Grape Seed Proanthocyanidins Induce Autophagy and Modulate Survivin in HepG2 Cells and Inhibit Xenograft Tumor Growth in Vivo



Abstract
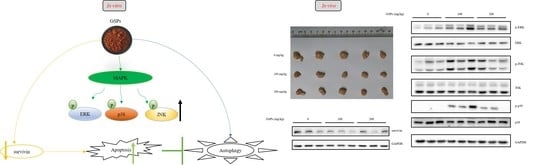
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qPCR) Analysis
2.4. Apoptosis Analysis with Flow Cytometry
2.5. Western Blot Analysis
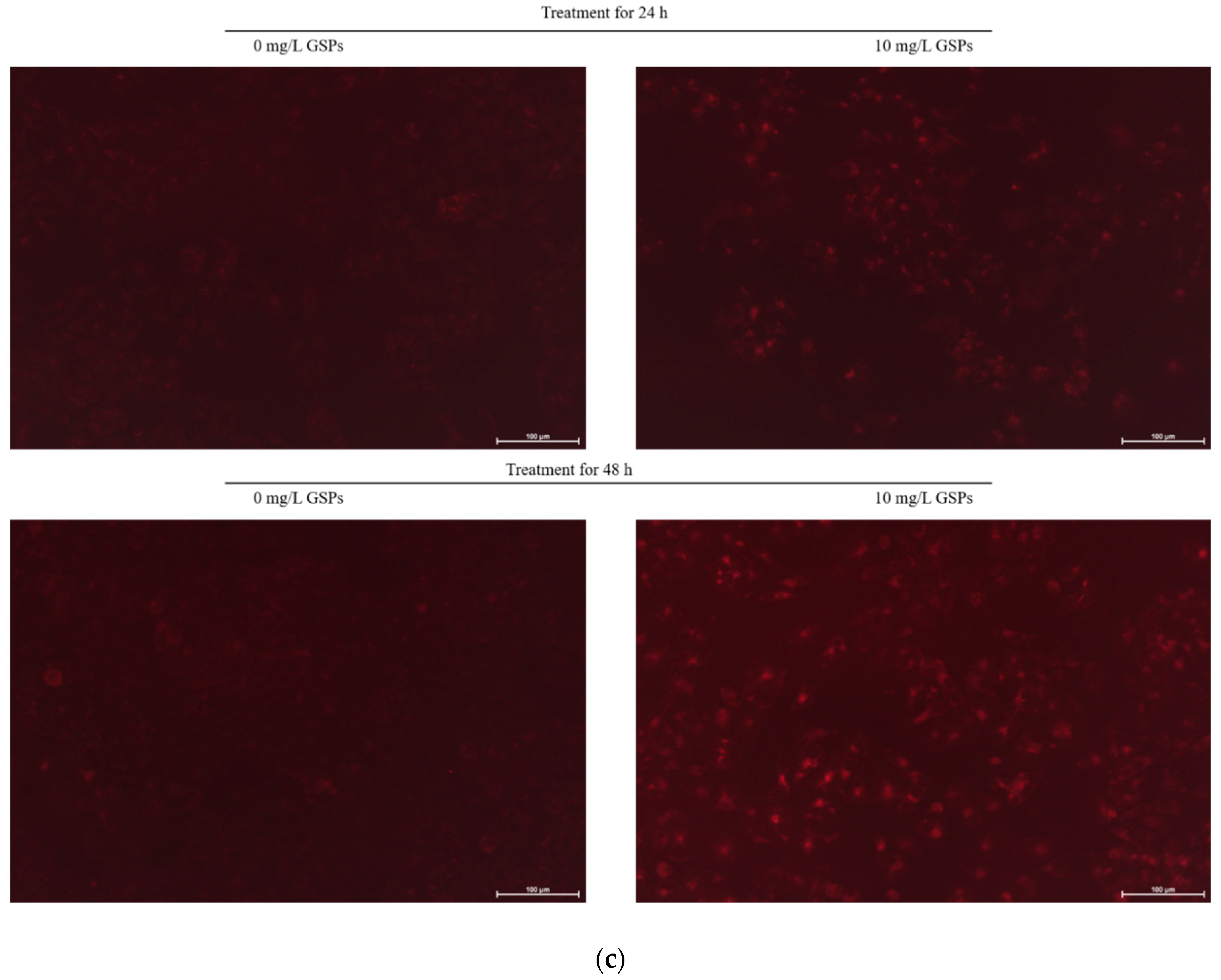
2.6. Observation of Acidic Vesicular Organelles (AVOs) Formation
2.7. Cell Transfection
2.8. HepG2-Derived Nude Mice Xenograft Model
2.9. Immunohistochemistry
2.10. Histopathological Examination
2.11. Statistical Analysis
3. Results
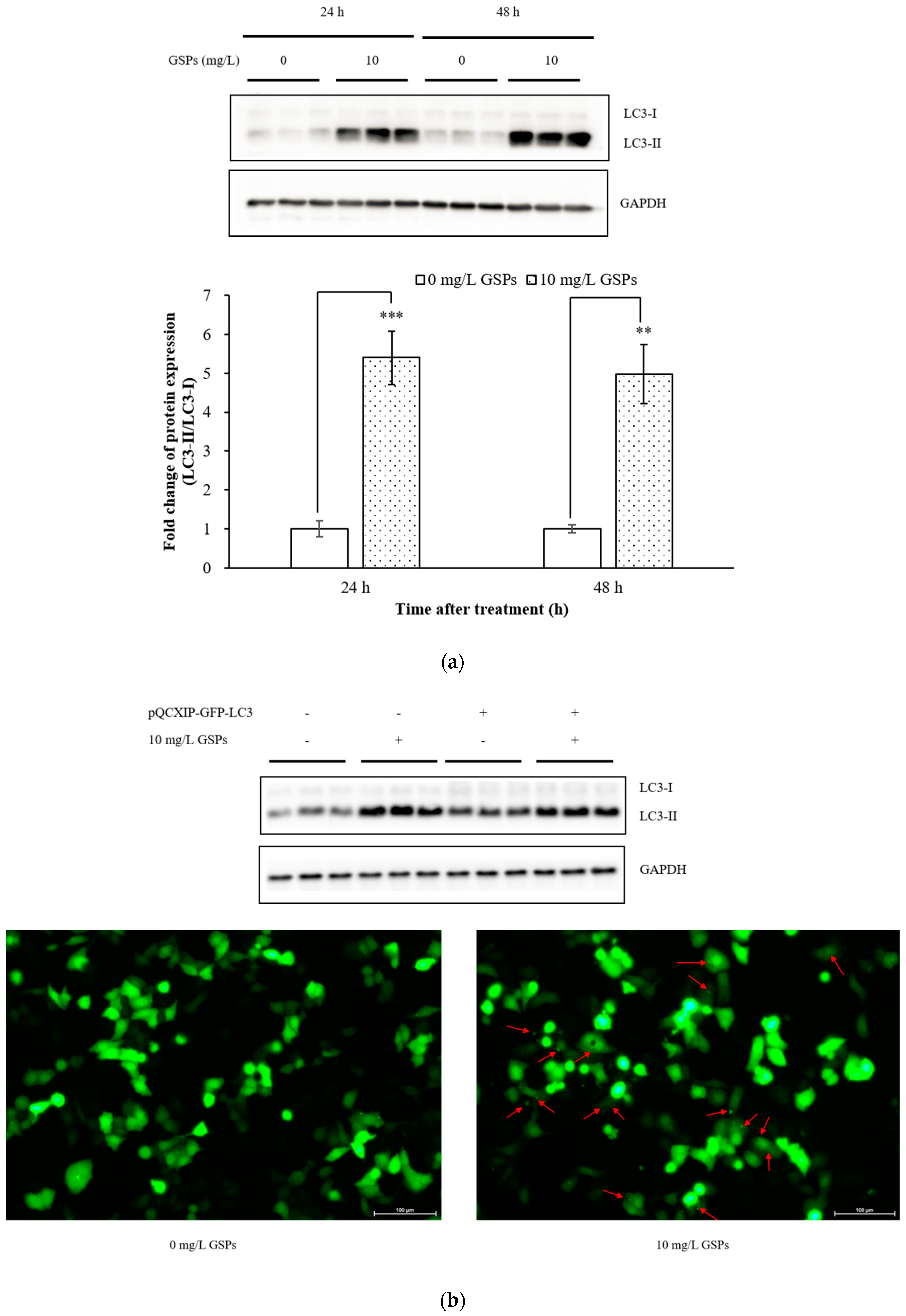
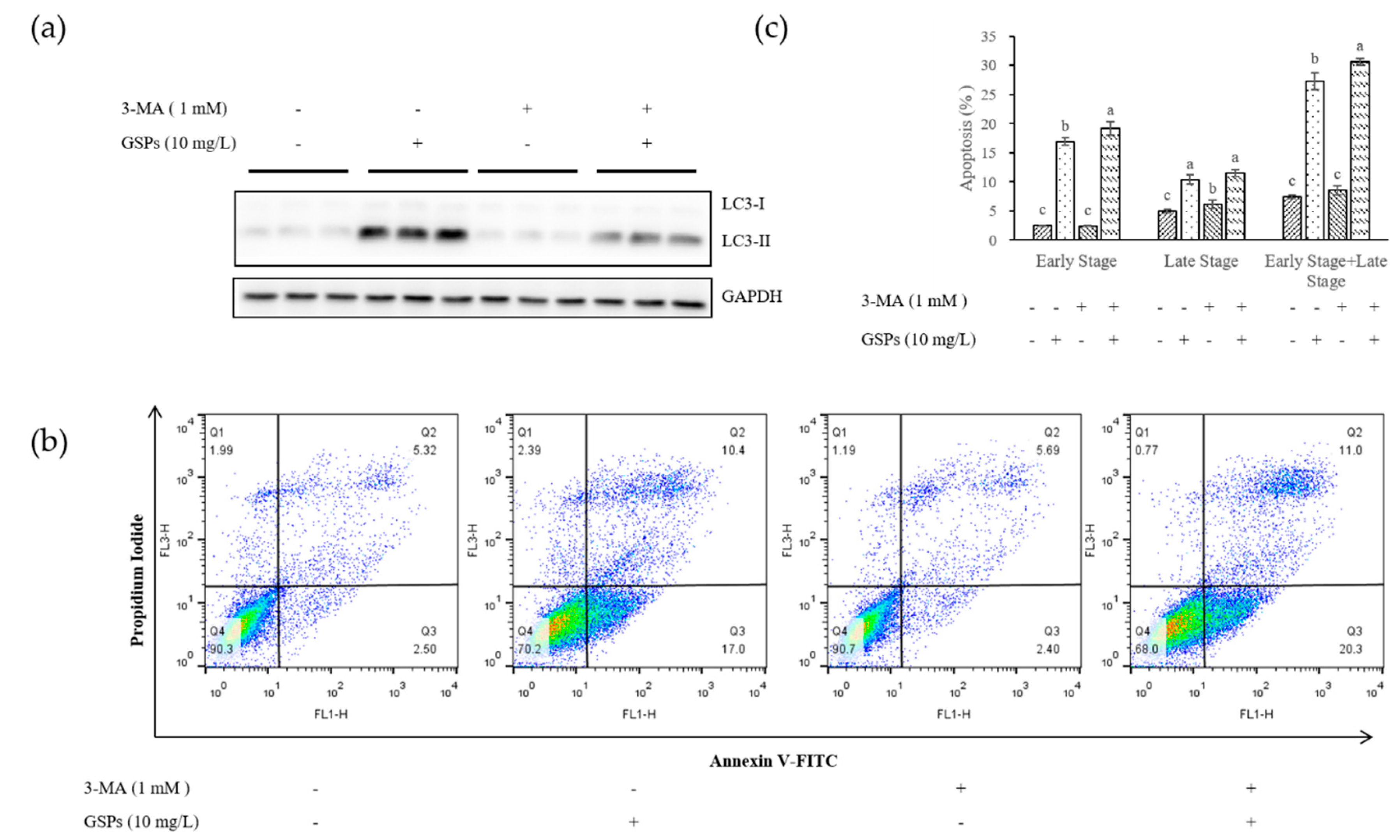
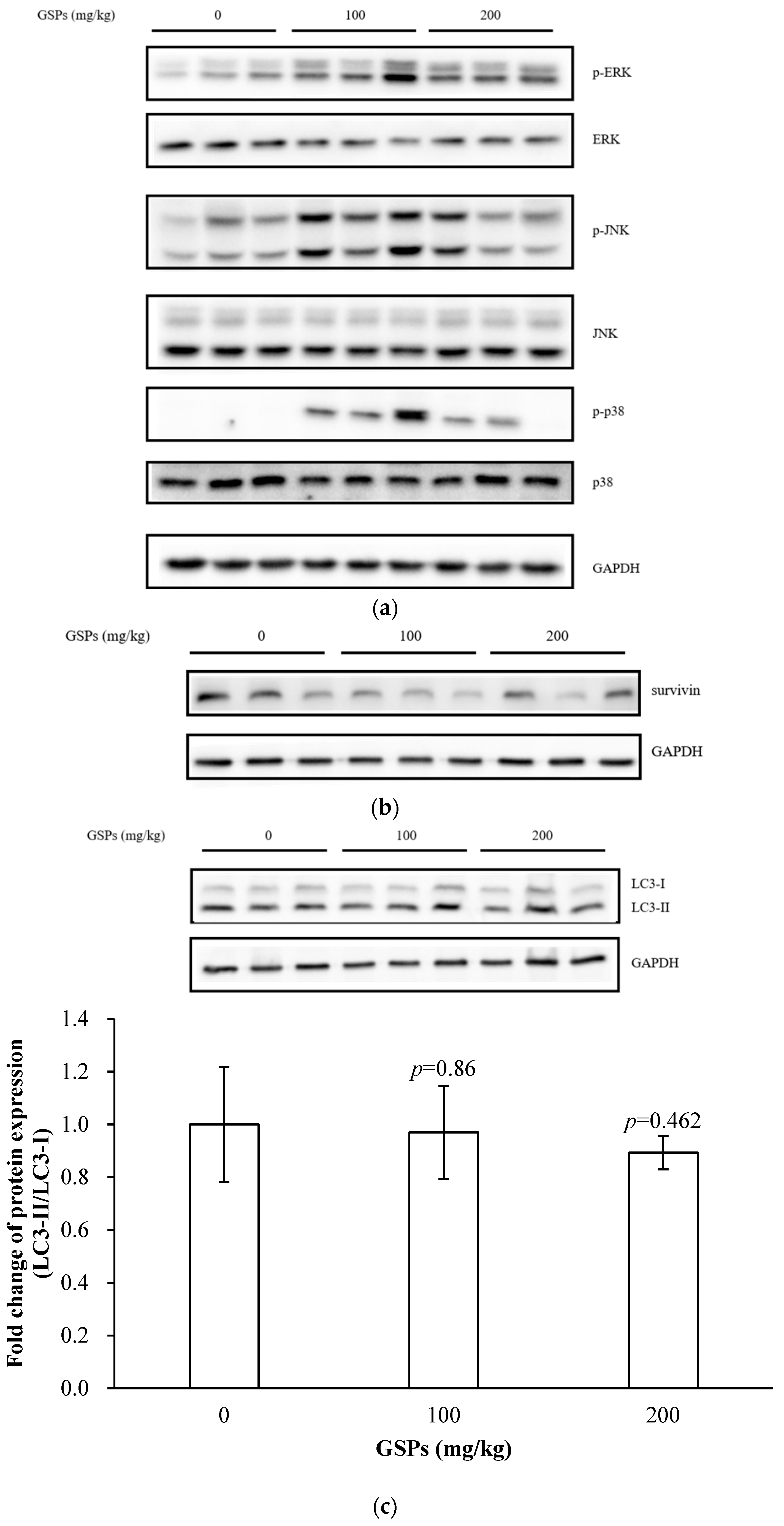
3.1. GSPs Induced Autophagy in HepG2 Cells
3.2. Inhibition of Autophagy Increased Early Stage Apoptosis of HepG2 Cells
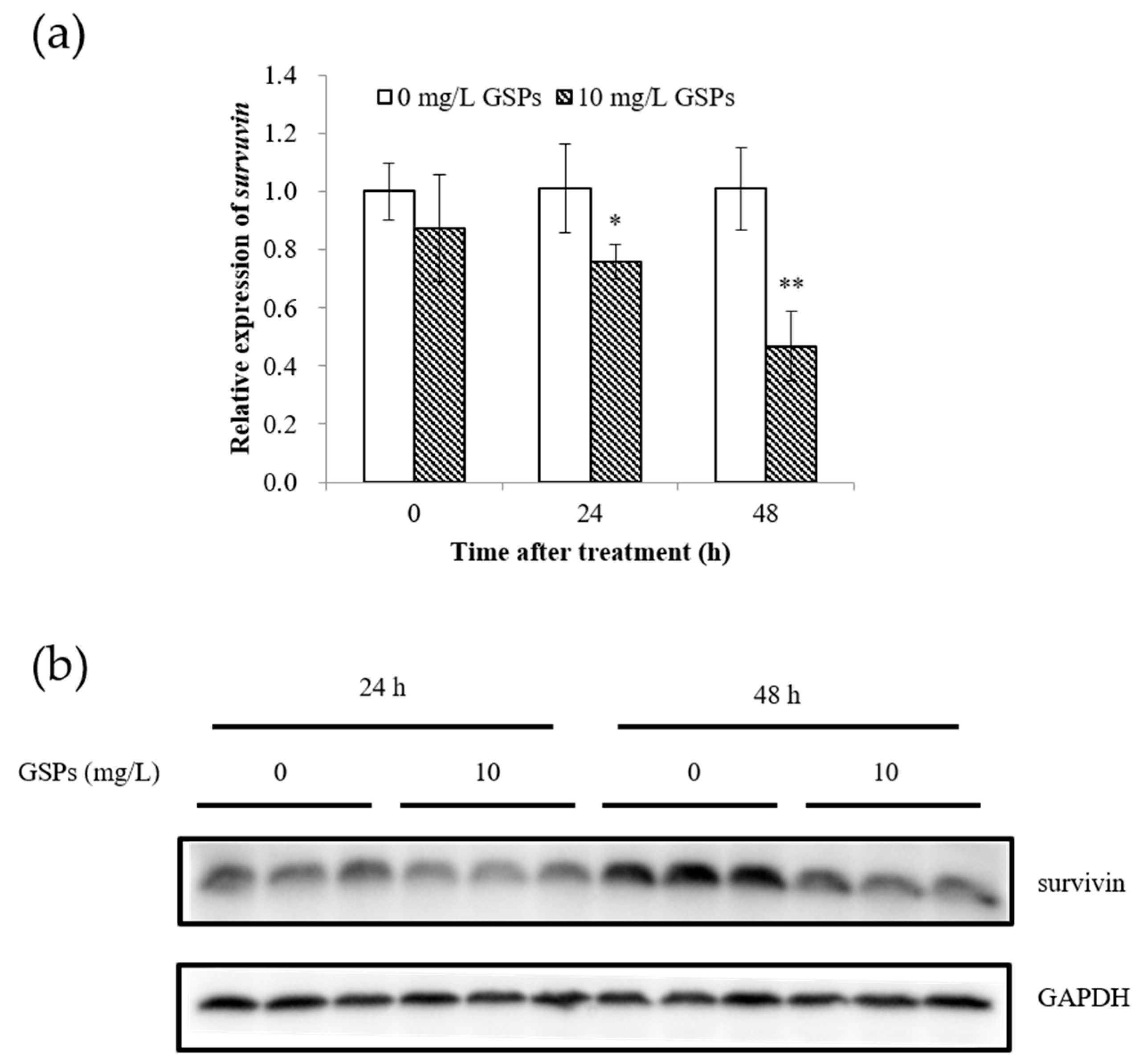
3.3. GSPs Significantly Reduced the Expression of Survivin in HepG2 Cells
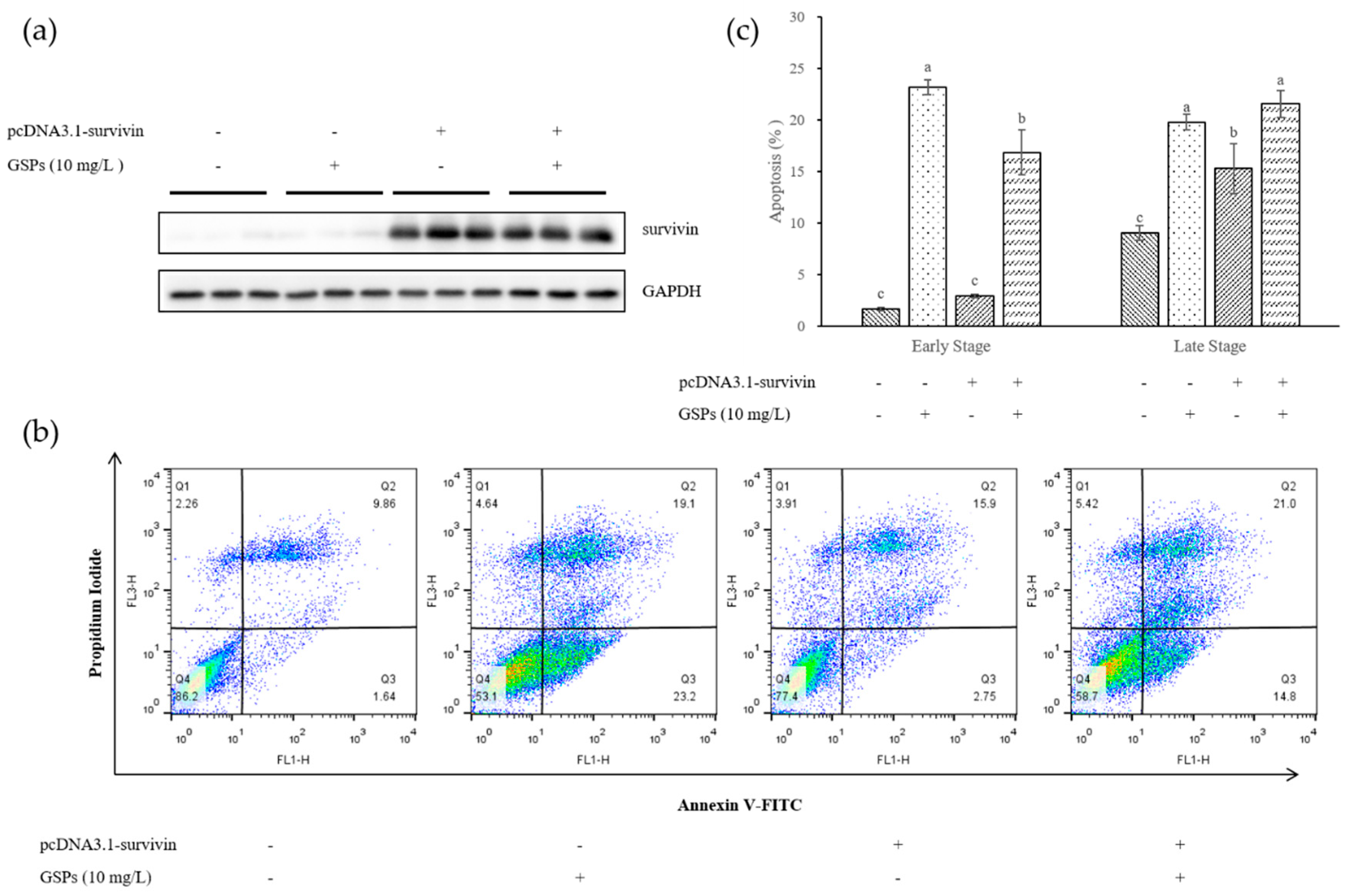
3.4. Survivin Was Involved in GSPs-induced Apoptosis
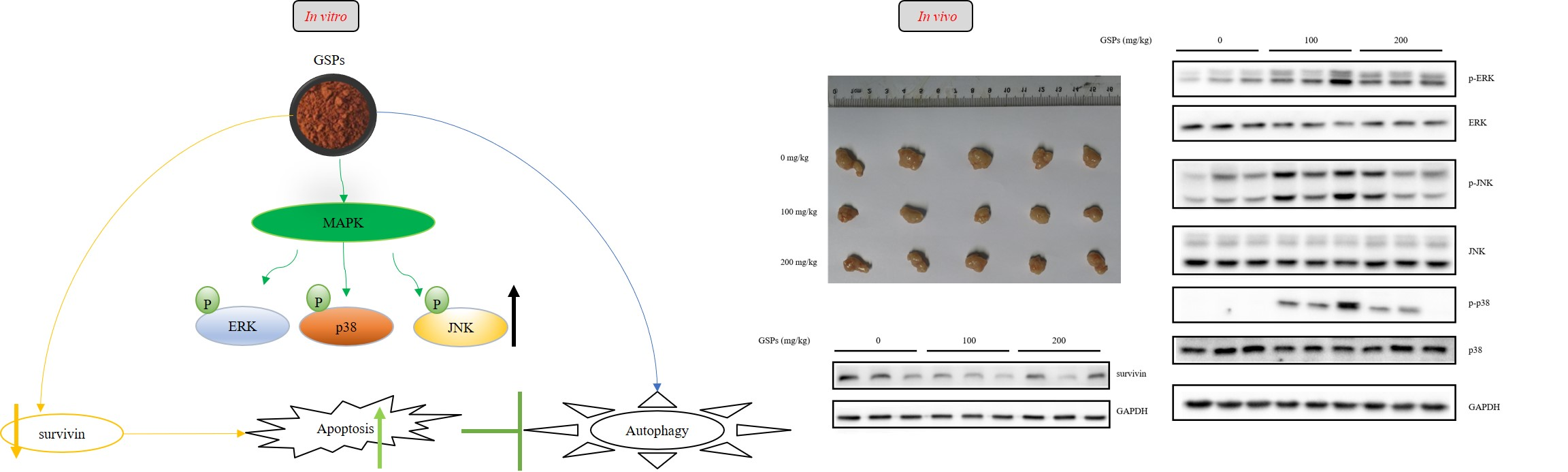
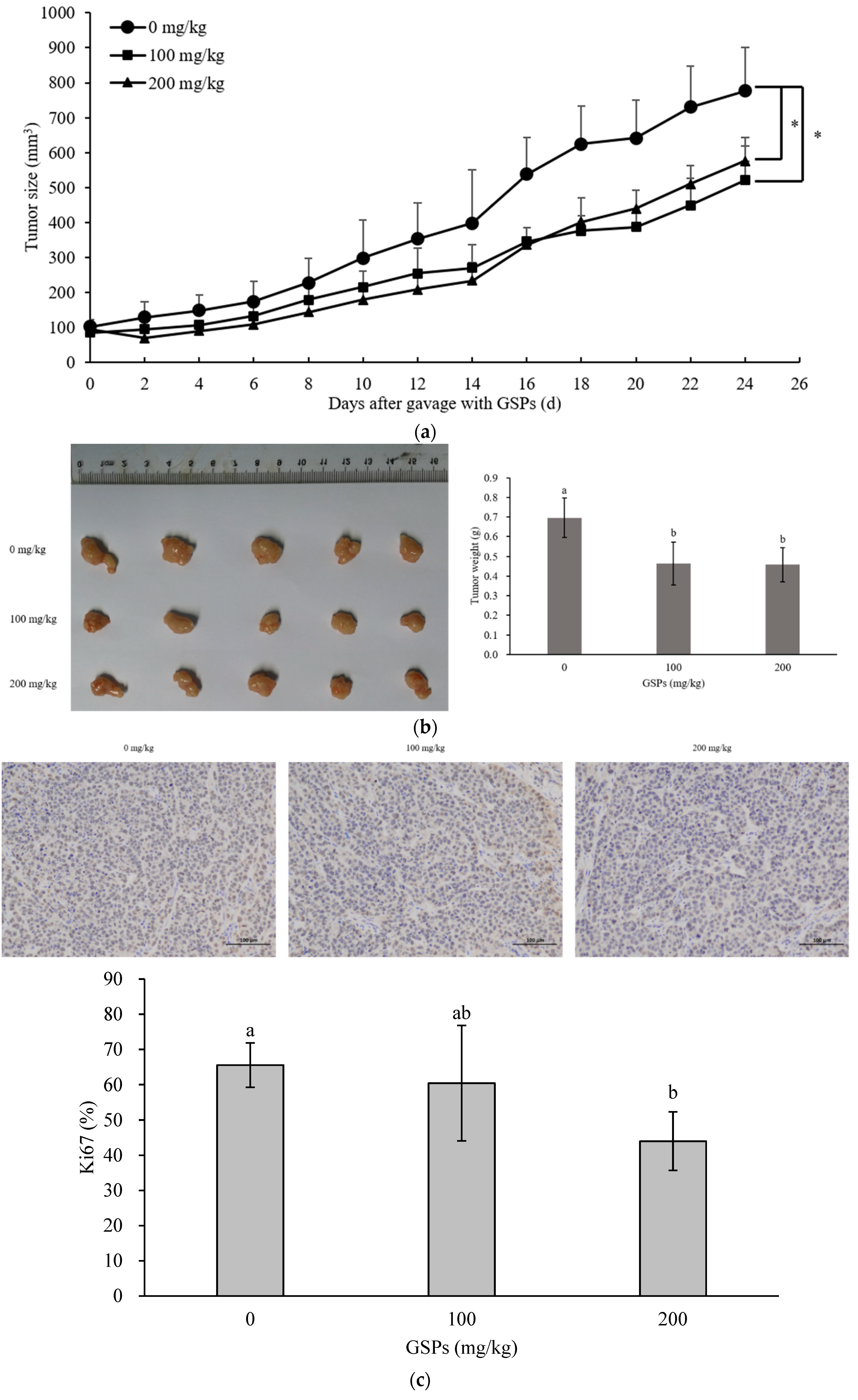
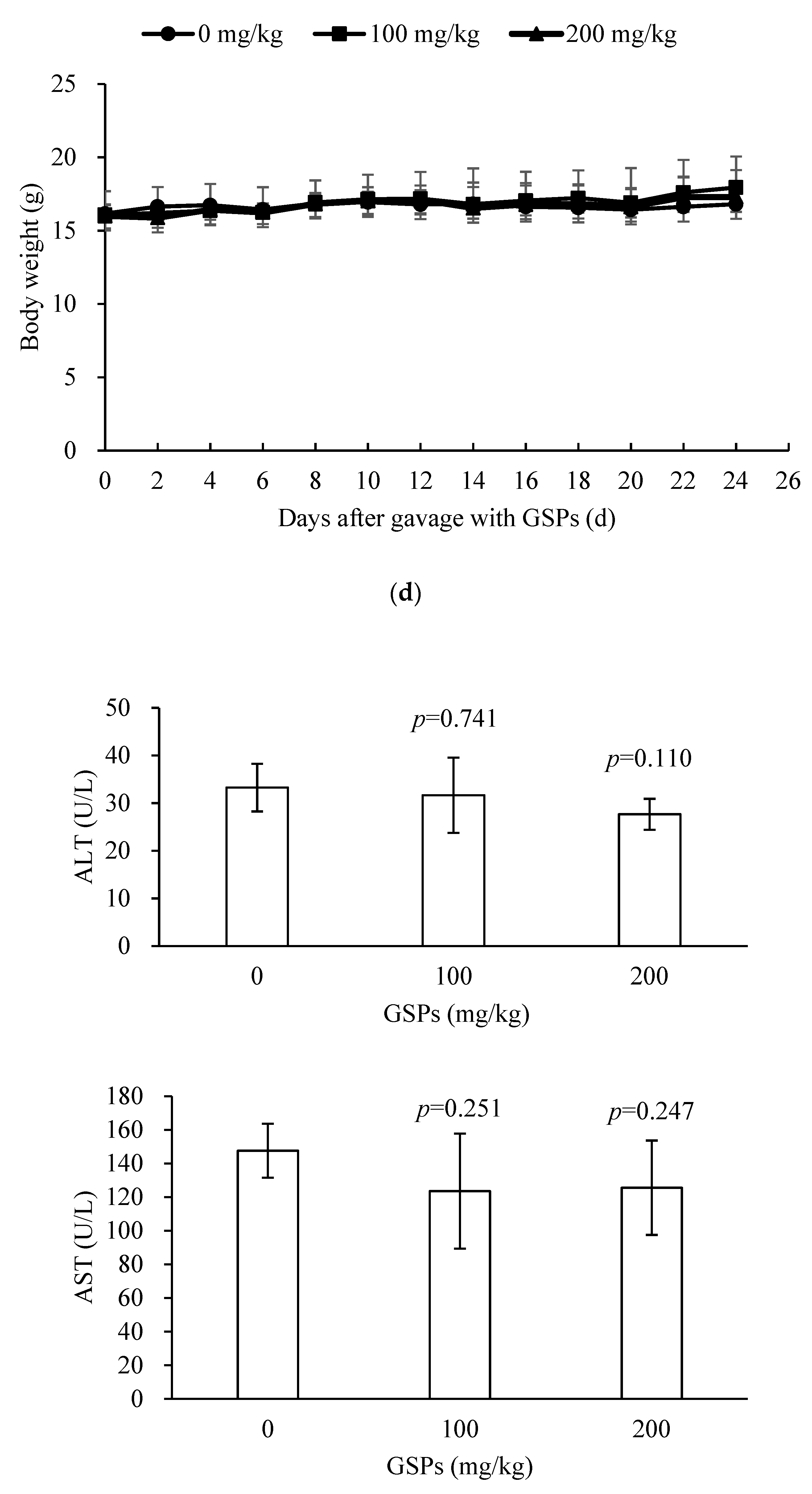
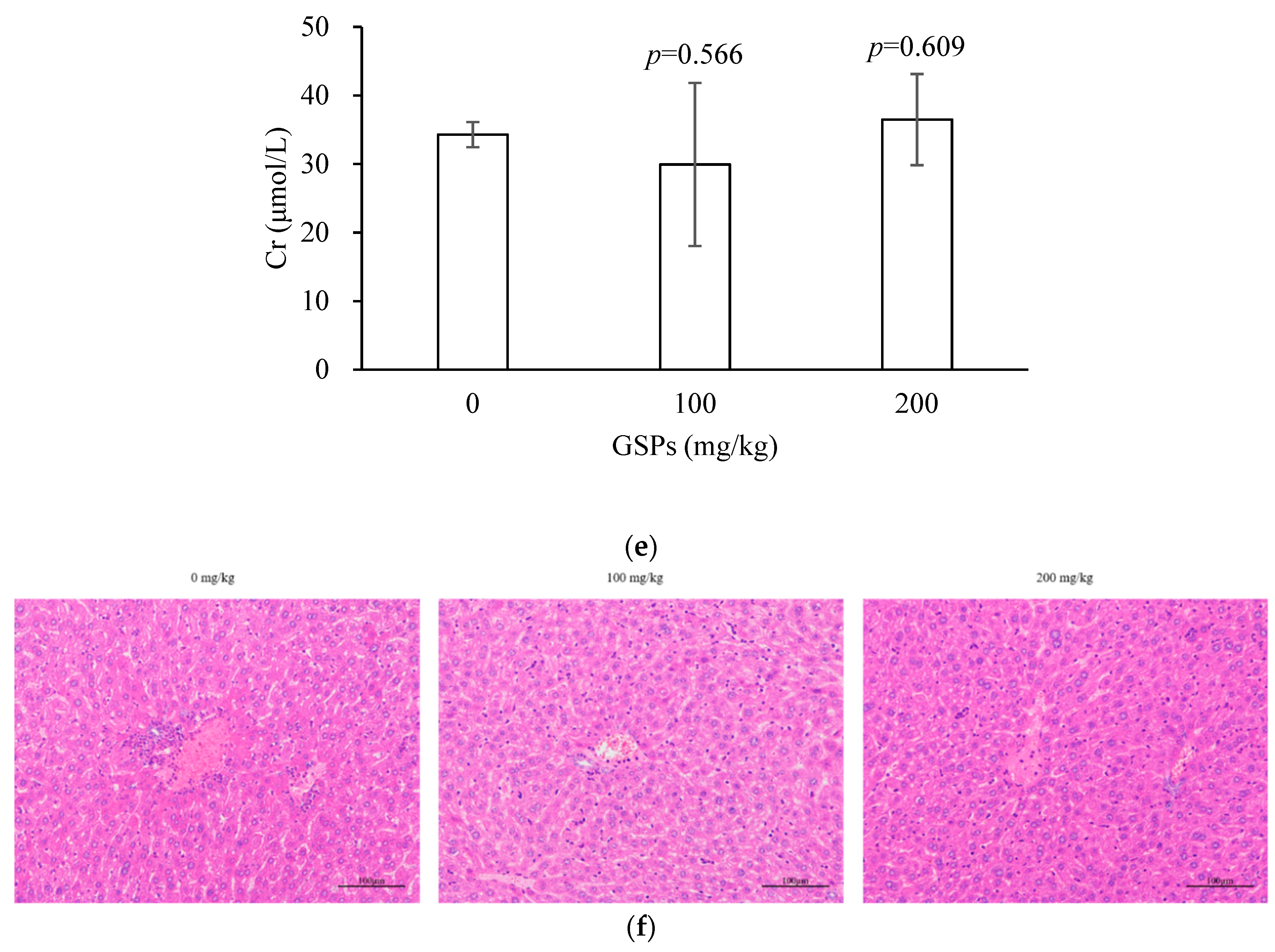
3.5. GSPs Inhibited the Growth of HepG2 Cells without Displaying Observable Toxicity in Nude Mice
3.6. GSPs Induced the Phosphorylation of the MAPK Pathway-Associated Proteins, and Decreased the Expression of Survivin
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sartorius, K.; Sartorius, B.; Aldous, C.; Govender, P.S.; Madiba, T.E. Global and country underestimation of hepatocellular carcinoma (HCC) in 2012 and its implications. Cancer Epidemiol. 2015, 39, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Daher, S.; Massarwa, M.; Benson, A.A.; Khoury, T. Current and future treatment of hepatocellular carcinoma: An updated comprehensive review. J. Clin. Transl. Hepatol. 2018, 6, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Mccormick, F. Cancer gene therapy: Fringe or cutting edge? Nat. Rev. Cancer 2001, 1, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, V.; Singh, T.; Katiyar, S.K. Multi-targeted prevention and therapy of cancer by proanthocyanidins. Cancer Lett. 2008, 269, 378–387. [Google Scholar] [CrossRef]
- Yamakoshi, J.; Saito, M.; Kataoka, S.; Kikuchi, M. Safety evaluation of proanthocyanidin-rich extract from grape seeds. Food Chem. Toxicol. 2002, 40, 599–607. [Google Scholar] [CrossRef]
- Kaur, M.; Mandair, R.; Agarwal, R.; Agarwal, C. Grape seed extract induces cell cycle arrest and apoptosis in human colon carcinoma cells. Nutr. Cancer 2008, 60, 2–11. [Google Scholar] [CrossRef]
- Kaur, M.; Singh, R.P.; Gu, M.; Agarwal, R.; Agarwal, C. Grape seed extract inhibits in vitro and in vivo growth of human colorectal carcinoma cells. Clin. Cancer Res. 2006, 12, 6194–6202. [Google Scholar] [CrossRef]
- Dinicola, S.; Mariggio, M.A.; Morabito, C.; Guarnieri, S.; Cucina, A.; Pasqualato, A.; D’Anselmi, F.; Proietti, S.; Coluccia, P.; Bizzarri, M. Grape seed extract triggers apoptosis in Caco-2 human colon cancer cells through reactive oxygen species and calcium increase: Extracellular signal-regulated kinase involvement. Br. J. Nutr. 2013, 110, 797–809. [Google Scholar] [CrossRef]
- Prasad, R.; Vaid, M.; Katiyar, S.K. Grape proanthocyanidin inhibit pancreatic cancer cell growth in vitro and in vivo through induction of apoptosis and by targeting the PI3K/Akt pathway. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Upanan, S.; Yodkeeree, S.; Thippraphan, P.; Punfa, W.; Wongpoomchai, R.; Limtrakul, P. The proanthocyanidin-rich fraction obtained from red rice germ and bran extract induces HepG2 hepatocellular carcinoma cell apoptosis. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Hamza, A.A.; Heeba, G.H.; Elwy, H.M.; Murali, C.; El-Awady, R.; Amin, A. Molecular characterization of the grape seeds extract’s effect against chemically induced liver cancer: In vivo and in vitro analyses. Sci. Rep. 2018, 8, 1270. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.D.; Meeran, S.M.; Katiyar, S.K. Proanthocyanidins inhibit in vitro and in vivo growth of human non-small cell lung cancer cells by inhibiting the prostaglandin E-2 and prostaglandin E-2 receptors. Mol. Cancer Ther. 2010, 9, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Sharma, S.D.; Katiyar, S.K. Grape Proanthocyanidins induce apoptosis by loss of mitochondrial membrane potential of human non-small cell lung cancer cells in vitro and in vivo. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Hah, Y.S.; Kim, J.G.; Cho, H.Y.; Park, J.S.; Heo, E.P.; Yoon, T.J. Procyanidins from Vitis vinifera seeds induce apoptotic and autophagic cell death via generation of reactive oxygen species in squamous cell carcinoma cells. Oncol. Lett. 2017, 14, 1925–1932. [Google Scholar] [CrossRef]
- Prasad, R.; Katiyar, S.K. Bioactive phytochemical proanthocyanidins inhibit growth of head and neck squamous cell carcinoma cells by targeting multiple signaling molecules. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Santos-Buelga, C.; Scalbert, A. Proanthocyanidins and tannin-like compounds-nature, occurrence, dietary intake and effects on nutrition and health. J. Sci. Food Agric. 2000, 80, 1094–1117. [Google Scholar] [CrossRef]
- Lan, C.Z.; Ding, L.; Su, Y.L.; Guo, K.; Wang, L.; Kan, H.W.; Ou, Y.R.; Gao, S. Grape seed proanthocyanidins prevent DOCA-salt hypertension-induced renal injury and its mechanisms in rats. Food Funct. 2015, 6, 2179–2186. [Google Scholar] [CrossRef]
- Wang, L.H.; Huang, W.D.; Zhan, J.C. Grape seed proanthocyanidins induce apoptosis and cell cycle arrest in HepG2 cells accompanied by induction of the MAPK signalling pathway and NAG-1. Unpublished Data. 2019. [Google Scholar]
- Wang, Y.; Yuan, S.; Li, L.; Yang, D.; Xu, C.; Wang, S.; Zhang, D. Novel proapoptotic agent SM-1 enhances the inhibitory effect of 5-fluorouracil on colorectal cancer cells in vitro and in vivo. Oncol. Lett. 2017, 13, 4762–4768. [Google Scholar] [CrossRef]
- Zhang, X.; Kang, Y.; Huo, T.; Tao, R.; Wang, X.; Li, Z.; Guo, Q.; Zhao, L. GL-V9 induced upregulation and mitochondrial localization of NAG-1 associates with ROS generation and cell death in hepatocellular carcinoma cells. Free Radic. Biol. Med. 2017, 112, 49–59. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ko, C.P.; Lin, C.W.; Chen, M.K.; Yang, S.F.; Chiou, H.L.; Hsieh, M.J. Pterostilbene induce autophagy on human oral cancer cells through modulation of Akt and mitogen-activated protein kinase pathway. Oral Oncol. 2015, 51, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Isitor, G.N.; Rao, S.; Nayak, S.B.; Sundaram, V. Autofluorescent vesicular structures in hematoxylin and eosin stained duodenal mucosa of the domestic cat. West Indian Vet. J. 2009, 9, 27–32. [Google Scholar]
- Cao, Y.J.; Luo, Y.C.; Zou, J.; Ouyang, J.; Cai, Z.H.; Zeng, X.; Ling, H.; Zeng, T.B. Autophagy and its role in gastric cancer. Clin. Chim. Acta 2019, 489, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of acridine orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Shanmugam, M.K.; Kumar, A.P.; Yap, C.T.; Sethi, G.; Bishayee, A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 2019, 125, 1228–1246. [Google Scholar] [CrossRef] [PubMed]
- Opipari, A.W.; Tan, L.J.; Boitano, A.E.; Sorenson, D.R.; Aurora, A.; Liu, J.R. Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res. 2004, 64, 696–703. [Google Scholar] [CrossRef]
- Ellington, A.A.; Berhow, M.; Singletary, K.W. Induction of macroautophagy in human colon cancer cells by soybean B-group triterpenoid saponins. Carcinogenesis 2005, 26, 159–167. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Bastholm, L.; Mathiasen, I.S.; Elling, F.; Jaattela, M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. 2005, 12, 1297–1309. [Google Scholar] [CrossRef]
- Manach, C.; Williamson, G.C.; Scalbert, A.; Remesy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef]
- Déprez, S.; Brezillon, C.; Rabot, S.; Philippe, C.; Mila, I.; Lapierre, C.; Scalbert, A. Polymeric proanthocyanidins are catabolized by human colonic microflora into low-molecular-weight phenolic acids. J. Nutr. 2000, 130, 2733–2738. [Google Scholar] [CrossRef]
- Rios, L.Y.; Gonthier, M.P.; Remesy, C.; Mila, L.; Lapierre, C.; Lazarus, S.A.; Williamson, G.; Scalbert, A. Chocolate intake increases urinary excretion of polyphenol-derived phenolic acids in healthy human subjects. Am. J. Clin. Nutr. 2003, 77, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perez, C.; Garcia-Villanova, B.; Guerra-Hernandez, E.; Verardo, V. Grape seeds proanthocyanidins: An overview of in vivo bioactivity in animal models. Nutrients 2019, 11, 2435. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Ashwell, J.D. IAPs: What’s in a Name? Mol. Cell 2008, 30, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Waligorska-Stachura, J.; Jankowska, A.; Wasko, R.; Liebert, W.; Biczysko, M.; Czarnywojtek, A.; Baszko-Blaszyk, D.; Shimek, V.; Ruchala, M. Survivin-prognostic tumor biomarker in human neoplasms-review. Ginekol. Pol. 2012, 83, 537–540. [Google Scholar] [CrossRef]
- Athanasoula, K.C.; Gogas, H.; Polonifi, K.; Vaiopoulos, A.G.; Polyzos, A.; Mantzourani, M. Survivin beyond physiology: Orchestration of multistep carcinogenesis and therapeutic potentials. Cancer Lett. 2014, 347, 175–182. [Google Scholar] [CrossRef]
- Altieri, D.C. Targeting survivin in cancer. Cancer Lett. 2013, 332, 225–228. [Google Scholar] [CrossRef]
- Li, F.Z.; Ackermann, E.J.; Bennett, C.F.; Rothermel, A.L.; Plescia, J.; Tognin, S.; Villa, A.; Marchisio, P.C.; Altieri, D.C. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat. Cell Biol. 1999, 1, 461–466. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, X.F.; Zheng, P.S. Grape seed proanthocyanidins (GSPs) inhibit the growth of cervical cancer by inducing apoptosis mediated by the mitochondrial pathway. PLoS ONE 2014, 9, e107045. [Google Scholar] [CrossRef]
- Ravindranathan, P.; Pasham, D.; Balaji, U.; Cardenas, J.; Gu, J.; Toden, S.; Goel, A. Mechanistic insights into anticancer properties of oligomeric proanthocyanidins from grape seeds in colorectal cancer. Carcinogenesis 2018, 39, 767–777. [Google Scholar] [CrossRef]
- Chen, M.; Yu, S. Lipophilic Grape seed proanthocyanidin exerts anti-proliferative and pro-apoptotic effects on PC3 human prostate cancer cells and suppresses PC3 xenograft tumor growth in vivo. J. Agric. Food Chem. 2019, 67, 229–235. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Huang, W.; Zhan, J. Grape Seed Proanthocyanidins Induce Autophagy and Modulate Survivin in HepG2 Cells and Inhibit Xenograft Tumor Growth in Vivo. Nutrients 2019, 11, 2983. https://doi.org/10.3390/nu11122983
Wang L, Huang W, Zhan J. Grape Seed Proanthocyanidins Induce Autophagy and Modulate Survivin in HepG2 Cells and Inhibit Xenograft Tumor Growth in Vivo. Nutrients. 2019; 11(12):2983. https://doi.org/10.3390/nu11122983
Chicago/Turabian StyleWang, Lihua, Weidong Huang, and Jicheng Zhan. 2019. "Grape Seed Proanthocyanidins Induce Autophagy and Modulate Survivin in HepG2 Cells and Inhibit Xenograft Tumor Growth in Vivo" Nutrients 11, no. 12: 2983. https://doi.org/10.3390/nu11122983
APA StyleWang, L., Huang, W., & Zhan, J. (2019). Grape Seed Proanthocyanidins Induce Autophagy and Modulate Survivin in HepG2 Cells and Inhibit Xenograft Tumor Growth in Vivo. Nutrients, 11(12), 2983. https://doi.org/10.3390/nu11122983