | Cross-sectional study Plasma levels were measured | Global DNA methylation via bisulfite pyrosequencing LINE-1 analysis | No association between vitamin B12 and global DNA methylation |
| Hubner, et al. [75] | Old adults Blood | Clinical trial 500 µg folic acid, 500 µg vitamin B12 and 50 mg vitamin B6 for 1 year | Global DNA methylation via bisulfite pyrosequencing LINE-1 analysis | Vitamin B supplementation had no effect on global DNA methylation in blood cells |
| Piyathilake et al. [76] | Women positive for human papilloma virus Exfoliated cervical cells | Cross-sectional study Plasma levels were measured | Gene-specific (HPV(human papilloma virus)-16) quantitative bisulfite pyrosequencing | Folate and vitamin B12, maintain a high degree of methylation at specific CpG sites in the HPV E6 gene and subsequently reduce the risk of cervical intraepithelial neoplasia |
| Choline and betaine | ||||
| Pauwels et al. [77] | Pregnant women Blood | MANOE (MAternal Nutrition and Offspring’s Epigenome) cohort study Estimated dietary intake via FFQ | Global DNA (hydroxy)methylation was measured in blood using LC-MS/MS (liquid chromatography-mass spectrometry/mass spectrometry) | Choline and betaine intake in the first weeks was negatively associated with DNA hydroxymethylation (a step that precedes demethylation) |
| Chiuve et al. [78] | Healthy women Blood | Cross-sectional study from The Nurses’ Health Study (NHS) Estimated dietary intake via FFQ | Plasma total homocysteine measurement via HPLC (high performance liquid chromatography) | Total choline + betaine intake was inversely associated with homocysteine (measured as a surrogate biomarker for effective methyl donation and DNMT activity) |
| Schwab et al. [79] | Obese adults Blood | Randomized, double-blind controlled trial Betaine supplements (6 gm/day) for 12 weeks | Plasma total homocysteine measurement via HPLC | Betaine supplementation decreased the plasma homocysteine concentration |
| Olthof et al. [80] | Healthy men Blood | Randomized, double-blind controlled trial Choline supplements (2.6 gm/day) for 2 weeks | Plasma total homocysteine measurement via HPLC | Choline supplementation decreased the plasma homocysteine concentration |
| Methionine | ||||
| Vineis et al. [67] | Details are in the folate section of the table | Methionine was associated with decreased methylation of RASSF1A gene | ||
| Pauwels et al. [77] | Details are in the choline and betaine section of the table | A high intake of methionine showed lower DNA hydroxymethylation (a step that precedes demethylation) | ||
| Perng et al. [81] | Healthy adults Blood | Multi-Ethnic Study of Atherosclerosis (MESA) Stress Study Estimated dietary intake via FFQ | Global DNA methylation via bisulfite pyrosequencing LINE-1 analysis | Dietary methionine was not associated with global DNA methylation |
| Tao et al. [82] | Breast cancer patients and control Breast cancer tissue | Cross-sectional study from the Western New York Exposures and Breast Cancer Study (WEB Study) Estimated dietary intake via FFQ | Methylation-specific PCR of E-cadherin, p16, and RAR-β(2) (retinoic acid receptor beta 2) genes | Dietary intake of methionine was not associated with promoter methylation of E-cadherin, p16, and RAR-β(2) genes |
| Authors | Population/Tissue | Study Design | Conclusion/Outcome |
|---|---|---|---|
| Folate | |||
| Giovannucci et al. [83] | Male and female adults | The Nurses’ Health Study, and the Health Professionals Follow-up Study Estimated dietary intake via FFQ | High dietary folate was inversely associated with risk of colorectal adenoma in women and men |
| Su et al. [84] | Male and female adults | The NHANES I Epidemiologic Follow-up Study (NHEFS) Estimated dietary intake via FFQ | Significant association between folate intake and lower risk of colon cancer among men and non-alcohol drinkers, but not women or alcohol drinkers |
| Fuchs et al. [85] | Female adult | The Nurses’ Health Study Estimated dietary intake via FFQ | Higher folate intake reduces the risk of colon cancer associated with a family history of the disease. |
| Stevens et al. [86] | Male adult | The American Cancer Society Cancer Prevention Study II Nutrition Cohort Estimated dietary intake via FFQ | Higher intake of folate was associated with a nonsignificant decrease in the risk of advanced prostate cancer |
| Gylling et al. [87] | Patients with colorectal cancer and matched controls | The Nurses’ Health Study | Low plasma levels of folate were associated with a reduced risk of colorectal cancer |
| Giovannucci et al. [88] | Female adult | A nested case-control study in the population-based Northern Sweden Health and Disease Study Estimated dietary intake via FFQ | Folate intake was associated with a lower risk for colon cancer |
| Konings et al. [89] | Male and female adults | The Netherlands Cohort Study Estimated dietary intake via FFQ | The study reported an inverse association between colon cancer risk and total dietary folate intake. |
| Terry et al. [90] | Patients with colorectal cancer and matched controls | A nested case-control study in the Canadian National Breast Screening Study Estimated dietary intake via FFQ | Folate intake was inversely associated with the risk of colorectal cancer |
| Wei et al. [91] | Patients with colorectal cancer and matched controls | A nested case-control study in the Nurses’ Health Study (NHS) and the Health Professionals Follow Up Study (HPFS) Estimated dietary intake via FFQ | Folate intake was associated with lower risk of colon cancer; however, rectal cancer cases tended to have slightly higher folate |
| Harnack et al. [92] | Female adults | Population-based Iowa Women’s Health Study cohort Estimated dietary intake via FFQ | There were no independent associations of folate with incidence of colon cancer; however, relative risk was lower among those who had a combined high folate and high vitamin B-12 or high folate and vitamin B6. |
| Benito et al. [93] | Colorectal cancer and matched controls | A case-control study Estimated dietary intake via FFQ | Folate intake was associated with reduced risk of colorectal cancer |
| Ferraroni et al. [94] | Colorectal cancer and matched controls | A case-control study Estimated dietary intake via FFQ | There was a trend of a protective effect of high folate intake against colorectal cancer development |
| Freudenheim et al. [95] | Colorectal cancer and matched controls | A case-control study Estimated dietary intake via FFQ | Folate intake was associated with a reduced risk of rectal cancer but not colon cancer |
| Glynn et al. [96] | Patients with colorectal cancer and matched control | A nested case-control study within the Alpha-Tocopherol Beta-Carotene Study cohort of male smokers Estimated dietary intake via FFQ and serum levels were measured | No association between serum folate and colorectal cancer. High dietary folate intake was protective against colorectal cancer. |
| La Vecchia et al. [97] | Patients with colorectal cancer and matched control | Case-control study Estimated dietary intake via FFQ | No association between dietary folate and risk of colorectal cancer |
| Le Marchand et al. [98] | Patients with colorectal cancer and matched control | Case-control study Estimated dietary intake via FFQ | Decreased risk of colorectal cancer in subjects who consume high levels of folate and vitamin B6 |
| Levi et al. [99] | Patients with colorectal cancer and matched control | Case-control study Estimated dietary intake via FFQ | No significant association between folate intake and colorectal cancer |
| Boutron-Ruault et al. [100] | Patients with colorectal cancer and matched control | Case-control study Estimated dietary intake via FFQ | Folate intake prevents adenoma formation and protective against adenoma growth associated with alcohol |
| Kato et al. [101] | Patients with colorectal cancer and matched control | A nested case-control study in the New York University Women’s Health Study cohort Serum levels were measured | The risk of colorectal cancer in the subjects in the highest quartile of serum folate concentrations was half that of those in the lowest quartile |
| Cole et al. [102] | Patients with colorectal adenoma | Randomized, double-blind controlled trial 1 mg/day of folic acid for 3 years | Folic acid at 1 mg/day does not reduce the risk of colorectal adenomas or their advancement to neoplastic lesions |
| Ebbing et al. [103] | Male and female adults with ischemic heart disease | Norwegian Vitamin Trial and Western Norway B Vitamin Intervention Trial folic acid (0.8 mg/day) plus vitamin B12 (0.4 mg/day) for 6–7 years | Folic acid plus vitamin B12 supplementations were associated with increased cancer outcomes and all-cause mortality in patients with ischemic heart disease |
| Vitamin B | |||
| Otani et al. [104] | Patients with colorectal cancer and matched control | Case-control study Estimated dietary intake via FFQ | Neither vitamin B2, vitamin B6, nor vitamin B12 were significantly associated with colorectal cancer |
| Hultdin et al. [105] | Patients with prostate cancer and matched control | Case-control study Serum levels were measured | Serum concentrations of vitamin B12 were associated with an up to three-fold increase in prostate cancer risk |
| Gylling et al. [87] | Details are in the folate section of the table | Plasma levels of vitamin B12 were inversely associated with rectal cancer risk | |
| Choline and Betaine | |||
| Du et al. [106] | Patients with breast cancer and matched control | A hospital-based case-control study Serum levels were measured | Serum betaine but not choline was inversely associated with risk of breast cancer development in subjects with below-median dietary folate intake |
| Lu et al. [107] | Patients with colorectal cancer and matched control | Case-control study Estimated dietary intake via FFQ | Total choline intake was inversely associated with colorectal cancer risk however no significant associations were observed for betaine or total choline plus betaine intakes |
| Zeng et al. [108] | Patients with nasopharyngeal cancer and matched control | Case-control study Estimated dietary intake via FFQ | Intakes of total choline, betaine, and combined choline and betaine were inversely associated with nasopharyngeal cancer |
| Zhou et al. [109] | Patients with liver cancer and matched control | Case-control study Estimated dietary intake via FFQ | Higher intake of choline and betaine was associated with a lower risk of liver cancer |
| Nitter et al. [110] | Patients with colorectal cancer and matched control | A nested case-control study within the European Prospective Investigation into Cancer and Nutrition (EPIC) Plasma concentrations were measured | Higher betaine and choline concentrations were associated with lower risk of colorectal cancer especially in subjects with lower folate concentrations |
| Methionine | |||
| Feigelson et al. [111] | Patients with prostate cancer and matched control | Case-control study Estimated dietary intake via FFQ | A direct association between higher methionine intake and prostate cancer risk was observed only in men who have at least one MTHFR A1298C allele |
| Giovannucci et al. [83] | Details are in the folate section of the table | Methionine intake was inversely associated with risk of having larger adenomas (1 cm or larger) | |
| Su et al. [84] | Details are in the folate section of the table | Significantly increased risk of colon cancer in men who consume low-methionine diet compared to those who consume high methionine diet | |
| Fuchs et al. [85] | Details are in the folate section of the table | Higher intake of methionine reduces the risk of colon cancer associated with a family history of the disease | |
| Nitter et al. [110] | Details are in the betaine and choline section of the table | Methionine concentrations were inversely associated with colorectal cancer risk with borderline significance | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmoud, A.M.; Ali, M.M. Methyl Donor Micronutrients that Modify DNA Methylation and Cancer Outcome. Nutrients 2019, 11, 608. https://doi.org/10.3390/nu11030608
Mahmoud AM, Ali MM. Methyl Donor Micronutrients that Modify DNA Methylation and Cancer Outcome. Nutrients. 2019; 11(3):608. https://doi.org/10.3390/nu11030608
Chicago/Turabian StyleMahmoud, Abeer M., and Mohamed M. Ali. 2019. "Methyl Donor Micronutrients that Modify DNA Methylation and Cancer Outcome" Nutrients 11, no. 3: 608. https://doi.org/10.3390/nu11030608
APA StyleMahmoud, A. M., & Ali, M. M. (2019). Methyl Donor Micronutrients that Modify DNA Methylation and Cancer Outcome. Nutrients, 11(3), 608. https://doi.org/10.3390/nu11030608