Lonicera caerulea Extract Attenuates Non-Alcoholic Fatty Liver Disease in Free Fatty Acid-Induced HepG2 Hepatocytes and in High Fat Diet-Fed Mice



Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Honeyberry Extract (HBE)
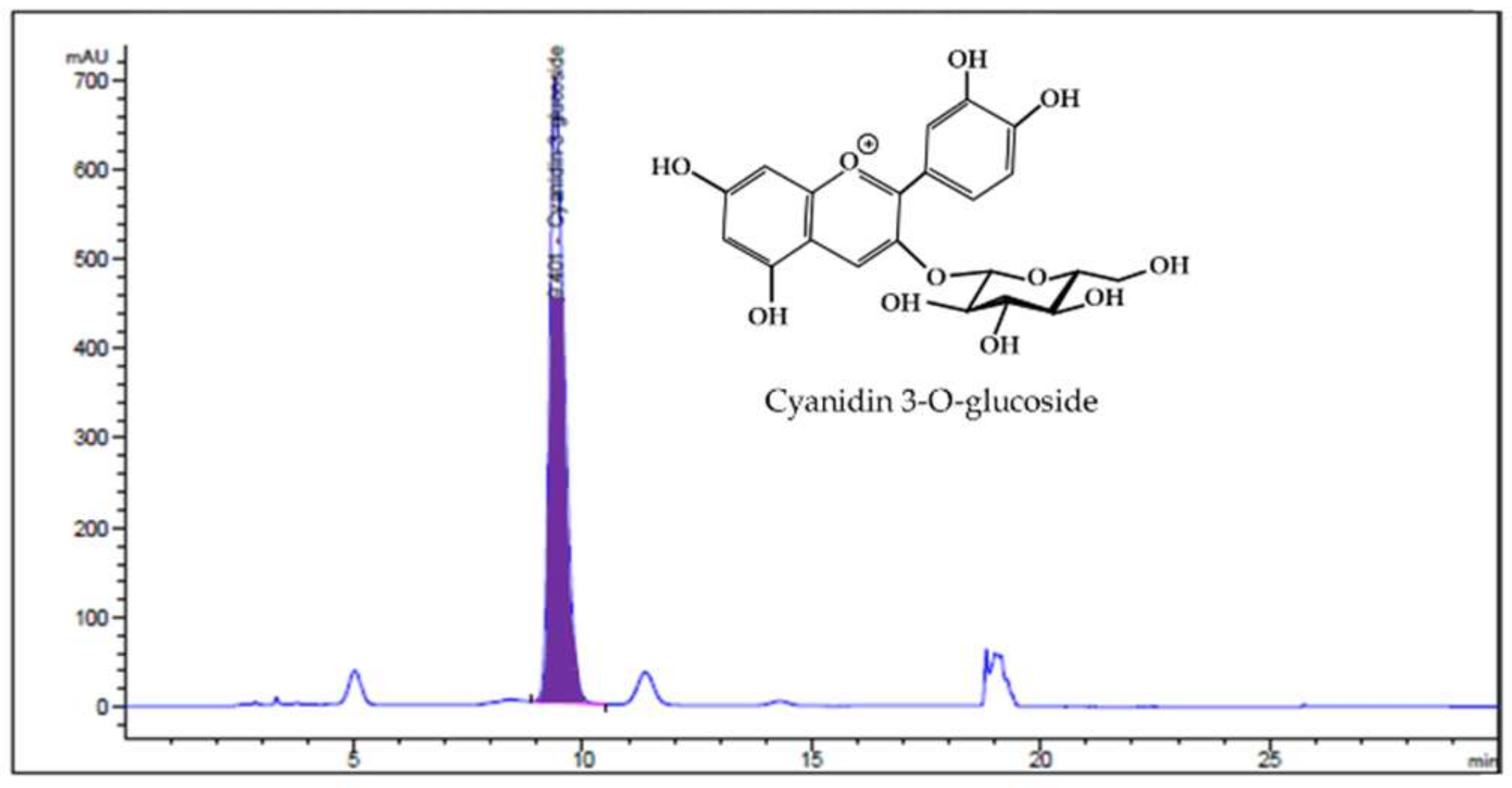
2.2. Analysis of HBE by HPLC
2.3. Cell Culture and Induction of Steatosis
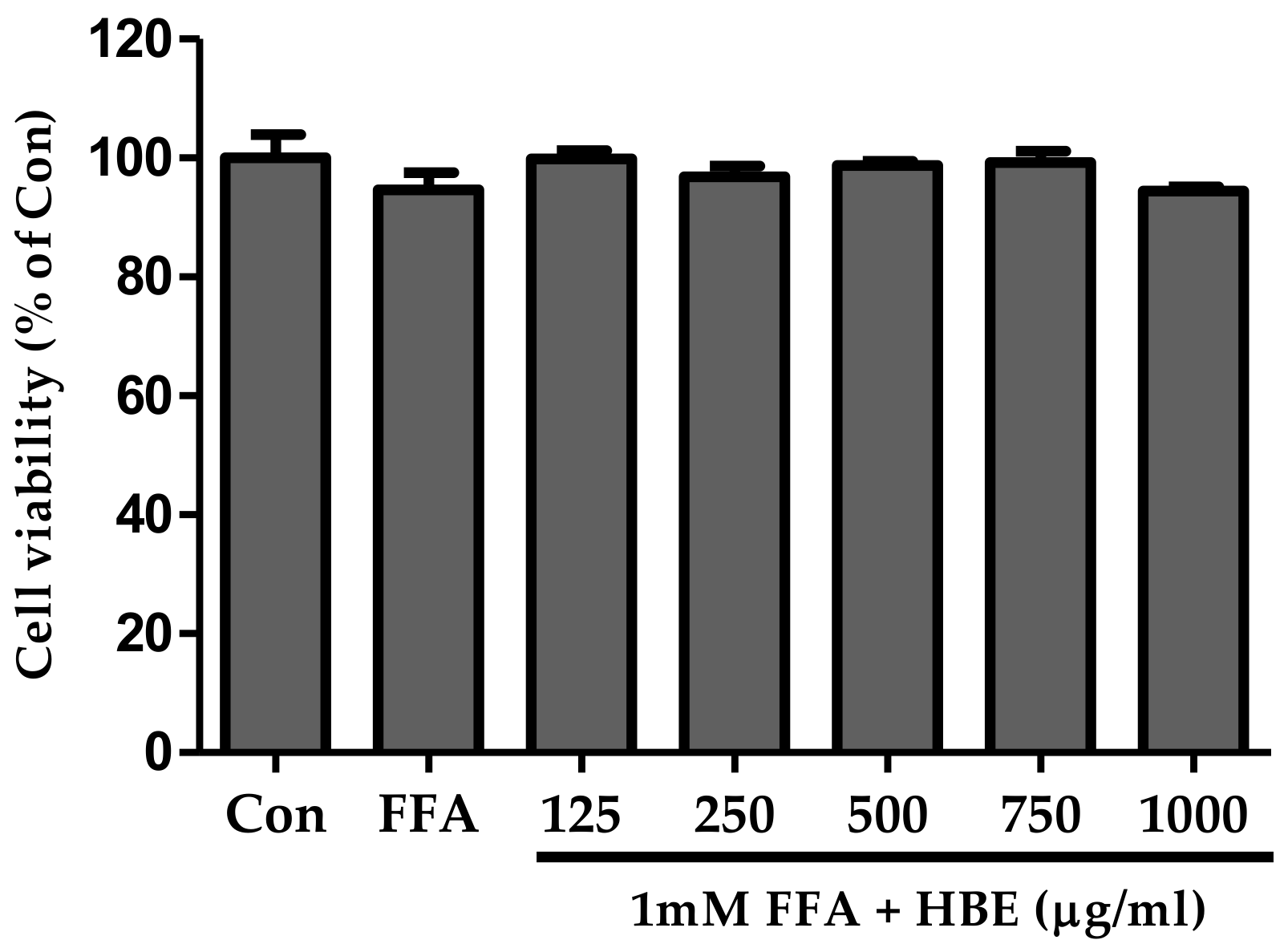
2.4. Effects of FFA on Cell Cytotoxicity
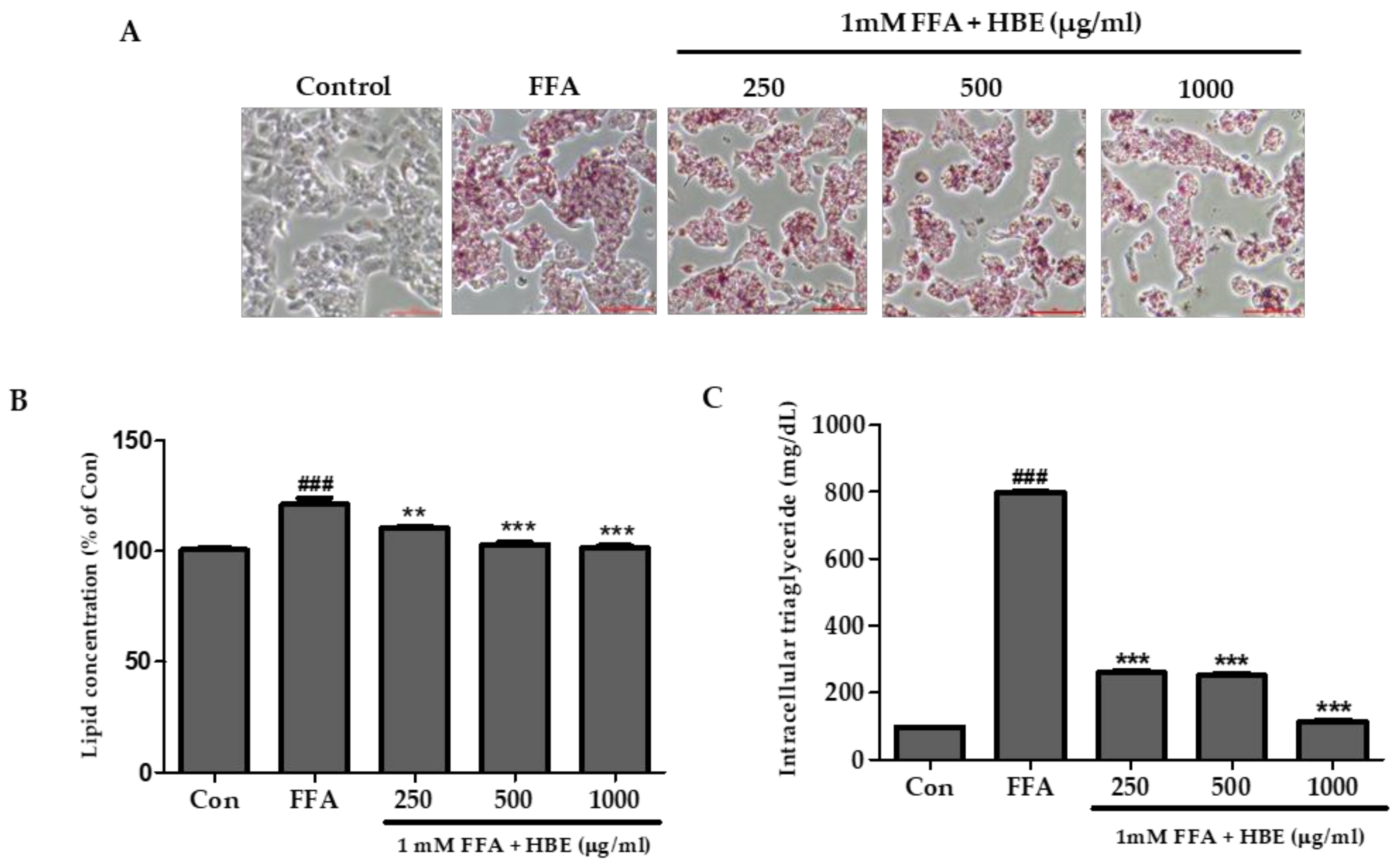
2.5. Oil Red O Staining
2.6. Triglyceride Colorimetric Assay (TG Assay)
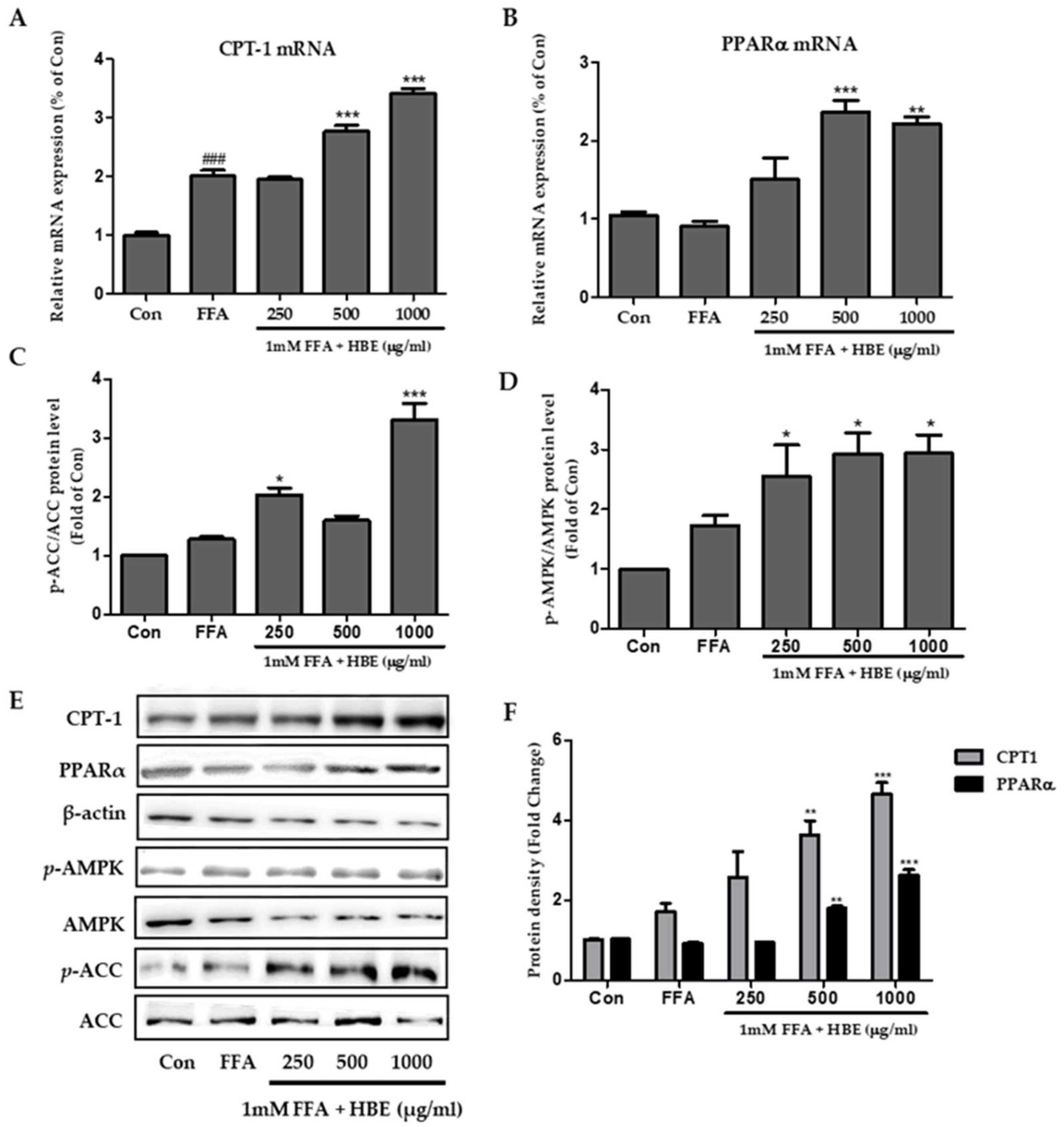
2.7. Quantification of Gene Expression Using Real-time PCR
2.8. Western Blot Analysis for HepG2 Cells
2.9. Animal Diets and Preparations of Tissue Sample
2.10. Liver Histology
2.11. Biochemical Assays
2.12. Western Blot Analysis of Liver Tissue
2.13. Data Analyses
3. Results
3.1. Cell Viability of HBE
3.2. Effects of HBE on FFA-Mediated Steatosis
3.3. Effects of HBE on Lipogenesis in FFA-Induced Steatosis
3.4. Effects of HBE on Phosphorylation of AMPK and ACC and Immunoblotting of CPT-1 and PPARα in HepG2 Cells
3.5. Effects of HBE on Blood and Liver in HFD-fed Mice
3.6. Effects of HBE on the Hepatic Steatosis in HFD-Fed Mice
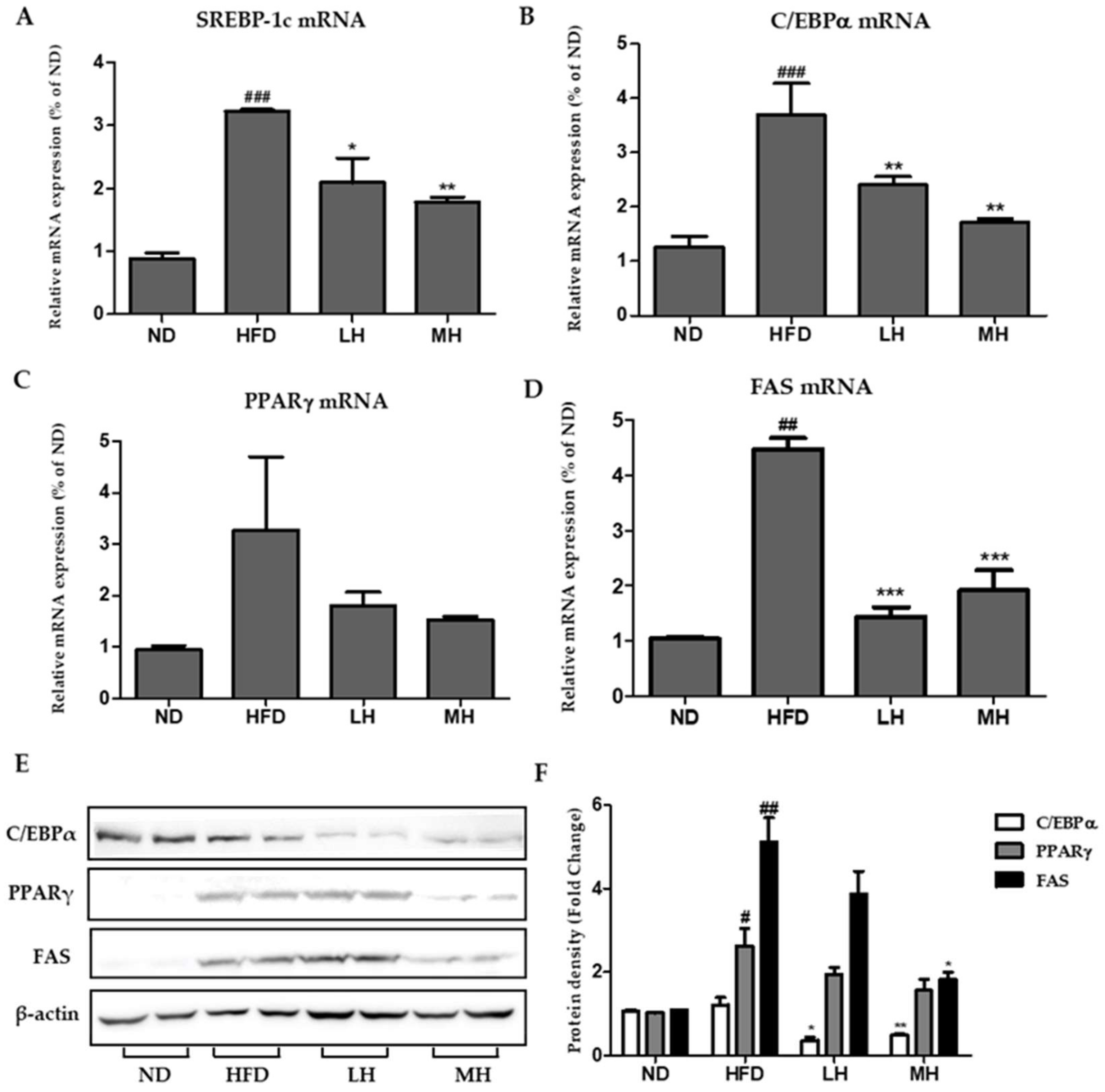
3.7. Effects of HBE on Hepatic Lipogenic Gene Expression of HFD-Fed Mice
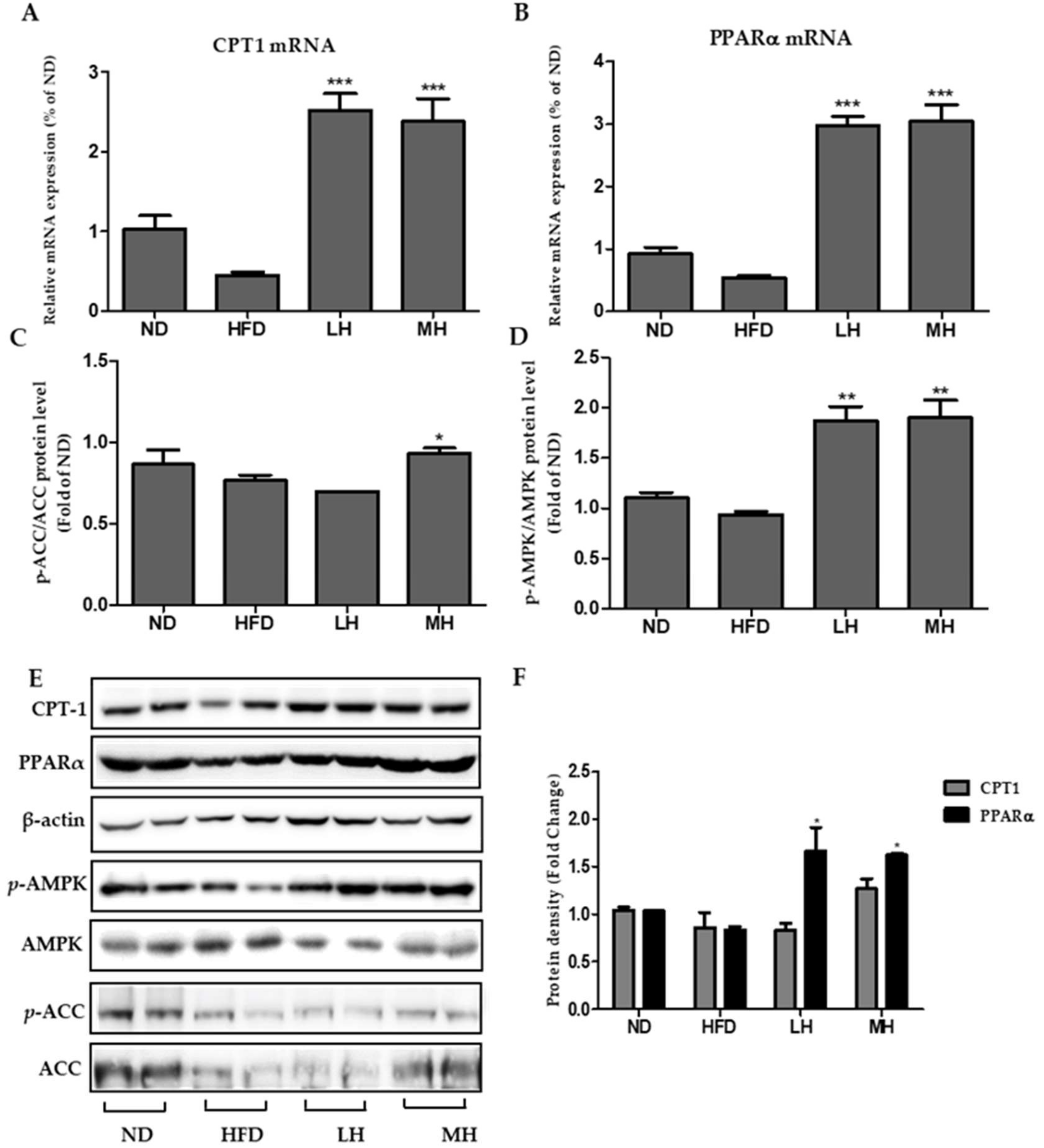
3.8. Effects of HBE on Phosphorylation of AMPK and ACC and Immunoblotting of CPT-1 and PPARα in HFD-Fed Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kang, Y.; Park, S.; Kim, S.; Koh, H. Normal serum alanine aminotransferase and non-alcoholic fatty liver disease among Korean adolescents: A cross-sectional study using data from KNHANES 2010–2015. BMC Pediatr. 2018, 18, 215. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, I.; Dominguez-Perez, M.; Bucio, L.; Souza, V.; Miranda, R.U.; Clemens, D.L.; Gomez-Quiroz, L.E.; Gutierrez-Ruiz, M.C. Free fatty acids enhance the oxidative damage induced by ethanol metabolism in an in vitro model. Food Chem. Toxicol. 2015, 76, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Kim, Y.; Lee, E.S.; Huh, J.H.; Chung, C.H. Caffeic acid ameliorates hepatic steatosis and decreased endoplasmic reticulum stress in high-fat diet-induced obese mice by regulating autophagy. Nutrition 2018, 55–56, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, H.; He, X.; Yuan, Y.; Wang, C.; Sun, J.; He, S.; Niu, J. Six-year changes in the prevalence of obesity and obesity-related diseases in Northeastern China from 2007 to 2013. Sci. Rep. 2017, 7, 41518. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.A.; Ellis, J.M. Peroxisomal acyl-CoA synthetases. Biochim. Biophys. Acta 2012, 1822, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.; Jonscher, K.R.; Friedman, J.E. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging 2011, 3, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Auzanneau, N.; Weber, P.; Kosinska-Cagnazzo, A.; Andlauer, W. Bioactive compounds and antioxidant capacity of Lonicera caerulea berries: Comparison of seven cultivars over three harvesting years. J. Food Compos. Anal. 2018, 66, 81–89. [Google Scholar] [CrossRef]
- Senica, M.; Stampar, F.; Mikulic-Petkovsek, M. Blue honeysuckle (Lonicera cearulea L. subs. edulis) berry; A rich source of Check for some nutrients and their differences among four different cultivars. Sci. Hortic.-Amsterdam. 2018, 238, 215–221. [Google Scholar] [CrossRef]
- Kowalska, K.; Olejnik, A.; Szwajgier, D.; Olkowicz, M. Inhibitory activity of chokeberry, bilberry, raspberry and cranberry polyphenol-rich extract towards adipogenesis and oxidative stress in differentiated 3T3-L1 adipose cells. PLoS ONE 2017, 12, e0188583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Gan, R.-Y.; Li, S.; Zhou, Y.; Li, A.-N.; Xu, D.-P.; Li, H.-B. Antioxidant Phytochemicals for the Prevention and Treatment of Chronic Diseases. Molecules 2015, 20, 19753. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, L.; Wang, D.; Huo, Y.; Ji, B. Anthocyanin-rich extracts from blackberry, wild blueberry, strawberry, and chokeberry:antioxidant activity and inhibitory effect onoleic acid-induce d hepatic steatosis in vitro. J. Sci. Food Agric. 2016. [Google Scholar] [CrossRef]
- Kowalska, K.; Olejnik, A. Current evidence on the health-beneficial effects of berry fruits in the prevention and treatment of metabolic syndrome. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xia, M.; Zou, T.; Ling, W.; Zhong, R.; Zhang, W. Cyanidin 3-glucoside attenuates obesity-associated insulin resistance and hepatic steatosis in high-fat diet-fed and db/db mice via the transcription factor FoxO1. J. Nutr. Biochem. 2012, 23, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Ham, J.; Kang, K.S.; Lee, H.J. Cyanidin 3-O-glucoside Isolated from Lonicera caerulea Fruit Improves Glucose Response in INS-1 Cells by Improving Insulin Secretion and Signaling. B Korean Chem. Soc. 2016, 37, 2015–2018. [Google Scholar] [CrossRef]
- Park, J.Y.; Kang, K.S.; Lee, H.J. Protection Effect of Cyanidin 3-O-Glucoside Against Oxidative Stress-induced HepG2 Cell Death Through Activation of Akt and Extracellular Signal-regulated Kinase Pathways. B Korean Chem. Soc. 2017, 38, 1316–1320. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Zhang, B.B.; Zhou, G.; Li, C. AMPK: An emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009, 9, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Parra-Vargas, M.; Sandoval-Rodriguez, A.; Rodriguez-Echevarria, R.; Dominguez-Rosales, J.; Santos-Garcia, A.; Armendariz-Borunda, J. Delphinidin Ameliorates Hepatic Triglyceride Accumulation in Human HepG2 Cells, but Not in Diet-Induced Obese Mice. Nutrients 2018, 10, 1060. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Kim, Y.M.; Yang, Y.M.; Kim, T.H.; Hwang, S.J.; Lee, J.R.; Kim, S.C.; Kim, S.G. Inhibition of SREBP-1c-mediated hepatic steatosis and oxidative stress by sauchinone, an AMPK-activating lignan in Saururus chinensis. Free Radic. Biol. Med. 2010, 48, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.J.; Hsu, M.J.; Huang, H.P.; Chung, D.J.; Chang, Y.C.; Wang, C.J. Mulberry anthocyanins inhibit oleic acid induced lipid accumulation by reduction of lipogenesis and promotion of hepatic lipid clearance. J. Agric. Food Chem. 2013, 61, 6069–6076. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Kim, J.Y.; Jun, H.J.; Kim, S.J.; Lee, J.H.; Hoang, M.H.; Kim, H.S.; Chang, H.I.; Hwang, K.Y.; Um, S.J.; et al. Cyanidin is an agonistic ligand for peroxisome proliferator-activated receptor-alpha reducing hepatic lipid. Biochim. Biophys. Acta 2013, 1831, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Ma, Y.; Wang, Y.; Du, Z.Y.; Shen, J.K.; Peng, H.L. Reduction of lipid accumulation in HepG2 cells by luteolin is associated with activation of AMPK and mitigation of oxidative stress. Phytother. Res. PTR 2011, 25, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-D.; Xu, S.-Y. Degradation kinetics of anthocyanins in blackberry juice and concentrate. J. Food Eng. 2007, 82, 271–275. [Google Scholar] [CrossRef]
- Wu, S.; He, X.; Wu, X.; Qin, S.; He, J.; Zhang, S.; Hou, D.-X. Inhibitory effects of blue honeysuckle (Lonicera caerulea L.) on adjuvant-induced arthritis in rats: Crosstalk of anti-inflammatory and antioxidant effects. J. Funct. Foods 2015, 17, 514–523. [Google Scholar] [CrossRef]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, H.; Ishiyama, M.; Ohseto, F.; Sasamoto, K.; Hamamoto, T.; Suzuki, K.; Watanabe, M. A water-soluble tetrazolium salt useful for colorimetric cell viability assay. Anal. Commun. 1999, 36, 47–50. [Google Scholar] [CrossRef]
- Takanezawa, Y.; Nakamura, R.; Sone, Y.; Uraguchi, S.; Kiyono, M. Atg5-dependent autophagy plays a protective role against methylmercury-induced cytotoxicity. Toxicol. Lett. 2016, 262, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Takikawa, M.; Inoue, S.; Horio, F.; Tsuda, T. Dietary anthocyanin-rich bilberry extract ameliorates hyperglycemia and insulin sensitivity via activation of AMP-activated protein kinase in diabetic mice. J. Nutr. 2010, 140, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Wedick, N.M.; Pan, A.; Cassidy, A.; Rimm, E.B.; Sampson, L.; Rosner, B.; Willett, W.; Hu, F.B.; Sun, Q.; van Dam, R.M. Dietary flavonoid intakes and risk of type 2 diabetes in US men and women. Am. J. Clin. Nutr. 2012, 95, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Mursu, J.; Virtanen, J.K.; Tuomainen, T.P.; Nurmi, T.; Voutilainen, S. Intake of fruit, berries, and vegetables and risk of type 2 diabetes in Finnish men: The Kuopio Ischaemic Heart Disease Risk Factor Study. The Am. J. Clin. Nutr. 2014, 99, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Rugina, D.; Diaconeasa, Z.; Coman, C.; Bunea, A.; Socaciu, C.; Pintea, A. Chokeberry Anthocyanin Extract as Pancreatic beta-Cell Protectors in Two Models of Induced Oxidative Stress. Oxidative Med. Cell. Longev. 2015, 2015, 429075. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Yan, Z.; Li, D.; Ma, Y.; Zhou, J.; Sui, Z. Antioxidant and Anti-Inflammatory Effects of Blueberry Anthocyanins on High Glucose-Induced Human Retinal Capillary Endothelial Cells. Oxidative Med. Cell. Longev. 2018, 2018, 1862462. [Google Scholar] [CrossRef] [PubMed]
- Smeriglio, A.; Barreca, D.; Bellocco, E.; Trombetta, D. Chemistry, Pharmacology and Health Benefits of Anthocyanins. Phytother. Res. PTR 2016, 30, 1265–1286. [Google Scholar] [CrossRef] [PubMed]
- Forbes-Hernández, T.; Giampieri, F.; Gasparrini, M.; Afrin, S.; Mazzoni, L.; Cordero, M.; Mezzetti, B.; Quiles, J.; Battino, M. Lipid Accumulation in HepG2 Cells Is Attenuated by Strawberry Extract through AMPK Activation. Nutrients 2017, 9, 621. [Google Scholar] [CrossRef] [PubMed]
- Li, H.B.; Ge, Y.K.; Zheng, X.X.; Zhang, L. Salidroside stimulated glucose uptake in skeletal muscle cells by activating AMP-activated protein kinase. Eur. J. Pharmacol. 2008, 588, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Savary, S.; Trompier, D.; Andreoletti, P.; Le Borgne, F.; Demarquoy, J.; Lizard, G. Fatty Acids–Induced Lipotoxicity and Inflammation. Curr. Drug Metab. 2012, 13, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′–3′) | Reverse (5′–3′) | |
|---|---|---|---|
| HepG2 | PPARγ | TGCAGGTGATCAAGAAGACG | AGTGCAACTGGAAGAAGGGA |
| C/EBPα | TGGACAAGAACAGCAACGAGTA | ATTGTCACTGGTCAGCTCCAG | |
| SREBP-1c | GCGCCTTGACAGGTGAAGTC | GCCAGGGAAGTCACTGTCTTG | |
| FAS | CCCCTGATGAAGAAGGATCA | ACTCCACAGGTGGGAACAAG | |
| CPT-1 | CCTCCGTAGCTGACTCGGTA | GGAGTGACCGTGAACTGAAA | |
| PPARα | ACGATTCGACTCAAGCTGGT | GTTGTGTGACATCCCGACAG | |
| β-actin | CTCTTCCAGCCTTCCTTCCT | AGCACTGTGTTGGCGTACAG | |
| Mouse Liver | PPARγ | CAGGAGAGCAGGGATTTGCA | CCTACGCTCAGCCCTCTTCAT |
| C/EBPα | TTACAACAGGCCAGGTTTCC | GGCTGGCGACATACAGTACA | |
| SREBP-1c | ATCGCAAACAAGCTGACCTG | AGATCCAGGTTTGAGGTGGG | |
| FAS | TTGCTGGCACTACAGAATGC | AACAGCCTCAGAGCGACAAT | |
| CPT-1 | CTCAGTGGGAGCGACTCTTCA | GGCCTCTGTGGTACACGACAA | |
| PPARα | CAGGAGAGCAGGGATTTGCA | CCTACGCTCAGCCCTCTTCAT | |
| β-actin | CTGTCCCTGTATGCCTCTG | ATGTCACGCACGATTTCC |
| Groups | ND | HFD | LH | MH |
|---|---|---|---|---|
| Food intake (g/day) | 3.8 ± 0.1 ns | 4.0 ± 0.1 | 3.9 ± 0.5 | 3.9 ± 0.3 |
| FER | 0.020 ± 0.00 b | 0.025 ± 0.00 a | 0.022 ± 0.00 ab | 0.022 ± 0.00 ab |
| Body mass (g) | ||||
| Initial (A) | 23.1 ± 0.6 ns | 22.8 ± 1.6 | 22.3 ± 1.6 | 22.5 ± 1.0 |
| Final (B) | 45.9 ± 3.7 c | 53.7 ± 3.0 a | 47.4 ± 1.4 bc | 48.2 ± 2.7 bc |
| Gains (B-A) | 22.7 ± 3.6 c | 30.9 ± 2.6 a | 25.1 ± 0.2 b | 25.7 ± 1.7 b |
| Relative organ weight (%) | ||||
| Liver | 3.6 ± 0.3 ns | 4.0 ± 1.6 | 3.8 ± 0.2 | 3.8 ± 0.4 |
| Abdominal adipose tissue | 0.9 ± 0.3 b | 1.7 ± 0.4 a | 1.3 ± 0.4 b | 1.2 ± 0.7 b |
| Epdidymal adipose tissue | 4.1 ± 0.3 b | 5.3 ± 0.7 a | 4.6 ± 0.5 b | 4.4 ± 0.4 b |
| Serum biochemical assays | ||||
| Adiponectin (ng/mL) | 4.7 ± 0.6 a | 3.1 ± 1.3 b | 4.1 ± 1.2 ab | 4.3 ± 1.2 ab |
| Leptin (pg/mL) | 0.4 ± 0.2 c | 1.5 ± 0.4 a | 0.9 ± 0.2 b | 1.0 ± 0.4 b |
| TG (mg/dL) | 148.6 ± 11.0 b | 184.2 ± 30.4 a | 171.4 ± 36.7 ab | 142.7 ± 20.9 b |
| TC (mg/dL) | 163.6 ± 23.2 ab | 191.3 ± 42.2 a | 158.4 ± 15.7 b | 179.9 ± 18.2 ab |
| HDL (mg/dL) | 83.5 ± 6.1 a | 71.2 ± 7.4 b | 82.1 ± 7.2 a | 84.2 ± 9.3 a |
| ALT (Karmen/mL) | 18.3 ± 2.1 b | 29.1 ± 7.5 a | 22.8 ± 8.6 ab | 19.9 ± 5.6 b |
| AST (Karmen/mL) | 54.0 ± 3.3 b | 65.1 ± 7.7 a | 55.4 ± 6.1 b | 56.8 ± 10.4 b |
| NO (µM) | 9.5 ± 2.3 b | 26.1 ± 9.4 a | 17.9 ± 6.3 b | 15.8 ± 5.0 b |
| Hepatic parameters | ||||
| MDA (nmol/g) | 35.1 ± 5.8 c | 66.5 ± 11.4 a | 51.1 ± 4.7 b | 38.4 ± 4.4c |
| NO (µM) | 38.9 ± 5.5 b | 53.1 ± 10.7 a | 42.0 ± 7.4 b | 41.9 ± 6.7 b |
| SOD (ug/mL) | 47.1 ± 1.9 b | 41.3 ± 2.2 b | 54.8 ± 11.5 a | 55.5 ± 4.1 a |
| GPx (ug/mL) | 1.4 ± 0.2 a | 1.0 ± 0.2 b | 1.3 ± 0.2 a | 1.3 ± 0.3 a |
| CAT (ng/mL) | 25.2 ± 3.1 a | 21.1 ± 1.0 b | 25.3 ± 5.7 a | 26.2 ± 2.7 a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, M.; Yoo, J.-H.; Lee, Y.-S.; Lee, H.-J. Lonicera caerulea Extract Attenuates Non-Alcoholic Fatty Liver Disease in Free Fatty Acid-Induced HepG2 Hepatocytes and in High Fat Diet-Fed Mice. Nutrients 2019, 11, 494. https://doi.org/10.3390/nu11030494
Park M, Yoo J-H, Lee Y-S, Lee H-J. Lonicera caerulea Extract Attenuates Non-Alcoholic Fatty Liver Disease in Free Fatty Acid-Induced HepG2 Hepatocytes and in High Fat Diet-Fed Mice. Nutrients. 2019; 11(3):494. https://doi.org/10.3390/nu11030494
Chicago/Turabian StylePark, Miey, Jeong-Hyun Yoo, You-Suk Lee, and Hae-Jeung Lee. 2019. "Lonicera caerulea Extract Attenuates Non-Alcoholic Fatty Liver Disease in Free Fatty Acid-Induced HepG2 Hepatocytes and in High Fat Diet-Fed Mice" Nutrients 11, no. 3: 494. https://doi.org/10.3390/nu11030494
APA StylePark, M., Yoo, J.-H., Lee, Y.-S., & Lee, H.-J. (2019). Lonicera caerulea Extract Attenuates Non-Alcoholic Fatty Liver Disease in Free Fatty Acid-Induced HepG2 Hepatocytes and in High Fat Diet-Fed Mice. Nutrients, 11(3), 494. https://doi.org/10.3390/nu11030494