Folic Acid and Vitamin B12 Administration in CKD, Why Not?


Abstract
1. Introduction
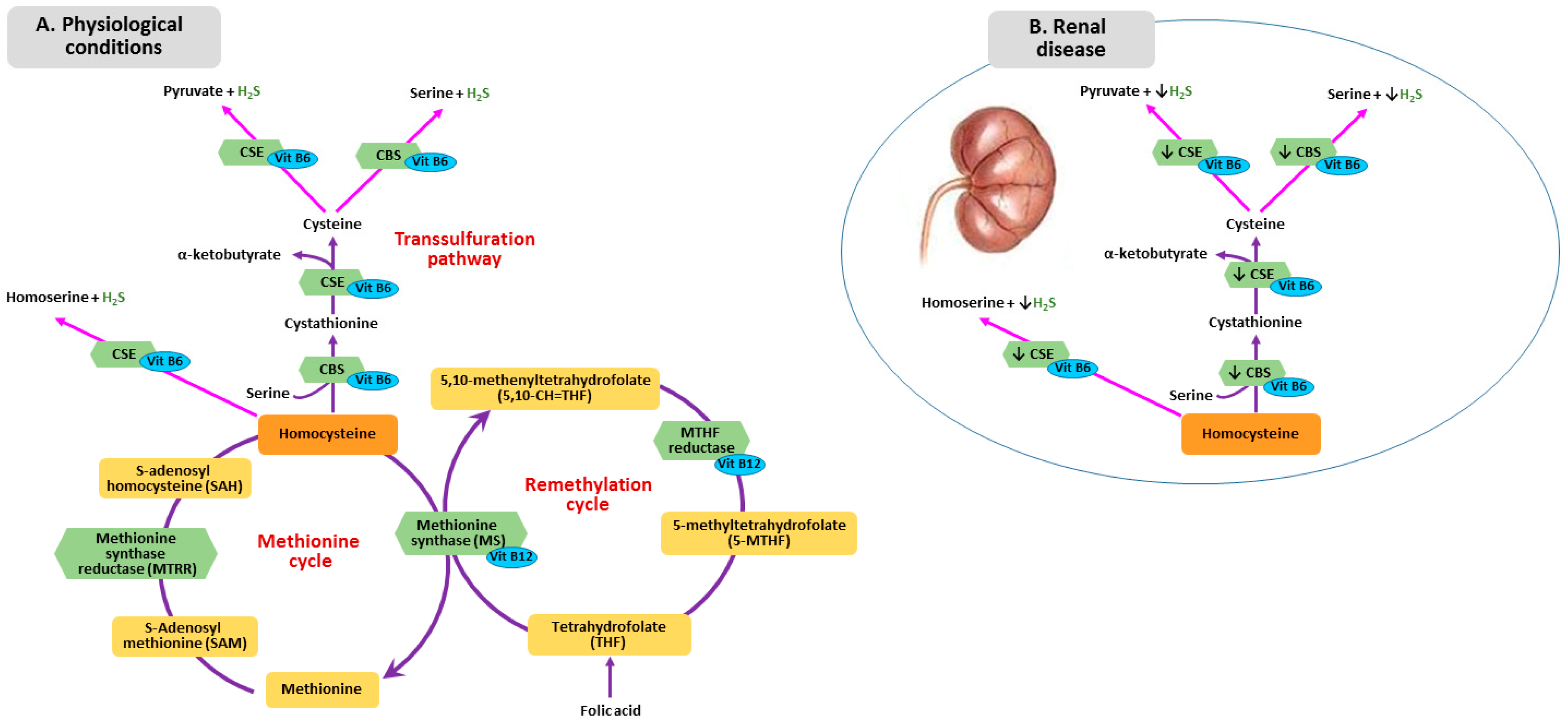
2. B Vitamins—Homocysteine Pathway
3. Metabolism of Homocysteine, Folic Acid and Vitamin B12 in CKD
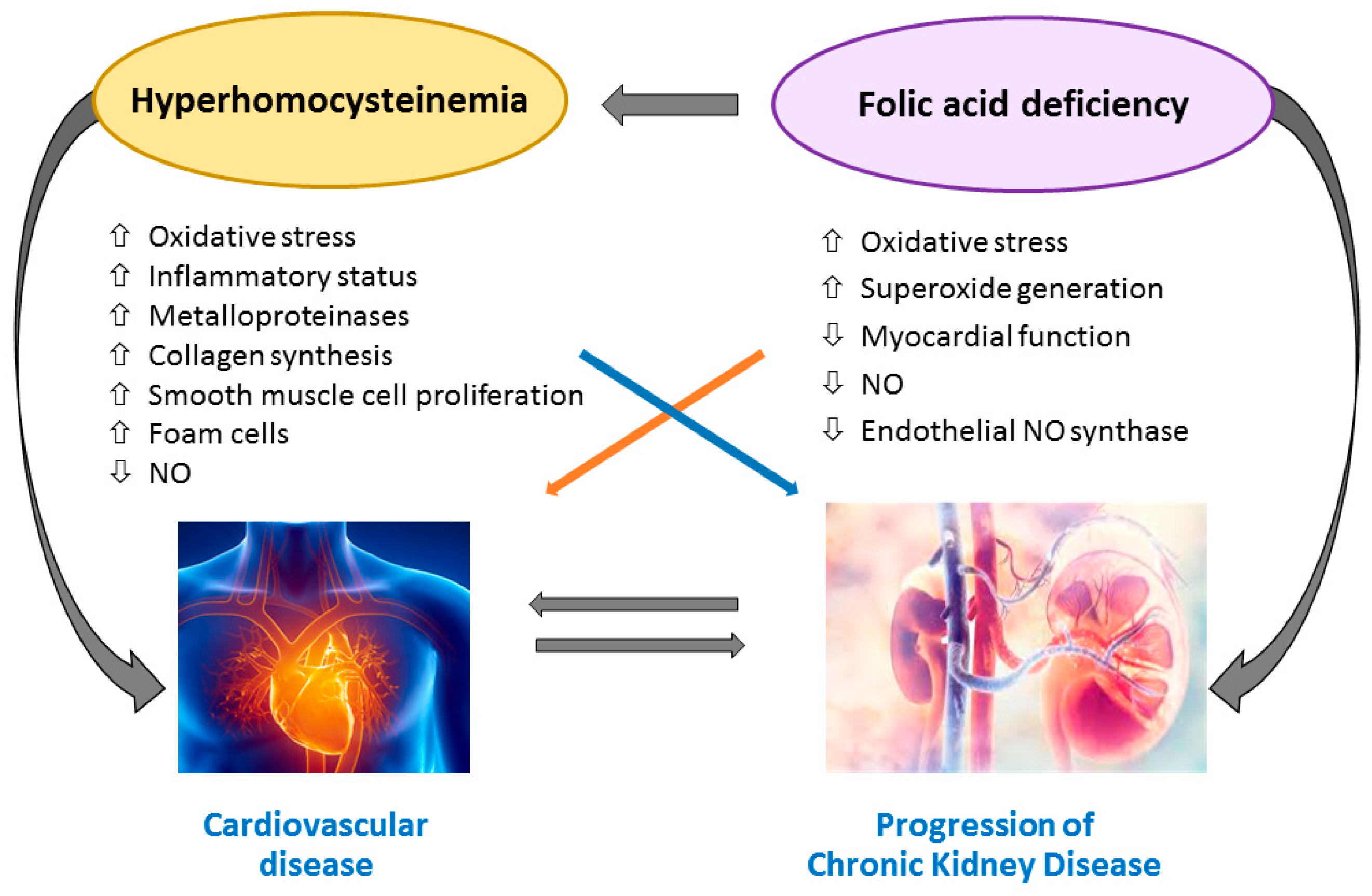
4. Homocysteine-Mediated Tissue Damage
5. Folic Acid and Vitamin B12 Impairment and Tissue Injury
6. MTHFR Gene Polymorphisms
7. Role of Folic Acid, Vitamin B12 and Homocysteine as Cardiovascular Risk Markers
8. Effect of Folic Acid and Vitamin B12 Supplementation on CVD and Mortality in CKD and ESRD
9. Folic Acid and Vitamin B12: Evidences on CKD Progression
10. Folic Acid and Vitamin B12 in Kidney Transplant Recipients
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Epidemiology of cardiovascular disease in chronic renal disease. J. Am. Soc. Nephrol. 1998, 9, S16–S23. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- McCullough, P.A.; Steigerwalt, S.; Tolia, K.; Chen, S.C.; Li, S.; Norris, K.C.; Whaley-Connell, A.; KEEP Investigators. Cardiovascular disease in chronic kidney disease: Data from the Kidney Early Evaluation Program (KEEP). Curr. Diab. Rep. 2011, 11, 47–55. [Google Scholar] [CrossRef]
- Ekdahl, K.; Soveri, I.; Hilborn, J.; Fellström, B.; Nilsson, B. Cardiovascular disease in hemodialysis: Role of the intravascular innate immune system. Nat. Rev. Nephrol. 2017, 13, 285–296. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Homocysteine and vascular disease. Nat. Med. 1996, 2, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Chrysant, S.G.; Chrysant, G.S. The current status of homocysteine as a risk factor for cardiovascular disease: A mini review. Expert. Rev. Cardiovasc. Ther. 2018, 16, 559–565. [Google Scholar] [CrossRef]
- Robinson, K. Renal disease, homocysteine, and cardiovascular complications. Circulation 2004, 109, 294–295. [Google Scholar] [CrossRef]
- Suliman, M.E.; Lindholm, B.; Barany, P.; Qureshi, A.R.; Stenvinkel, P. Homocysteine-lowering is not a primary target for cardiovascular disease prevention in chronic kidney disease patients. Semin. Dial. 2007, 20, 523–529. [Google Scholar] [CrossRef]
- Heinz, J.; Kropf, S.; Luley, C.; Dierkes, J. Homocysteine as a risk factor for cardiovascular disease in patients treated by dialysis: A meta-analysis. Am. J. Kidney Dis. 2009, 54, 478–489. [Google Scholar] [CrossRef]
- Cianciolo, G.; La Manna, G.; Colì, L.; Donati, G.; D’Addio, F.; Persici, E.; Comai, G.; Wratten, M.; Dormi, A.; Mantovani, V.; et al. 5-methyltetrahydrofolate administration is associated with prolonged survival and reduced inflammation in ESRD patients. Am. J. Nephrol. 2008, 28, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo, G.; De Pascalis, A.; Di Lullo, L.; Ronco, C.; Zannini, C.; La Manna, G. Folic Acid and Homocysteine in Chronic Kidney Disease and Cardiovascular Disease Progression: Which Comes First? Cardiorenal. Med. 2017, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Marti, F.; Vollenweider, P.; Marques-Vidal, P.M.; Mooser, V.; Waeber, G.; Paccaud, F.; Bochud, M. Hyperhomocysteinemia is independently associated with albuminuria in the population-based CoLaus study. BMC Public Health. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Ponte, B.; Pruijm, M.; Marques-Vidal, P.; Martin, P.Y.; Burnier, M.; Paccaud, F.; Waeber, G.; Vollenweider, P.; Bochud, M. Determinants and burden of chronic kidney disease in the population-based CoLaus study: A cross-sectional analysis. Nephrol. Dial. Transpl. 2013, 28, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Soohoo, M.; Ahmadi, S.F.; Qader, H.; Streja, E.; Obi, Y.; Moradi, H.; Rhee, C.M.; Kim, T.H.; Kovesdy, C.P.; Kalantar-Zadeh, K. Association of serum vitamin B12 and folate with mortality in incident hemodialysis patients. Nephrol. Dial. Transpl. 2017, 32, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Wong, P.W.K.; Malinow, M.R. Hyperhomocyst(e)inemia as a risk factor for occlusive vascular disease. Annu. Rev. Nutr. 1992, 12, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Randaccio, L.; Geremia, S.; Demitri, N.; Wuerges, J. Vitamin B12: Unique metalorganic compounds and the most complex vitamins. Molecules 2010, 15, 3228–3259. [Google Scholar] [CrossRef]
- Long, Y.; Nie, J. Homocysteine in Renal Injury. Kidney Dis. 2016, 2, 80–87. [Google Scholar] [CrossRef]
- Van Guldener, C.; Stam, F.; Stehouwer, C.D. Hyperhomocysteinaemia in chronic kidney disease: Focus on transmethylation. Clin. Chem. Lab. Med. 2005, 43, 1026–1031. [Google Scholar] [CrossRef]
- Jacobsen, D.W. Homocysteine and vitamins in cardiovascular disease. Clin. Chem. 1998, 44, 1833–1843. [Google Scholar]
- Cianciolo, G.; Cappuccilli, M.; La Manna, G. The Hydrogen Sulfide-Vitamin B12-Folic Acid Axis: An Intriguing Issue in Chronic Kidney Disease. A Comment on Toohey JI: “Possible Involvement of Hydrosulfide in B12-Dependent Methyl Group Transfer”. Molecules 2017, 22, 1216. [Google Scholar] [CrossRef] [PubMed]
- Toohey, J.I. Possible Involvement of Hydrosulfide in B12-Dependent Methyl Group Transfer. Molecules 2017, 22, 582. [Google Scholar] [CrossRef] [PubMed]
- Welch, G.N.; Loscalzo, J. Homocysteine and atherothrombosis. N. Engl. J. Med. 1998, 338, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Sepe, I.; Lanza, D.; Capasso, R.; Di Marino, V.; De Santo, N.G.; Ingrosso, D. The gasotransmitter hydrogen sulfide in hemodialysis patients. J. Nephrol. 2010, 23, S92–S96. [Google Scholar] [PubMed]
- Li, H.; Feng, S.J.; Zhang, G.Z.; Wang, S.X. Correlation of lower concentrations of hydrogen sulfide with atherosclerosis in chronic hemodialysis patients with diabetic nephropathy. Blood Purif. 2014, 38, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Van Guldener, C.; Stehouwer, C.D. Homocysteine metabolism in renal disease. Clin. Chem. Lab. Med. 2003, 41, 1412–1417. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; Satta, E.; Lombardi, C.; Acanfora, F.; De Santo, N.G. Homocysteine metabolism in renal failure. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 53–57. [Google Scholar] [CrossRef]
- Langan, R.C.; Goodbred, A.J. Vitamin B12 Deficiency: Recognition and Management. Am. Fam. Physician 2017, 96, 384–389. [Google Scholar]
- Brattström, L.; Wilcken, D.E. Homocysteine and cardiovascular disease: Cause or effect? Am. J. Clin. Nutr. 2000, 72, 315–323. [Google Scholar] [CrossRef]
- Van Guldener, C.; Kulik, W.; Berger, R.; Dijkstra, D.A.; Jakobs, C.; Reijngoud, D.J.; Donker, A.J.; Stehouwer, C.D.; De Meer, K. Homocysteine and methionine metabolism in ESRD: A stable isotope study. Kidney Int. 1999, 56, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Zha, Y.; Qian, Q. Protein Nutrition and Malnutrition in CKD and ESRD. Nutrients 2017, 27, 208. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Goldman, I.D. Inhibition of the membrane transport of folate by anions retained in uremia. J. Lab. Clin. Med. 1975, 86, 834–843. [Google Scholar] [PubMed]
- Bamonti-Catena, F.; Buccianti, G.; Porcella, A.; Valenti, G.; Como, G.; Finazzi, S.; Maiolo, A.T. Folate measurements in patients on regular hemodialysis treatment. Am. J. Kidney Dis. 1999, 33, 492–497. [Google Scholar] [CrossRef]
- McMahon, G.M.; Hwang, S.J.; Tanner, R.M.; Jacques, P.F.; Selhub, J.; Muntner, P.; Fox, C.S. The association between vitamin B12, albuminuria and reduced kidney function: An observational cohort study. BMC Nephrol. 2015, 16. [Google Scholar] [CrossRef]
- Ermens, A.A.; Vlasveld, L.T.; Lindemans, J. Significance of elevated cobalamin (vitamin B12) levels in blood. Clin. Biochem. 2003, 36, 585–590. [Google Scholar] [CrossRef]
- Andres, E.; Serraj, K.; Zhu, J.; Vermorken, A.J. The pathophysiology of elevated vitamin B12 in clinical practice. QJM 2013, 106, 505–515. [Google Scholar] [CrossRef]
- Koyama, K.; Yoshida, A.; Takeda, A.; Morozumi, K.; Fujinami, T.; Tanaka, N. Abnormal cyanide metabolism in uraemic patients. Nephrol. Dial. Transpl. 1997, 12, 1622–1628. [Google Scholar] [CrossRef]
- Poddar, R.; Sivasubramanian, N.; DiBello, P.M.; Robinson, K.; Jacobsen, D.W. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: Implications for vascular disease. Circulation 2001, 103, 2717–2723. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, H.; Liu, N.; Chen, J.; Gu, Y.; Chen, J.; Yang, K. Role of Hyperhomocysteinemia and Hyperuricemia in Pathogenesis of Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2017, 26, 2695–2699. [Google Scholar] [CrossRef] [PubMed]
- Au-Yeung, K.K.; Woo, C.W.; Sung, F.L.; Yip, J.C.; Siow, Y.L.; O, K. Hyperhomocysteinemia activates nuclear factor-kappaB in endothelial cells via oxidative stress. Circ. Res. 2004, 94, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lewis, A.; Brodsky, S.; Rieger, R.; Iden, C.; Goligorsky, M.S. Homocysteine induces 3-hydroxy-3-methylglutaryl coenzyme a reductase in vascular endothelial cells: A mechanism for development of atherosclerosis? Circulation 2002, 105, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Pecoits-Filho, R.; Lindholm, B.; Stenvinkel, P. The malnutrition, inflammation, and atherosclerosis (MIA) syndrome – the heart of the matter. Nephrol. Dial. Transpl. 2002, 17, 28–31. [Google Scholar] [CrossRef]
- Colì, L.; Donati, G.; Cappuccili, M.L.; Cianciolo, G.; Comai, G.; Cuna, V.; Carretta, E.; La Manna, G.; Stefoni, S. Role of the hemodialysis vascular access type in inflammation status and monocyte activation. Int. J. Art. Organs. 2011, 34, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.C.; Perrella, M.A.; Yoshizumi, M.; Hsieh, C.M.; Haber, E.; Schlegel, R.; Lee, M.E. Promotion of vascular smooth muscle cell growth by homocysteine: A link to atherosclerosis. Proc. Natl. Acad. Sci. USA 1994, 91, 6369–6373. [Google Scholar] [CrossRef]
- Bottiger, A.K.; Hurtig-Wennlof, A.; Sjostrom, M.; Yngve, A.; Nilsson, T.K. Association of total plasma homocysteine with methylenetetrahydrofolate reductase genotypes 677C>T, 1298A>C, and 1793G>A and the corresponding haplotypes in Swedish children and adolescents. Int. J. Mol. Med. 2007, 19, 659–665. [Google Scholar] [CrossRef]
- Sen, U.; Mishra, P.K.; Tyagi, N.; Tyagi, S.C. Homocysteine to hydrogen sulfide or hypertension. Cell Biochem. Biophys. 2010, 57, 49–58. [Google Scholar] [CrossRef]
- Zhang, C.; Cai, Y.; Adachi, M.T.; Oshiro, S.; Aso, T.; Kaufman, R.J.; Kitajima, S. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J. Biol. Chem. 2001, 276, 35867–53874. [Google Scholar] [CrossRef]
- Baydas, G.; Reiter, R.J.; Akbulut, M.; Tuzcu, M.; Tamer, S. Melatonin inhibits neural apoptosis induced by homocysteine in hippocampus of rats via inhibition of cytochrome c translocation and caspase-3 activation and by regulating pro- and antiapoptotic protein levels. Neuroscience 2005, 135, 879–886. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; Violetti, E.; Luciano, M.G.; Sepe, I.; Lanza, D.; Capasso, R.; Ascione, E.; Raiola, I.; Lombardi, C.; et al. Hyperhomocysteinemia in uremia—A red flag in a disrupted circuit. Semin. Dial. 2009, 22, 351–356. [Google Scholar] [CrossRef]
- Deussen, A.; Pexa, A.; Loncar, R.; Stehr, S.N. Effects of homocysteine on vascular and tissue adenosine: A stake in homocysteine pathogenicity? Clin. Chem. Lab. Med. 2005, 43, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Santini, E.; Ferrannini, E. Effect of short-term folic acid supplementation on insulin sensitivity and inflammatory markers in overweight subjects. Int. J. Obes. (Lond) 2006, 30, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Prasad, S.K.; Pal, S.; Maji, B.; Syamal, A.K.; Mukherjee, S. Synergistic protective effect of folic acid and vitamin B12 against nicotine-induced oxidative stress and apoptosis in pancreatic islets of the rat. Pharm. Biol. 2016, 54, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.G.; Li, W.H.; Lin, Z.Q.; Wang, L.X. Effects of folic acid on cardiac myocyte apoptosis in rats with streptozotocin-induced diabetes mellitus. Cardiovasc. Drugs Ther. 2008, 22, 299–304. [Google Scholar] [CrossRef]
- Verhaar, M.C.; Stroes, E.; Rabelink, T.J. Folates and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 6–13. [Google Scholar] [CrossRef]
- Antoniades, C.; Shirodaria, C.; Warrick, N.; Cai, S.; de Bono, J.; Lee, J.; Leeson, P.; Neubauer, S.; Ratnatunga, C.; Pillai, R.; et al. 5-Methyltetrahydrofolate Rapidly Improves Endothelial Function and Decreases Superoxide Production in Human Vessels Effects on Vascular Tetrahydrobiopterin Availability and Endothelial Nitric Oxide Synthase Coupling. Circulation 2006, 114, 1193–1201. [Google Scholar] [CrossRef]
- Doshi, S.N.; McDowell, I.F.; Moat, S.J.; Payne, N.; Durrant, H.J.; Lewis, M.J.; Goodfellow, J. Folic Acid Improves Endothelial Function in Coronary Artery Disease via Mechanisms Largely Independent of Homocysteine Lowering. Circulation 2002, 105, 22–26. [Google Scholar] [CrossRef]
- Title, L.M.; Cummings, P.M.; Giddens, K.; Genest, J.J., Jr.; Nassar, B.A. Effect of folic acid and antioxidant vitamins on endothelial dysfunction in patients with coronary artery disease. J. Am. Coll. Cardiol. 2000, 36, 758–765. [Google Scholar] [CrossRef]
- Chambers, J.C.; Ueland, P.M.; Obeid, O.A.; Wrigley, J.; Refsum, H.; Kooner, J.S. Improved vascular endothelial function after oral B vitamins: An effect mediated through reduced concentrations of free plasma homocysteine. Circulation 2000, 102, 2479–2483. [Google Scholar] [CrossRef] [PubMed]
- Doshi, S.N.; McDowell, I.F.; Moat, S.J.; Lang, D.; Newcombe, R.G.; Kredan, M.B.; Lewis, M.J.; Goodfellow, J. Folate improves endothelial function in coronary artery disease: An effect mediated by reduction of intracellular superoxide? Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Liu, H.; Gao, F.; Luo, H.; Lin, H.; Meng, L.; Jiang, C.; Guo, Y.; Chi, J.; Guo, H. Folic acid delays development of atherosclerosis in lowdensity lipoprotein receptor-deficient mice. J. Cell Mol. Med. 2018, 22, 3183–3191. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Ahmadi, S.F.; Streja, E.; Molnar, M.Z.; Flegal, K.M.; Gillen, D.; Kovesdy, C.P.; Kalantar-Zadeh, K. Obesity paradox in end-stage kidney disease patients. Prog. Cardiovasc. Dis. 2014, 56, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Obi, Y.; Qader, H.; Kovesdy, C.P.; Kalantar-Zadeh, K. Latest consensus and update on proteinenergy wasting in chronic kidney disease. Curr. Opin. Clin. Nutr. Metab. Care. 2015, 18, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Salles, N.; Herrmann, F.; Sakbani, K.; Rapin, C.H.; Sieber, C. High vitamin B12 level: A strong predictor of mortality in elderly inpatients. J. Am. Geriatr. Soc. 2005, 53, 917–918. [Google Scholar] [CrossRef]
- Seetharam, B.; Li, N. Transcobalamin II and its cell surface receptor. Vitam. Horm. 2000, 59, 337–366. [Google Scholar]
- Sviri, S.; Khalaila, R.; Daher, S.; Bayya, A.; Linton, D.M.; Stav, I.; van Heerden, P.V. Increased Vitamin B12 levels are associated with mortality in critically ill medical patients. Clin. Nutr. 2012, 31, 53–59. [Google Scholar] [CrossRef]
- Bamgbola, O.F. Pattern of resistance to erythropoietin-stimulating agents in chronic kidney disease. Kidney Int. 2011, 80, 464–474. [Google Scholar] [CrossRef]
- Saifan, C.; Samarneh, M.; Shtaynberg, N.; Nasr, R.; El-Charabaty, E.; El-Sayegh, S. Treatment of confirmed B12 deficiency in hemodialysis patients improves Epogen requirements. Int. J. Nephrol. Renovasc. Dis. 2013, 6, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Brustolin, S.; Giugliani, R.; Félix, T.M. Genetics of homocysteinemetabolism and associated disorders. Braz. J. Med. Biol. Res. 2010, 43, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Homberger, A.; Linnebank, M.; Winter, C.; Willenbring, H.; Marquardt, T.; Harms, E.; Koch, H.G. Genomic structure and transcript variants of the human methylenetetrahydrofolate reductase gene. Eur. J. Hum. Genet. 2000, 8, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Cristalli, C.P.; Zannini, C.; Comai, G.; Baraldi, O.; Cuna, V.; Cappuccilli, M.; Mantovani, V.; Natali, N.; Cianciolo, G.; La Manna, G. Methylenetetrahydrofolate reductase, MTHFR, polymorphisms and predisposition to different multifactorial disorders. Genes Genomics 2017, 39, 689–699. [Google Scholar] [CrossRef]
- Rady, P.L.; Szucs, S.; Grady, J.; Hudnall, S.D.; Kellner, L.H.; Nitowsky, H.; Tyring, S.K.; Matalon, R.K. Genetic polymorphisms of methylenetetrahydrofolate reductase (MTHFR) and methionine synthase reductase (MTRR) in ethnic populations in Texas; a report of a novel MTHFR polymorphic site, G1793A. Am. J. Med. Genet. 2002, 107, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Wrone, E.M.; Hornberger, J.M.; Zehnder, J.L.; McCann, L.M.; Coplon, N.S.; Fortmann, S.P. Randomized trial of folic acid for prevention of cardiovascular events in end-stage renal disease. J. Am. Soc. Nephrol. 2004, 15, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Trovato, F.M.; Catalano, D.; Ragusa, A.; Martines, G.F.; Pirri, C.; Buccheri, M.A.; Di Nora, C.; Trovato, G.M. Relationship of MTHFR gene polymorphisms with renal and cardiac disease Send to World. J. Nephrol. 2015, 4, 127–137. [Google Scholar]
- Bostom, A.G.; Shemin, D.; Lapane, K.L.; Hume, A.L.; Yoburn, D.; Nadeau, M.R.; Bendich, A.; Selhub, J.; Rosenberg, I.H. High-dose-B vitamin treatment of hyperhomocysteinemia in dialysis patients. Kidney Int. 1996, 49, 147–152. [Google Scholar] [CrossRef]
- Achour, O.; Elmtaoua, S.; Zellama, D.; Omezzine, A.; Moussa, A.; Rejeb, J.; Boumaiza, I.; Bouacida, L.; Rejeb, N.B.; Achour, A.; et al. The C677T MTHFR genotypes influence the efficacy of B9 and B12 vitamins supplementation to lowering plasma total homocysteine in hemodialysis. J. Nephrol. 2016, 29, 691–698. [Google Scholar] [CrossRef]
- Huo, Y.; Li, J.; Qin, X.; Huang, Y.; Wang, X.; Gottesman, R.F.; Tang, G.; Wang, B.; Chen, D.; He, M.; et al. Efficacy of Folic Acid Therapy in Primary Prevention of Stroke among Adults with Hypertension in China: The CSPPT randomized clinical trial. JAMA 2015, 313, 1325–1335. [Google Scholar] [CrossRef]
- Bostom, A.G.; Shemin, D.; Bagley, P.; Massy, Z.A.; Zanabli, A.; Christopher, K.; Spiegel, P.; Jacques, P.F.; Dworkin, L.; Selhub, J. Controlled comparison of L-5- methyltetrahydrofolate versus folic acid for the treatment of hyperhomocysteinemia in hemodialysis patients. Circulation 2000, 101, 2829–2832. [Google Scholar] [CrossRef] [PubMed]
- Dierkes, J.; Domröse, U.; Ambrosch, A.; Schneede, J.; Guttormsen, A.B.; Neumann, K.H.; Luley, C. Supplementation with vitamin B12 decreases homocystein and methylmalonic acid but also serum folate in patients with end stage renal disease. Metabolism 1999, 48, 631–635. [Google Scholar] [CrossRef]
- Pastore, A.; De Angelis, S.; Casciani, S.; Ruggia, R.; Di Giovamberardino, G.; Noce, A.; Splendiani, G.; Cortese, C.; Federici, G.; Dessì, M. Effects of folic acid before and after vitamin B12 on plasma homocysteine concentrations in hemodialysis patients with known MTHFR genotypes. Clin. Chem. 2006, 52, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, R.; Bonnardeaux, A.; Geadah, D.; Busque, L.; Lebrun, M.; Ouimet, D.; Leblanc, M. Hyperhomocysteinemia in hemodialysis patients: Effects of 12-month supplementation with hydrosoluble vitamins. Kidney Int. 2000, 58, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Kuhlmann, M.K.; Köhler, H.; Herrmann, W. Response of homocysteine, cystathionine, and methylmalonic acid to vitamin treatment in dialysis patients. Clin. Chem. 2005, 51, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Malinow, M.R.; Nieto, F.J.; Kruger, W.D.; Duell, P.B.; Hess, D.L.; Gluckman, R.A.; Block, P.C.; Holzgang, C.R.; Anderson, P.H.; Seltzer, D.; et al. The effects of folic acid supplementation on plasma total homocysteine are modulated by multivitamin use and methylenetetrahydrofolate reductase genotypes. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qin, X.; Li, Y.; Sun, D.; Wang, J.; Liang, M.; Wang, B.; Huo, Y.; Hou, F.F.; Investigators of the Renal Substudy of the China Stroke Primary Prevention Trial (CSPPT). Efficacy of folic acid therapy on the progression of chronic kidney disease: The Renal Substudy of the China Stroke Primary Prevention Trial. JAMA Intern. Med. 2016, 176, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Hickey, S.E.; Curry, C.J.; Toriello, H.V. ACMG Practice Guideline: Lack of evidence for MTHFR polymorphism testing. Genet Med. 2013, 15, 153–156. [Google Scholar] [CrossRef]
- Wald, D.S.; Law, M.; Morris, J.K. Homocysteine and cardiovascular disease: Evidence on causality from a metaanalysis. BMJ 2002, 325, 1202. [Google Scholar] [CrossRef]
- Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: A meta-analysis. JAMA 2002, 288, 2015–2022. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Block, G.; Humphreys, M.H.; Kopple, J.D. Reverse epidemiology of cardiovascular risk factors in maintenance dialysis patients. Kidney Int. 2003, 63, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Ducloux, D.; Klein, A.; Kazory, A.; Devillard, N.; Chalopin, J.M. Impact of malnutrition-inflammation on the association between homocysteine and mortality. Kidney Int. 2006, 69, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Suliman, M.; Stenvinkel, P.; Qureshi, A.R.; Kalantar-Zadeh, K.; Bárány, P.; Heimbürger, O.; Vonesh, E.F.; Lindholm, B. The reverse epidemiology of plasma total homocysteine as a mortality risk factor is related to the impact of wasting and inflammation. Nephrol. Dial. Transpl. 2007, 22, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhang, Q.; Li, Y.; Wang, C.; Zhang, J.; Ma, X.; Peng, H.; Lou, T. High Prevalence of Hyperhomocysteinemia and Its Association with Target Organ Damage in Chinese Patients with Chronic Kidney Disease. Nutrients 2016, 8, 645. [Google Scholar] [CrossRef] [PubMed]
- Anan, F.; Takahashi, N.; Shimomura, T.; Imagawa, M.; Yufu, K.; Nawata, T.; Nakagawa, M.; Yonemochi, H.; Eshima, N.; Saikawa, T.; et al. Hyperhomocysteinemia is a significant risk factor for silent cerebral infarction in patients with chronic renal failure undergoing hemodialysis. Metabolism 2006, 55, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.P.; Nemirovsky, D.; Kim, M.; Geer, E.B.; Farkouh, M.E.; Winston, J.; Halperin, J.L.; Robbins, M.J. Elevated homocysteine levels in patients with end-stage renal disease. Mt. Sinai. J. Med. 2005, 72, 365–373. [Google Scholar] [PubMed]
- London, G.M.; Pannier, B.; Agharazii, M.; Guerin, A.P.; Verbeke, F.H.; Marchais, S.J. Forearm reactive hyperemia and mortality in end-stage renal disease. Kidney Int. 2004, 65, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Block, G.; Humphreys, M.H.; McAllister, C.J.; Kopple, J.D. A low rather than a high, total plasma homocysteine is an indicator of poor outcome in hemodialysis patients. J. Am. Soc. Nephrol. 2004, 15, 442–453. [Google Scholar] [CrossRef]
- Buccianti, G.; Baragetti, I.; Bamonti, F.; Furiani, S.; Dorighet, V.; Patrosso, C. Plasma homocysteine levels and cardiovascular mortality in patients with end-stage renal disease. J. Nephrol. 2004, 17, 405–410. [Google Scholar]
- Bayés, B.; Pastor, M.C.; Bonal, J.; Juncà, J.; Hernandez, J.M.; Riutort, N.; Foraster, A.; Romero, R. Homocysteine, C-reactive protein, lipid peroxidation and mortality in haemodialysis patients. Nephrol. Dial. Transpl. 2003, 18, 106–112. [Google Scholar] [CrossRef]
- Mallamaci, F.; Zoccali, C.; Tripepi, G.; Fermo, I.; Benedetto, F.A.; Cataliotti, A.; Bellanuova, I.; Malatino, L.S.; Soldarini, A.; CREED Investigators. Hyperhomocysteinemia predicts cardiovascular outcomes in hemodialysis patients. Kidney Int. 2002, 61, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Ducloux, D.; Bresson-Vautrin, C.; Kribs, M.; Abdelfatah, A.; Chalopin, J.M. C-reactive protein and cardiovascular disease in peritoneal dialysis patients. Kidney Int. 2002, 62, 1417–1422. [Google Scholar] [CrossRef] [PubMed]
- Haraki, T.; Takegoshi, T.; Kitoh, C.; Kajinami, K.; Wakasugi, T.; Hirai, J.; Shimada, T.; Kawashiri, M.; Inazu, A.; Koizumi, J.; et al. Hyperhomocysteinemia, diabetes mellitus, and carotid atherosclerosis independently increase atherosclerotic vascular disease outcome in Japanese patients with end-stage renal disease. Clin. Nephrol. 2001, 56, 132–139. [Google Scholar] [PubMed]
- Wrone, E.M.; Zehnder, J.L.; Hornberger, J.M.; McCann, L.M.; Coplon, N.S.; Fortmann, S.P. An MTHFR variant, homocysteine, and cardiovascular comorbidity in renal disease. Kidney Int. 2001, 60, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Dierkes, J.; Domröse, U.; Westphal, S.; Ambrosch, A.; Bosselmann, H.P.; Neumann, K.H.; Luley, C. Cardiac troponin T predicts mortality in patients with end-stage renal disease. Circulation 2000, 102, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Suliman, M.E.; Qureshi, A.R.; Bárány, P.; Stenvinkel, P.; Filho, J.C.; Anderstam, B.; Heimbürger, O.; Lindholm, B.; Bergström, J. Hyperhomocysteinemia, nutritional status, and cardiovascular disease in hemodialysis patients. Kidney Int. 2000, 57, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Kunz, K.; Petitjean, P.; Lisri, M.; Chantrel, F.; Koehl, C.; Wiesel, M.L.; Cazenave, J.P.; Moulin, B.; Hannedouche, T.P. Cardiovascular morbidity and endothelial dysfunction in chronic haemodialysis patients: Is homocyst(e)ine the missing link? Nephrol. Dial. Transpl. 1999, 14, 1934–1942. [Google Scholar] [CrossRef]
- Manns, B.J.; Burgess, E.D.; Hyndman, M.E.; Parsons, H.G.; Schaefer, J.P.; Scott-Douglas, N.W. Hyperhomocyst(e)inemia and the prevalence of atherosclerotic vascular disease in patients with end-stage renal disease. Am. J. Kidney Dis. 1999, 34, 669–677. [Google Scholar] [CrossRef]
- Sirrs, S.; Duncan, L.; Djurdjev, O.; Nussbaumer, G.; Ganz, G.; Frohlich, J.; Levin, A. Homocyst(e)ine and vascular access complications in haemodialysis patients: Insights into a complex metabolic relationship. Nephrol. Dial. Transpl. 1999, 14, 738–743. [Google Scholar] [CrossRef]
- Moustapha, A.; Naso, A.; Nahlawi, M.; Gupta, A.; Arheart, K.L.; Jacobsen, D.W.; Robinson, K.; Dennis, V.W. Prospective study of hyperhomocysteinemia as an adverse cardiovascular risk factor in end-stage renal disease. Circulation 1998, 97, 138–141. [Google Scholar] [CrossRef]
- Vychytil, A.; Födinger, M.; Wölfl, G.; Enzenberger, B.; Auinger, M.; Prischl, F.; Buxbaum, M.; Wiesholzer, M.; Mannhalter, C.; Hörl, W.H.; et al. Major determinants of hyperhomocysteinemia in peritoneal dialysis patients. Kidney Int. 1998, 53, 1775–1782. [Google Scholar] [CrossRef] [PubMed]
- Bostom, A.G.; Shemin, D.; Verhoef, P.; Nadeau, M.R.; Jacques, P.F.; Selhub, J.; Dworkin, L.; Rosenberg, I.H. Elevated fasting total plasma homocysteine levels and cardiovascular disease outcomes in maintenance dialysis patients. A. prospective study. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2554–2558. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Gupta, A.; Dennis, V.; Arheart, K.; Chaudhary, D.; Green, R.; Vigo, P.; Mayer, E.L.; Selhub, J.; Kutner, M.; et al. Hyperhomocysteinemia confers an independent increased risk of atherosclerosis in end-stage renal disease and is closely linked to plasma folate and pyridoxine concentrations. Circulation 1996, 94, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, J.; Tepel, M.; Raidt, H.; Riezler, R.; Graefe, U.; Langer, K.; Zidek, W. Hyperhomocysteinemia and the risk for vascular disease in hemodialysis patients. J. Am. Soc. Nephrol. 1995, 6, 121–125. [Google Scholar] [PubMed]
- Bostom, A.G.; Shemin, D.; Lapane, K.L.; Miller, J.W.; Sutherland, P.; Nadeau, M.; Seyoum, E.; Hartman, W.; Prior, R.; Wilson, P.W.; et al. Hyperhomocysteinemia and traditional cardiovascular disease risk factors in end-stage renal disease patients on dialysis: A case-control study. Atherosclerosis 1995, 114, 93–103. [Google Scholar] [CrossRef]
- Wu, C.C.; Zheng, C.M.; Lin, Y.F.; Lo, L.; Liao, M.T.; Lu, K.C. Role of homocysteine in end-stage renal disease. Clin. Biochem. 2012, 45, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Righetti, M.; Ferrario, G.M.; Milani, S.; Serbelloni, P.; La Rosa, L.; Uccellini, M.; Sessa, A. Effects of folic acid treatment on homocysteine levels and vascular disease in hemodialysis patients. Med. Sci. Monit. 2003, 9, 19–24. [Google Scholar]
- Zoungas, S.; McGrath, B.P.; Branley, P.; Kerr, P.G.; Muske, C.; Wolfe, R.; Atkins, R.C.; Nicholls, K.; Fraenkel, M.; Hutchison, B.G.; et al. Cardiovascular Morbidity and Mortality in the Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) in Chronic Renal Failure. J. Am. Coll. Cardiol. 2006, 47, 1108–1116. [Google Scholar] [CrossRef]
- Jamison, R.L.; Hartigan, P.; Kaufman, J.S.; Goldfarb, D.S.; Warren, S.R.; Guarino, P.D.; Gaziano, J.M.; Veterans Affairs Site Investigators. Effect of Homocysteine Lowering on Mortality and Vascular Disease in Advanced Chronic Kidney Disease and End-stage Renal Disease. JAMA 2007, 298, 1163–1170. [Google Scholar] [CrossRef]
- Heinz, J.; Kropf, S.; Domröse, U.; Westphal, S.; Borucki, K.; Luley, C.; Neumann, K.H.; Dierkes, J. B Vitamins and the Risk of Total Mortality and Cardiovascular Disease in End-Stage Renal Disease Results of a Randomized Controlled Trial. Circulation 2010, 121, 1432–1438. [Google Scholar] [CrossRef]
- Qin, X.; Huo, Y.; Langman, C.B.; Hou, F.; Chen, Y.; Matossian, D.; Xu, X.; Wang, X. Folic acid therapy and cardiovascular disease in ESRD or advanced chronic kidney disease: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2011, 6, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Tan, S.; Xu, Y.; Chandra, A.; Shi, C.; Song, B.; Qin, J.; Gao, Y. Vitamin B supplementation, homocysteine levels, and the risk of cerebrovascular disease: A meta-analysis. Neurology 2013, 81, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Guo, L.L.; Cai, L.L.; Zhu, X.J.; Shu, J.L.; Liu, X.L.; Jin, H.M. Homocysteine-lowering therapy does not lead to reduction in cardiovascular outcomes in chronic kidney disease patients: A meta-analysis of randomised, controlled trials. Br. J. Nutr. 2012, 108, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Nigwekar, S.U.; Kang, A.; Zoungas, S.; Cass, A.; Gallagher, M.P.; Kulshrestha, S.; Navaneethan, S.D.; Perkovic, V.; Strippoli, G.F.M.; Jardine, M.J. Interventions for lowering plasma homocysteine levels in dialysis patients. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- House, A.A.; Eliasziw, M.; Cattran, D.C.; Churchill, D.N.; Oliver, M.J.; Fine, A.; Dresser, G.K.; Spence, J.D. Effect of B-vitamin therapy on progression of diabetic nephropathy: A randomized controlled trial. JAMA 2010, 303, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.; Sheridan, P.; McQueen, M.J.; Held, C.; Arnold, J.M.; Fodor, G.; Yusuf, S.; Lonn, E.M. HOPE-2 investigators. Homocysteine lowering with folic acid and B vitamins in people with chronic kidney disease—results of the renal Hope-2 study. Nephrol. Dial. Transpl. 2008, 23, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Vianna, A.C.; Mocelin, A.J.; Matsuo, T.; Morais-Filho, D.; Largura, A.; Delfino, V.A.; Soares, A.E.; Matni, A.M. Uremic hyperhomocysteinemia: A randomized trial of folate treatment for the prevention of cardiovascular events. Hemodial. Int. 2007, 11, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Righetti, M.; Serbelloni, P.; Milani, S.; Ferrario, G. Homocysteine-lowering vitamin B treatment decreases cardiovascular events in hemodialysis patients. Blood Purif. 2006, 24, 379–386. [Google Scholar] [CrossRef]
- Xie, D.; Yuan, Y.; Guo, J.; Yang, S.; Xu, X.; Wang, Q.; Li, Y.; Qin, X.; Tang, G.; Huo, Y.; et al. Hyperhomocysteinemia predicts renal function decline: A prospective study in hypertensive adults. Sci. Rep. 2015, 5, 16268. [Google Scholar] [CrossRef]
- La Manna, G.; Cappuccilli, M.L.; Cianciolo, G.; Conte, D.; Comai, G.; Carretta, E.; Scolari, M.P.; Stefoni, S. Cardiovascular disease in kidney transplant recipients. The prognostic value of inflammatory cytokine genotypes. Transplantation 2010, 89, 1001–1008. [Google Scholar] [CrossRef]
- Friedman, A.N.; Rosenberg, I.H.; Selhub, J.; Levey, A.S.; Bostom, A.G. Hyperhomocysteinemia in renal transplant recipients. Am. J. Transpl. 2002, 2, 308–313. [Google Scholar] [CrossRef]
- Bostom, A.G.; Gohh, R.Y.; Liaugaudas, G.; Beaulieu, A.J.; Han, H.; Jacques, P.F.; Dworkin, L.; Rosenberg, I.H.; Selhub, J. Prevalence of mild fasting hyperhomocysteinemia in renal transplant versus coronary artery disease patients after fortification of cereal grain flour with folic acid. Atherosclerosis 1999, 145, 221–224. [Google Scholar] [CrossRef]
- Bostom, A.G.; Gohh, R.Y.; Beaulieu, A.J.; Nadeau, M.R.; Hume, A.L.; Jacques, P.F.; Selhub, J.; Rosenberg, I.H. Treatment of hyperhomocysteinemia in renal transplant recipients. A randomized placebo-controlled trial. Ann. Intern. Med. 1997, 127, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, A.J.; Gohh, R.Y.; Han, H.; Hakas, D.; Jacques, P.F.; Selhub, J.; Bostom, A.G. Enhanced reduction of fasting total homocysteine levels with supraphysiological versus standard multivitamin dose folic acid supplementation in renal transplant recipients. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2918–2921. [Google Scholar] [CrossRef]
- Bostom, A.G.; Carpenter, M.A.; Kusek, J.W.; Levey, A.S.; Hunsicker, L.; Pfeffer, M.A.; Selhub, J.; Jacques, P.F.; Cole, E.; Gravens-Mueller, L.; et al. Homocysteine-lowering and cardiovascular disease outcomes in kidney transplant recipients: Primary results from the Folic Acid for Vascular Outcome Reduction in Transplantation trial. Circulation 2011, 123, 1763–1770. [Google Scholar] [CrossRef]
- Scott, T.M.; Rogers, G.; Weiner, D.E.; Livingston, K.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; Troen, A.M. B-Vitamin Therapy for Kidney Transplant Recipients Lowers Homocysteine and Improves Selective Cognitive Outcomes in the Randomized FAVORIT Ancillary Cognitive Trial. J. Prev. Alzheimers Dis. 2017, 4, 174–182. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study | Design | Participants, n | Case Definition/Outcome | Results |
|---|---|---|---|---|
| Soohoo et al., 2017 [15] | Retrospective | 9517 (folate group), 12968 (B12 group) HD | Mortality | Lower folic acid predicts mortality |
| Ye et al., 2016 [94] | Cross-sectional | 1042 CKD stage 1–5 | CVD | HHcy associated with CKD severity, LVH, LVD and vascular disease |
| Anan et al., 2006 [95] | Retrospective case-control | 44 HD | Silent cerebral infarction | HHcy predicted outcome |
| Ducloux et al., 2006 [92] | Prospective observational | 459 HD | Mortality and fatal CVD | HHcy predicted outcome only in patients without CIMS |
| Nair et al., 2005 [96] | Retrospective case-control | 146 HD | MI, heart surgery | HHcy did not predict CVD risk |
| London et al., 2004 [97] | Prospective observational | 78 HD | Mortality | HHcy did not predict outcome |
| Kalantar-Zadeh et al., [98] | Prospective observational | 367 HD | Mortality | Lower Hcy levels predicted mortality |
| Buccianti et al., 2004 [99] | Prospective observational | 77 HD | Fatal CVD | HHcy predicted outcome |
| Bayès et al., 2003 [100] | Prospective observational | 94 HD | Mortality, fatal CVD | HHcy did not predict outcome |
| Mallamaci et al., 2002 [101] | Prospective observational | 175 HD | Mortality, fatal CVD | HHcy predicted outcome |
| Ducloux et al., 2002 [102] | Prospective observational | 240 PD | Fatal and nonfatal CVD | HHcy did not predict outcome |
| Haraki et al.,2001 (retrospective part) [103] | Retrospective case-control | 43 HD/PD | Coronary, cerebral and peripheral vascular disease | HHcy CVD risk factor |
| Haraki et al., 2001 (prospective part) [103] | Prospective observational | 55 HD/PD | Fatal and nonfatal CVD | HHcy predicted outcome |
| Wrone et al., 2001 [104] | Retrospective case-control | 459 HD/PD | MI, stroke, TIA, carotid endarterectomy. | HHcy did not predict CVD risk |
| Dierkes et al., 2000 [105] | Prospective observational | 102 HD | Mortality, fatal/nonfatal CVD | HHcy predicted outcome |
| Suliman et al., 2000 [106] | Retrospective case-control | 117 HD | Coronary, cerebral and peripheral vascular disease | HHcy did not predict CVD risk |
| Kunz et al., 1999 [107] | Retrospective case-control | 63 HD | Coronary, cerebral and peripheral vascular disease | HHcy cardiovascular risk factor |
| Manns et al., 1999 [108] | Retrospective case-control | 218 HD | Coronary, cerebral and peripheral vascular disease | HHcy cardiovascular risk factor only in males |
| Sirrs et al., 1999 [109] | Prospective observational | 88 HD | Mortality and CVD events | Lower Hcy levels predicted mortality |
| Moustapha et al., 1998 [110] | Prospective observational | 167 HD/PD | Mortality and CVD events | HHcy predicted outcome |
| Vychytil et al., 1998 [111] | Retrospective case-control | 154 PD | Coronary, cerebral and peripheral vascular disease | HHcy did not predict CVD risk |
| Bostom et al., 1997 [112] | Prospective observational | 73 HD/PD | CVD events | HHcy predicted outcome |
| Robinson et al., 1996 [113] | Retrospective case-control | 176 HD/PD | Coronary, cerebral and peripheral vascular disease | HHcy cardiovascular risk factor |
| Bachmann et al., 1995 [114] | Retrospective case-control | 45 HD | Coronary, cerebral and peripheral vascular disease | HHcy cardiovascular risk factor |
| Bostom et al., 1995 [115] | Retrospective case-control | 24 HD/PD | Coronary, cerebral and peripheral vascular disease | HHcy did not predict CVD risk |
| Study | Design/Intervention | Participants, n | End Point | Follow-up, Years | Results |
|---|---|---|---|---|---|
| Xu et al., 2016 [87] | Double blind RCT: enalapril 10 mg versus enalapril 10 mg plus folic acid | 15,104 (eGFR ≥ 30 mL/min). No folic acid fortification | CKD progression | 4.4 | Enalapril plus folic acid delayed CKD progression |
| House et al., 2010 [125] | Double blind RCT: folic acid 2.5 mg + Vitamin B6 25 mg + Vitamin B12 1 mg versus placebo | 238 (diabetic nephropathy with eGFR > 30 mL/min). Folic acid fortification | CKD progression | 2.6 | Greater GFR decrease and more CVD events in treatment group |
| Heinz et al., 2010 [120] | Double blind RCT: folic acid 5 mg, vitamin B12 50 µg, vitamin B6 20 mg versus placebo 3 times a week | 650 hemodialysis patients | All-cause mortality, cardiovascular events | 2 | No differences |
| Mann et al., 2008 [126] | Double blind RCT: folic acid 2.5 mg + vitamin B6 50 mg + vitamin B12 1 mg versus placebo | 619 CKD (eGFR <60 mL/min) | All-cause mortality, cardiovascular events | 5 | No differences |
| Cianciolo et al., 2008 [11] | Open label randomized trial: 5-MTHF intravenous. three times a week versus folic acid 5 mg oral daily | 314 hemodialysis patients | All-cause mortality | 4.5 | Less mortality risk in 5-MTHF group (independent of homocysteine) |
| Jamison et al., 2007 [119] | Double blind RCT (HOST): folic acid 40 mg + vitamin B6 100 mg + vitamin B12 2 mg versus placebo | 2056 CKD (eGFR ≤ 30) or hemodialysis (folic acid fortification) | All-cause mortality, CKD progression | 3.2 | No differences |
| Vianna et al., 2007 [127] | Double blind RCT: folic acid 5 mg versus placebo | 97 hemodialysis patients | Cardiovascular events | 2 | No differences |
| Zoungas et al., 2006 [118] | Double blind RCT (ASFAST): folic acid 15 mg versus placebo | 315 CKD (eGFR < 25 mL/min), hemodialysis and peritoneal dialysis | Cardiovascular events and mortality | 3.6 | No differences |
| Righetti et al., 2006 [128] | Open prospective trial: folic acid 5 mg versus untreated | 114 hemodialysis patients | Cardiovascular events | 2.4 | Folic acid decreases CVD events |
| Wrone et al., 2004 [76] | Three arms, double blind RCT: folic acid 1 mg or 5 mg or 15 mg | 510 hemodialysis patients | Cardiovascular events and mortality | 2 | No differences |
| Righetti et al., 2003 [117] | Placebo-controlled, non-blinded RCT: folic acid 5, 15, 25 mg or placebo | 81 hemodialysis patients | Cardiovascular mortality | 1 | No differences |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capelli, I.; Cianciolo, G.; Gasperoni, L.; Zappulo, F.; Tondolo, F.; Cappuccilli, M.; La Manna, G. Folic Acid and Vitamin B12 Administration in CKD, Why Not? Nutrients 2019, 11, 383. https://doi.org/10.3390/nu11020383
Capelli I, Cianciolo G, Gasperoni L, Zappulo F, Tondolo F, Cappuccilli M, La Manna G. Folic Acid and Vitamin B12 Administration in CKD, Why Not? Nutrients. 2019; 11(2):383. https://doi.org/10.3390/nu11020383
Chicago/Turabian StyleCapelli, Irene, Giuseppe Cianciolo, Lorenzo Gasperoni, Fulvia Zappulo, Francesco Tondolo, Maria Cappuccilli, and Gaetano La Manna. 2019. "Folic Acid and Vitamin B12 Administration in CKD, Why Not?" Nutrients 11, no. 2: 383. https://doi.org/10.3390/nu11020383
APA StyleCapelli, I., Cianciolo, G., Gasperoni, L., Zappulo, F., Tondolo, F., Cappuccilli, M., & La Manna, G. (2019). Folic Acid and Vitamin B12 Administration in CKD, Why Not? Nutrients, 11(2), 383. https://doi.org/10.3390/nu11020383