Hormonal Therapy Resistance and Breast Cancer: Involvement of Adipocytes and Leptin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study of Breast Cancer Cells
2.1.1. Cell Culture
2.1.2. Influence of Adipose Secretome
2.1.3. Evaluation of Cell Cell Interactions
2.1.4. Annexin V–FITC/PI Apoptosis Assay
2.1.5. Quantitative Real-Time PCR (qPCR) Assays
2.2. Study of Cancer Stem Cells (CSCs)
2.2.1. Mammosphere Formation
2.2.2. Staining of Side-Population Cells (SP) by Flow Cytometry
2.2.3. Phenotyping of SP Cells
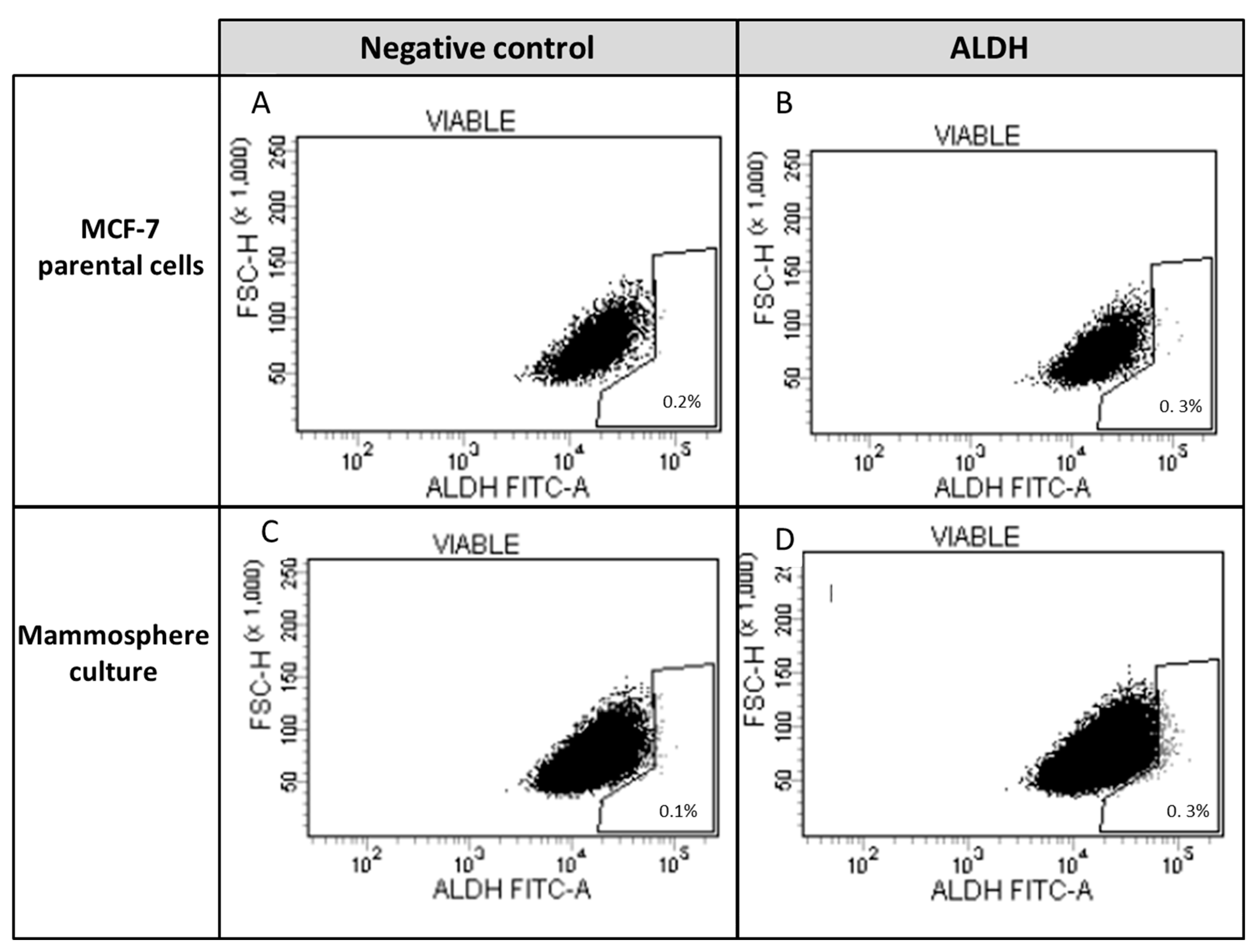
2.2.4. Identification of ALDH-1-Expressing Cells
2.2.5. Cytometry Analysis, Cell Sorting
2.2.6. Proliferation
2.3. Statistics
3. Results
3.1. Study of Breast Cancer Cells
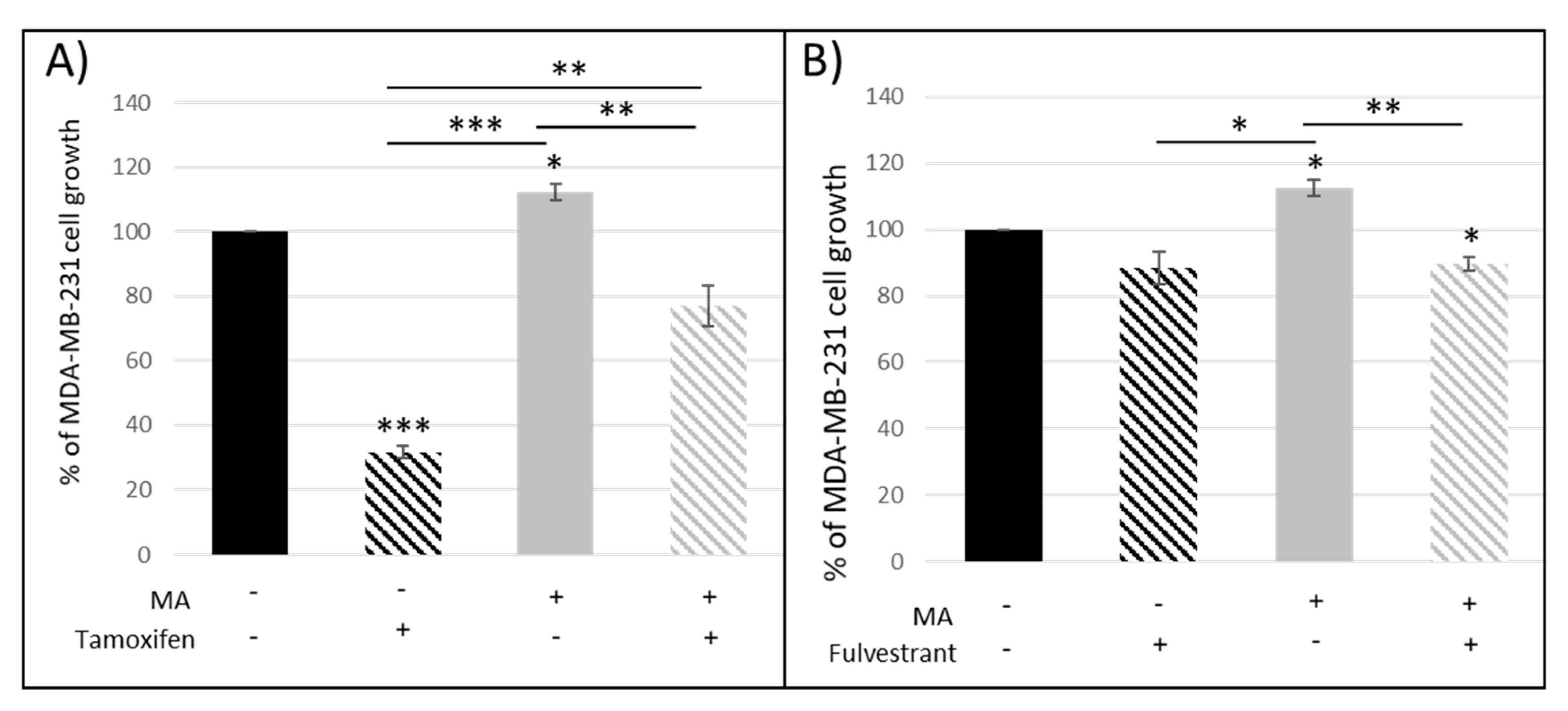
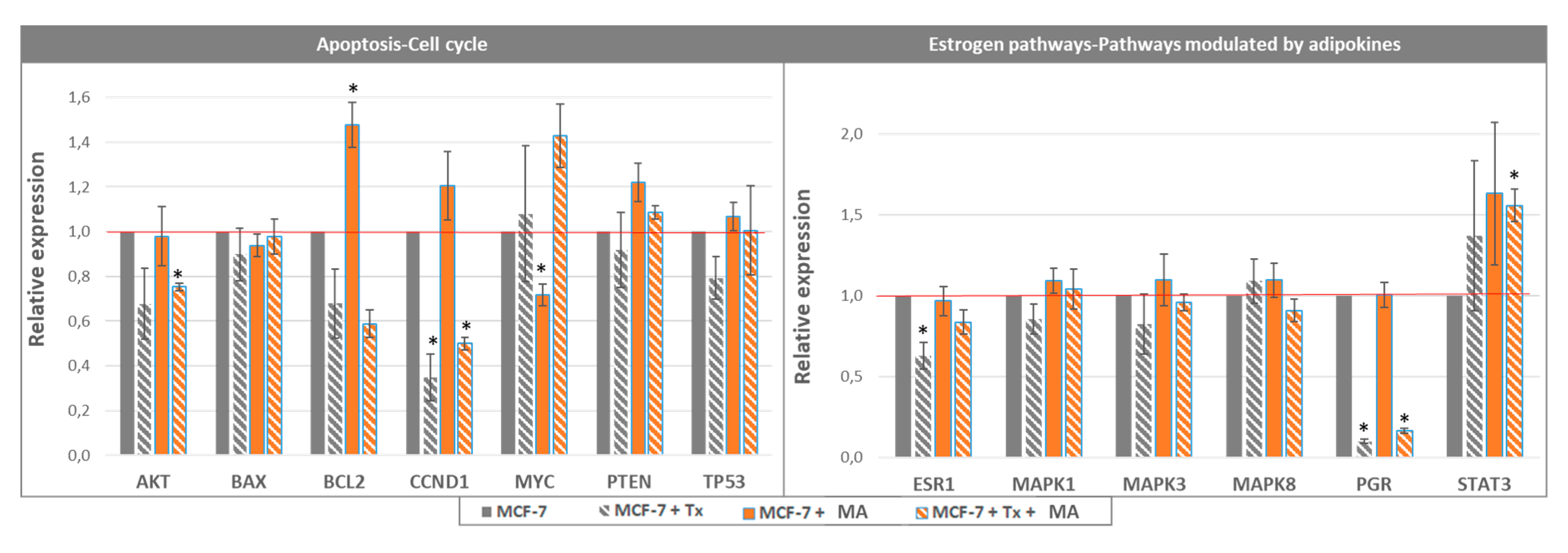
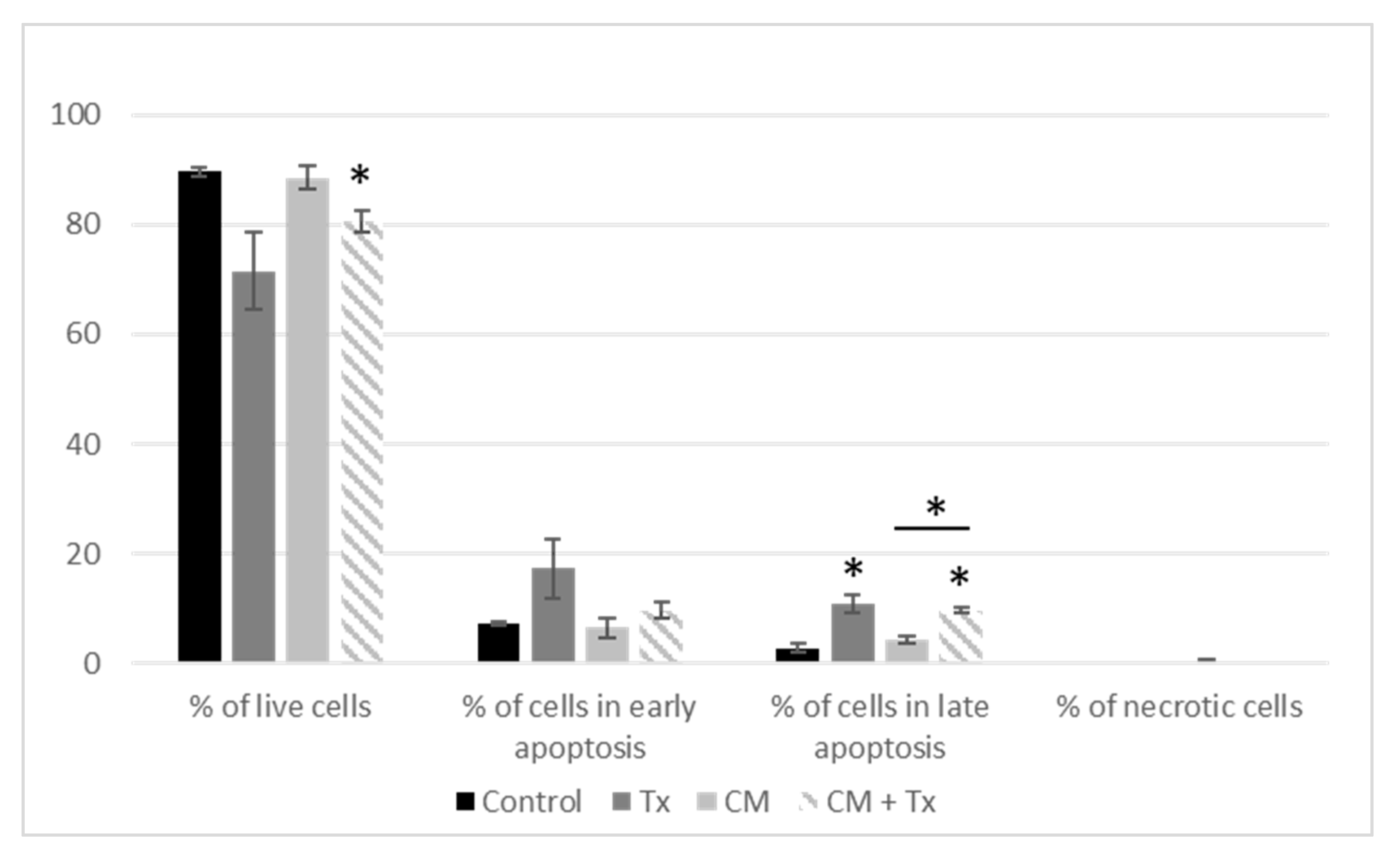
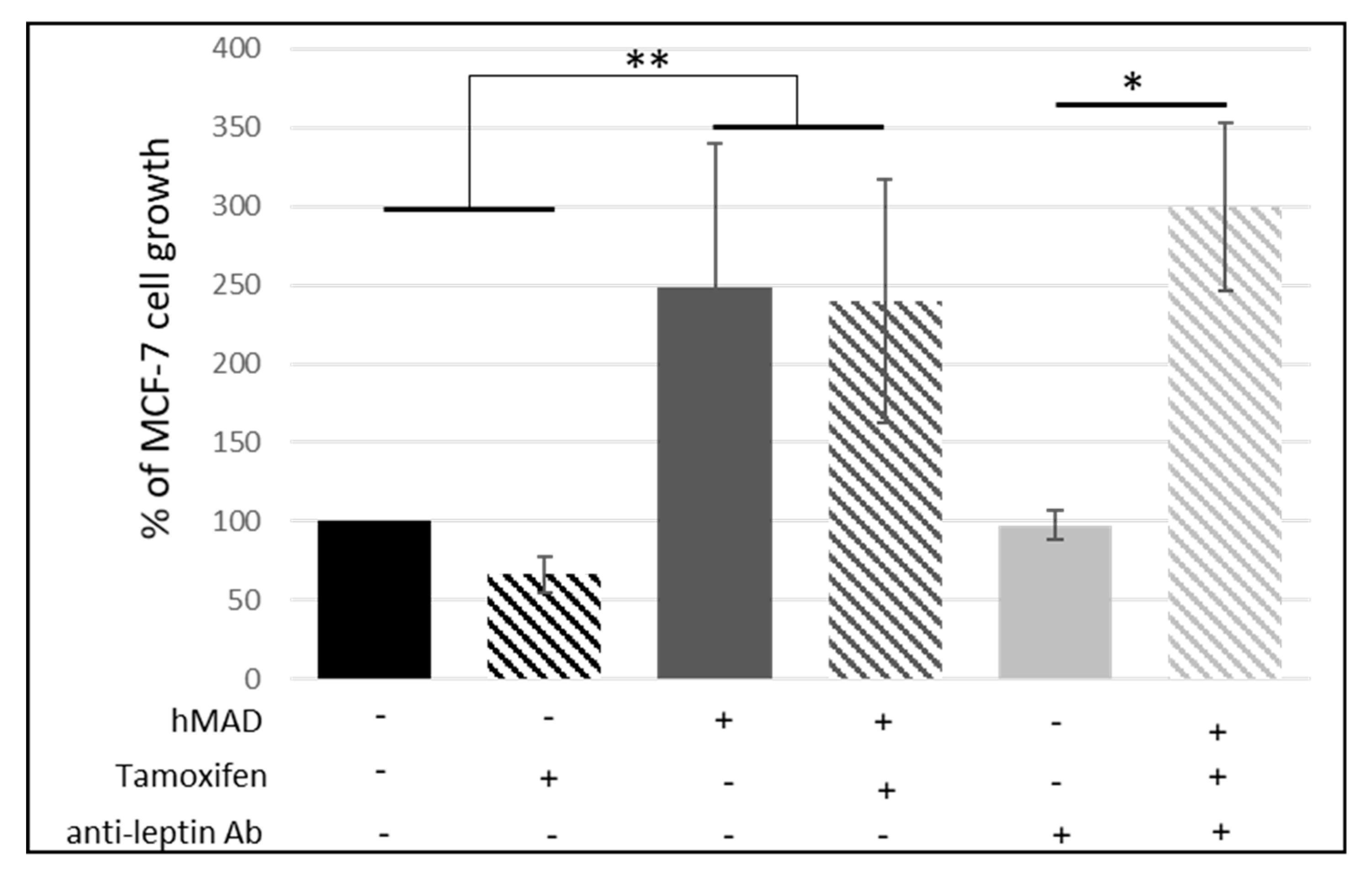
3.1.1. Crosstalk between Mammary Cancer Cells and Adipocytes Is a Key Element in the Resistance of Antiestrogen Therapy and Can Be Mediated by the Indirect Estrogen Pathway
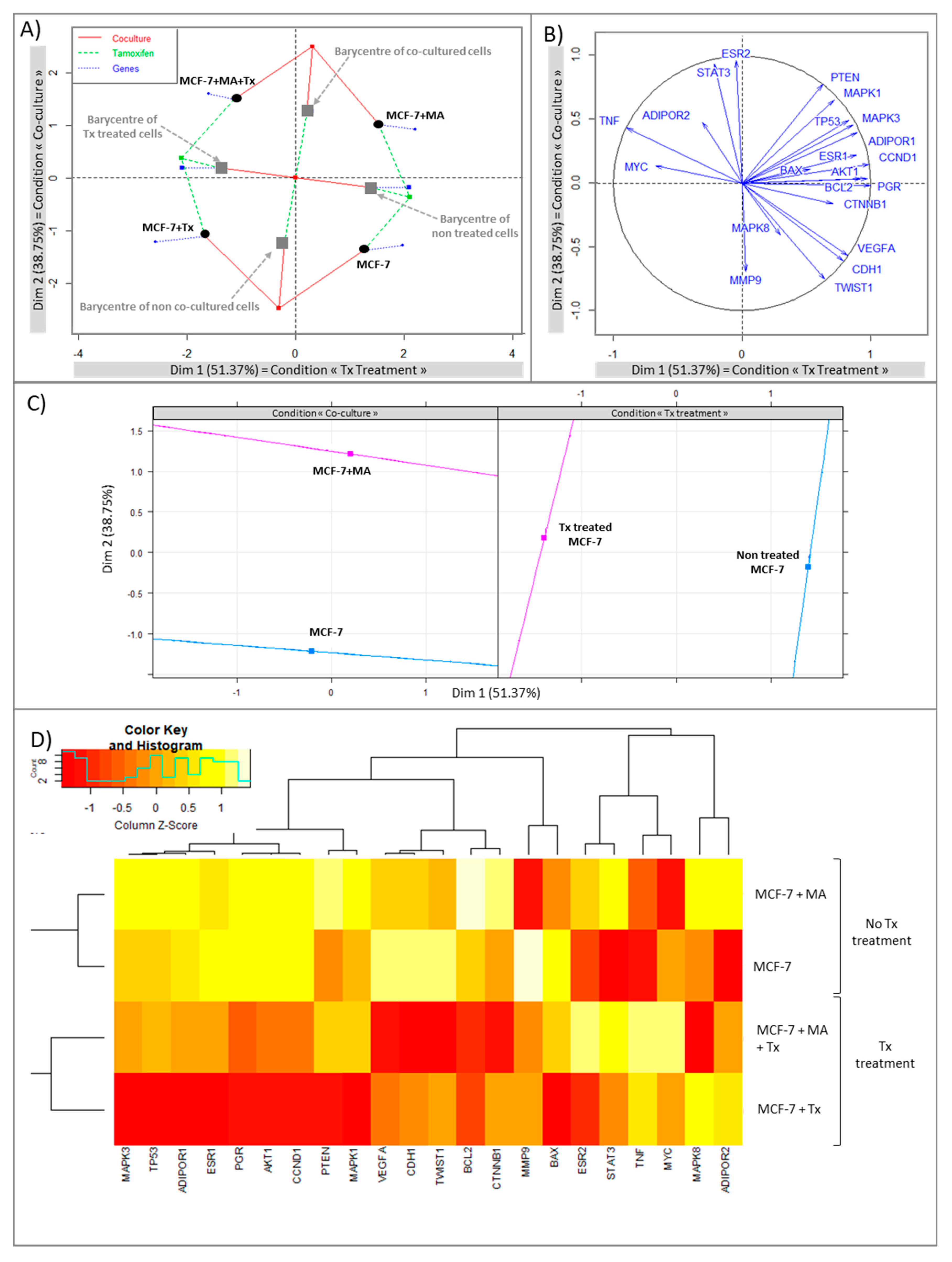
3.1.2. STAT3 Could Be a Target in Tx Resistance in the Presence of an Adipose Microenvironment
3.1.3. Leptin Does Not Seem to Be Involved in the Reduction of the Antiproliferative Effect of Tamoxifen
3.2. Study of Cancer Stem Cells (CSCs)
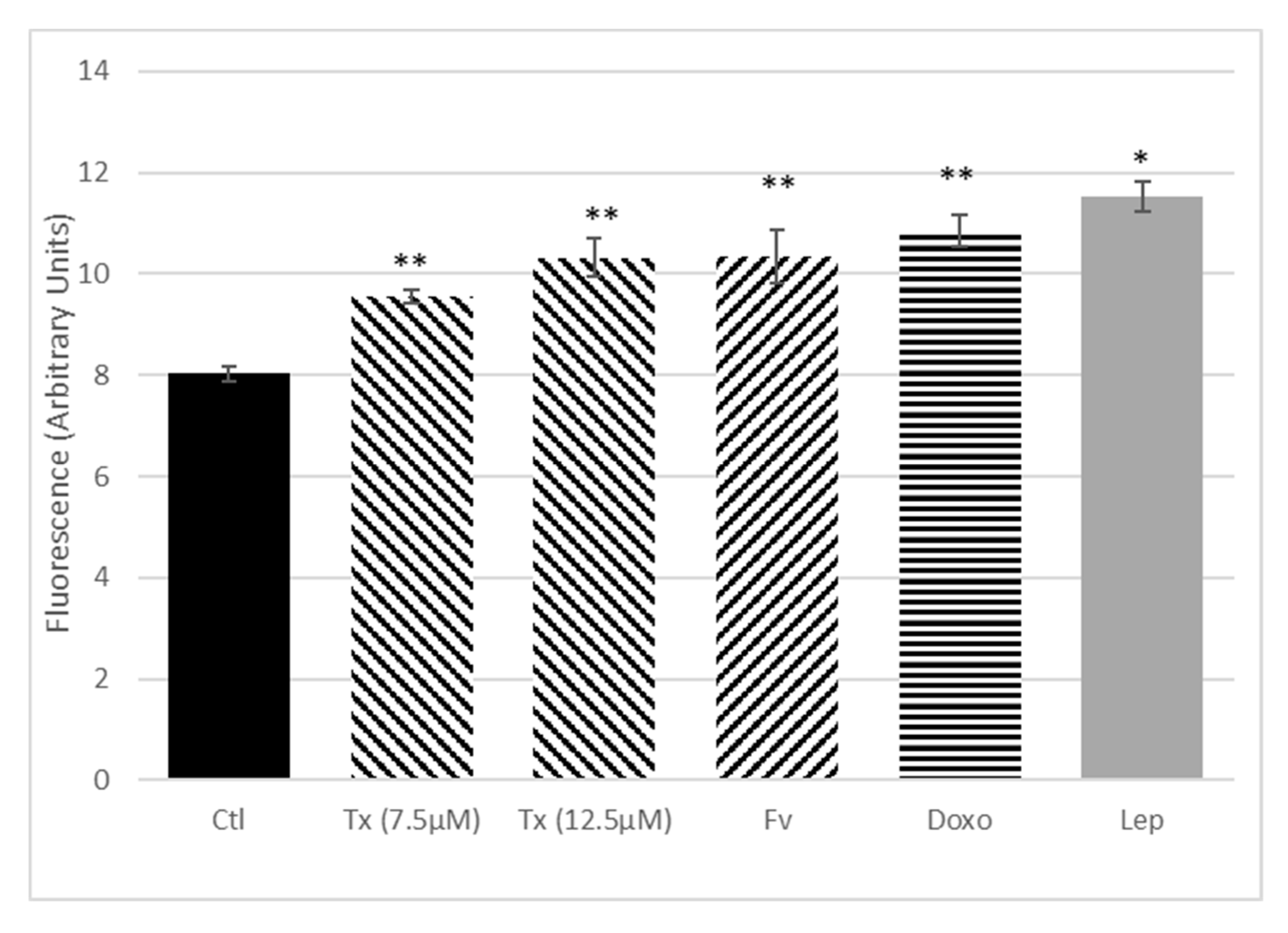
Anticancer Treatment and Adipokines Increase the Proliferation of Isolated SP Cells
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CM | Conditioned media |
| ER | Estrogen receptor |
| Fv | Fulvestrant |
| MA | Mature adipocyte |
| Tx | Tamoxifen |
References
- World Cancer Research Fund (WCRF) Diet and Cancer. Available online: https://www.wcrf.org/dietandcancer (accessed on 6 November 2018).
- Picon-Ruiz, M.; Morata-Tarifa, C.; Valle-Goffin, J.J.; Friedman, E.R.; Slingerland, J.M. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA Cancer J. Clin. 2017, 67, 378–397. [Google Scholar] [CrossRef] [PubMed]
- Thivat, E.; Thérondel, S.; Lapirot, O.; Abrial, C.; Gimbergues, P.; Gadéa, E.; Planchat, E.; Kwiatkowski, F.; Mouret-Reynier, M.A.; Chollet, P.; et al. Weight change during chemotherapy changes the prognosis in non metastatic breast cancer for the worse. BMC Cancer 2010, 10, 648. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Liang, S.; Ghosh, S.; Hornsby, P.J.; Li, R. Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene 2009, 28, 2745–2755. [Google Scholar] [CrossRef] [PubMed]
- Place, A.E.; Jin Huh, S.; Polyak, K. The microenvironment in breast cancer progression: Biology and implications for treatment. Breast Cancer Res. 2011, 13, 227. [Google Scholar] [CrossRef]
- Wolters, R.; Schwentner, L.; Regierer, A.; Wischnewsky, M.; Kreienberg, R.; Wöckel, A. Endocrine therapy in obese patients with primary breast cancer: Another piece of evidence in an unfinished puzzle. Breast Cancer Res. Treat. 2012, 131, 925–931. [Google Scholar] [CrossRef]
- Litton, J.K.; Gonzalez-Angulo, A.M.; Warneke, C.L.; Buzdar, A.U.; Kau, S.-W.; Bondy, M.; Mahabir, S.; Hortobagyi, G.N.; Brewster, A.M. Relationship between obesity and pathologic response to neoadjuvant chemotherapy among women with operable breast cancer. J. Clin. Oncol. 2008, 26, 4072–4077. [Google Scholar] [CrossRef]
- Hayes, E.L.; Lewis-Wambi, J.S. Mechanisms of endocrine resistance in breast cancer: An overview of the proposed roles of noncoding RNA. Breast Cancer Res. 2015, 17, 40. [Google Scholar] [CrossRef]
- Riggins, R.B.; Schrecengost, R.S.; Guerrero, M.S.; Bouton, A.H. Pathways to tamoxifen resistance. Cancer Lett. 2007, 256, 1–24. [Google Scholar] [CrossRef]
- Lee, K.; Kruper, L.; Dieli-Conwright, C.M.; Mortimer, J.E. The Impact of Obesity on Breast Cancer Diagnosis and Treatment. Curr. Oncol. Rep. 2019, 21, 41. [Google Scholar] [CrossRef]
- Rondón-Lagos, M.; Villegas, V.E.; Rangel, N.; Sánchez, M.C.; Zaphiropoulos, P.G. Tamoxifen Resistance: Emerging Molecular Targets. Int. J. Mol. Sci. 2016, 17, 1357. [Google Scholar] [CrossRef]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.M.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Klonisch, T.; Wiechec, E.; Hombach-Klonisch, S.; Ande, S.R.; Wesselborg, S.; Schulze-Osthoff, K.; Los, M. Cancer stem cell markers in common cancers—Therapeutic implications. Trends Mol. Med. 2008, 14, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Raffo, D.; Berardi, D.E.; Pontiggia, O.; Todaro, L.; de Kier Joffé, E.B.; Simian, M. Tamoxifen selects for breast cancer cells with mammosphere forming capacity and increased growth rate. Breast Cancer Res. Treat. 2013, 142, 537–548. [Google Scholar] [CrossRef]
- Lequeux, C.; Auxenfans, C.; Mojallal, A.; Sergent, M.; Damour, O. Optimization of a culture medium for the differentiation of preadipocytes into adipocytes in a monolayer. Biomed. Mater. Eng. 2009, 19, 283–291. [Google Scholar]
- Goodell, M.A.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar] [CrossRef]
- Bougaret, L.; Delort, L.; Billard, H.; Le Huede, C.; Boby, C.; De la Foye, A.; Rossary, A.; Mojallal, A.; Damour, O.; Auxenfans, C.; et al. Adipocyte/breast cancer cell crosstalk in obesity interferes with the anti-proliferative efficacy of tamoxifen. PLoS ONE 2018, 13, e0191571. [Google Scholar] [CrossRef]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- Caldefie-Chézet, F.; Dubois, V.; Delort, L.; Rossary, A.; Vasson, M.-P. Leptin: Involvement in the pathophysiology of breast cancer. Ann. Endocrinol. 2013, 74, 90–101. [Google Scholar] [CrossRef]
- Delort, L.; Rossary, A.; Farges, M.-C.; Vasson, M.-P.; Caldefie-Chézet, F. Leptin, adipocytes and breast cancer: Focus on inflammation and anti-tumor immunity. Life Sci. 2015, 140, 37–48. [Google Scholar] [CrossRef]
- Dubois, V.; Jarde, T.; Delort, L.; Billard, H.; Bernard-Gallon, D.J.; Berger, E.; Geloen, A.; Vasson, M.P.; Caldefie-Chezet, F. Leptin induces a proliferative response in breast cancer cells but not in normal breast cells. Nutr. Cancer 2014, 66, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Delort, L.; Billard, H.; Vasson, M.-P.; Caldefie-Chezet, F. Breast cancer and obesity: In vitro interferences between adipokines and proangiogenic features and/or antitumor therapies? PLoS ONE 2013, 8, e58541. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Deng, L.-L.; Cui, J.-Q.; Shi, L.; Yang, Y.-C.; Luo, J.-H.; Qin, D.; Wang, L. Association between serum leptin levels and breast cancer risk: An updated systematic review and meta-analysis. Medicine 2018, 97, e11345. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Gyamfi, J.; Lee, Y.-H.; Eom, M.; Choi, J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci. Rep. 2018, 8, 8859. [Google Scholar] [CrossRef]
- Celis, J.E.; Moreira, J.M.; Cabezon, T.; Gromov, P.; Friis, E.; Rank, F.; Gromova, I. Identification of extracellular and intracellular signaling components of the mammary adipose tissue and its interstitial fluid in high risk breast cancer patients: Toward dissecting the molecular circuitry of epithelial-adipocyte stromal cell interactions. Mol. Cell Proteom. 2005, 4, 492–522. [Google Scholar] [CrossRef]
- Pistone Creydt, V.; Fletcher, S.J.; Giudice, J.; Bruzzone, A.; Chasseing, N.A.; Gonzalez, E.G.; Sacca, P.A.; Calvo, J.C. Human adipose tissue from normal and tumoral breast regulates the behavior of mammary epithelial cells. Clin. Transl. Oncol. 2013, 15, 124–131. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Lehuédé, C.; Laurent, V.; Dirat, B.; Dauvillier, S.; Bochet, L.; Le Gonidec, S.; Escourrou, G.; Valet, P.; Muller, C. Adipose tissue and breast epithelial cells: A dangerous dynamic duo in breast cancer. Cancer Lett. 2012, 324, 142–151. [Google Scholar] [CrossRef]
- Fletcher, S.J.; Sacca, P.A.; Pistone-Creydt, M.; Coló, F.A.; Serra, M.F.; Santino, F.E.; Sasso, C.V.; Lopez-Fontana, C.M.; Carón, R.W.; Calvo, J.C.; et al. Human breast adipose tissue: Characterization of factors that change during tumor progression in human breast cancer. J. Exp. Clin. Cancer Res. 2017, 36, 26. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Scherer, P.E. Leptin and cancer: From cancer stem cells to metastasis. Endocr. Relat. Cancer 2011, 18, C25–C29. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.E.; Chen, C.; Punj, V.; Tsukamoto, H.; Machida, K. Pluripotency factor-mediated expression of the leptin receptor (OB-R) links obesity to oncogenesis through tumor-initiating stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 829–834. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delort, L.; Bougaret, L.; Cholet, J.; Vermerie, M.; Billard, H.; Decombat, C.; Bourgne, C.; Berger, M.; Dumontet, C.; Caldefie-Chezet, F. Hormonal Therapy Resistance and Breast Cancer: Involvement of Adipocytes and Leptin. Nutrients 2019, 11, 2839. https://doi.org/10.3390/nu11122839
Delort L, Bougaret L, Cholet J, Vermerie M, Billard H, Decombat C, Bourgne C, Berger M, Dumontet C, Caldefie-Chezet F. Hormonal Therapy Resistance and Breast Cancer: Involvement of Adipocytes and Leptin. Nutrients. 2019; 11(12):2839. https://doi.org/10.3390/nu11122839
Chicago/Turabian StyleDelort, Laetitia, Lauriane Bougaret, Juliette Cholet, Marion Vermerie, Hermine Billard, Caroline Decombat, Céline Bourgne, Marc Berger, Charles Dumontet, and Florence Caldefie-Chezet. 2019. "Hormonal Therapy Resistance and Breast Cancer: Involvement of Adipocytes and Leptin" Nutrients 11, no. 12: 2839. https://doi.org/10.3390/nu11122839
APA StyleDelort, L., Bougaret, L., Cholet, J., Vermerie, M., Billard, H., Decombat, C., Bourgne, C., Berger, M., Dumontet, C., & Caldefie-Chezet, F. (2019). Hormonal Therapy Resistance and Breast Cancer: Involvement of Adipocytes and Leptin. Nutrients, 11(12), 2839. https://doi.org/10.3390/nu11122839