ER-Negative Breast Cancer Is Highly Responsive to Cholesterol Metabolite Signalling

 and
and 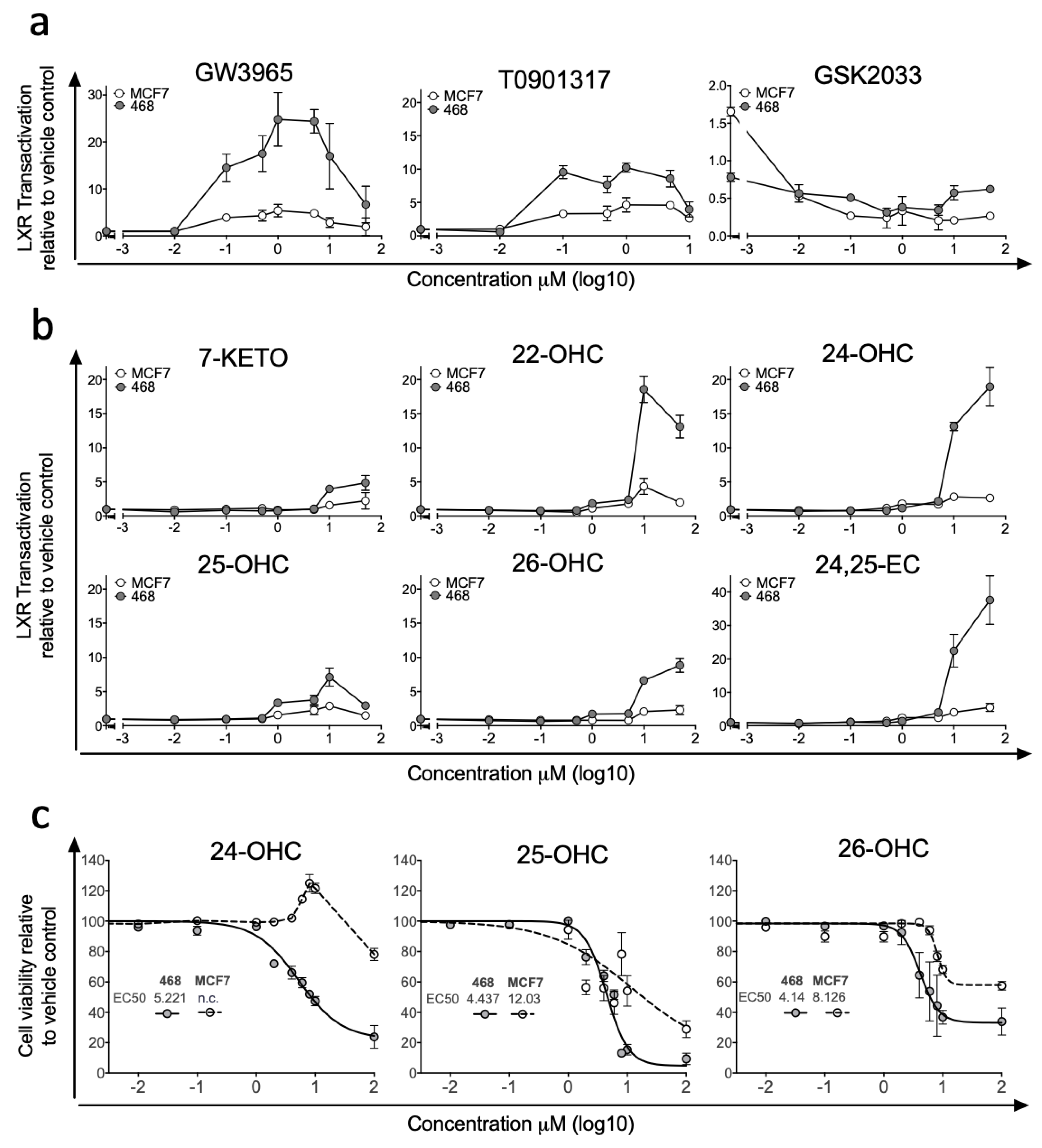{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfections
2.2. Drugs and Reagents
2.3. Luciferase Reporter Assay
2.4. qPCR
2.5. MTT Assays
2.6. The Cancer Genome Atlas Gene Expression Analysis
2.7. Transcription Factor-Target Gene Correlation Analysis
3. Results
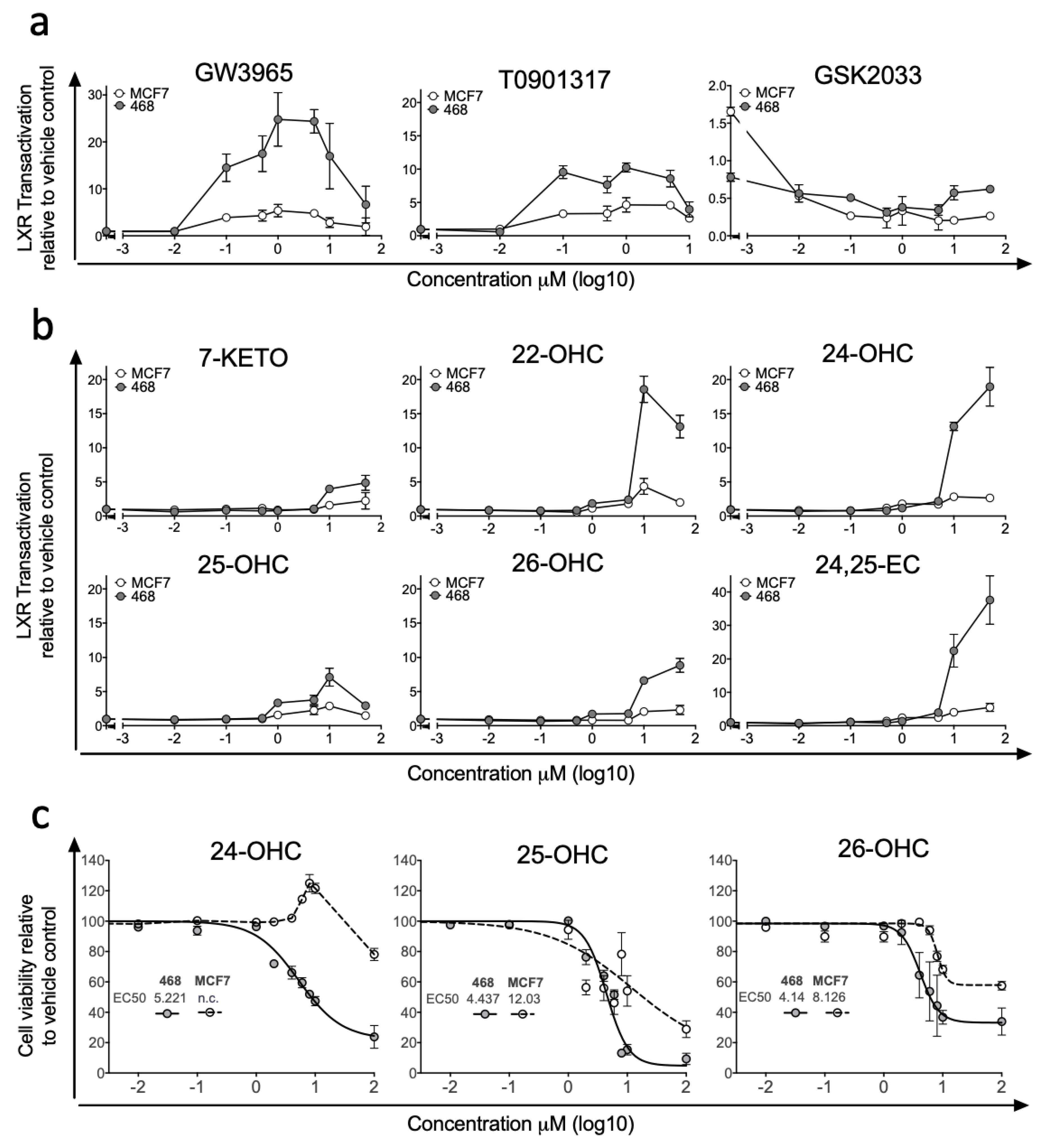
3.1. LXR Activation Potential Is Retained in ER-Negative Disease but Dampened in ER-Positive Disease
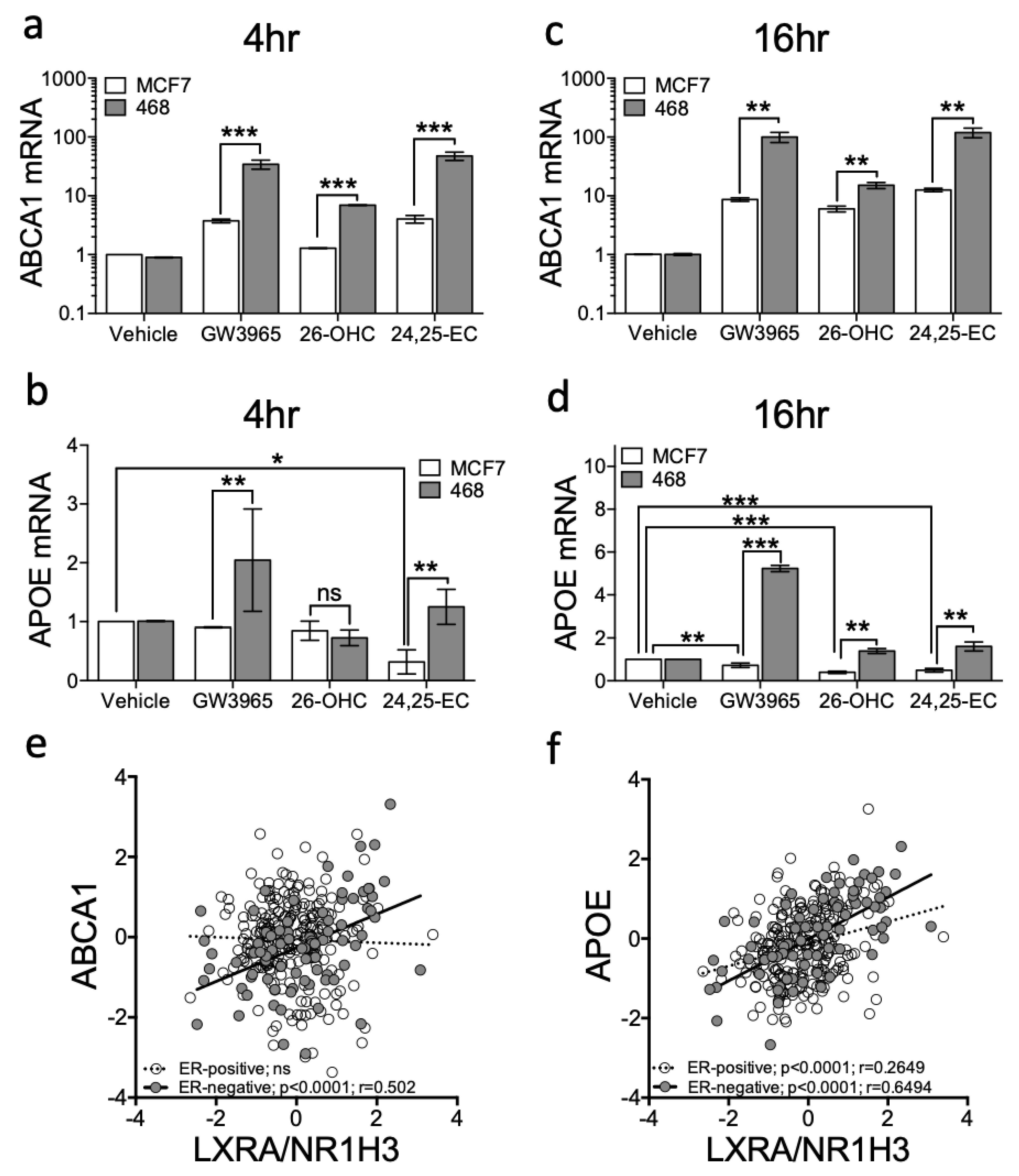
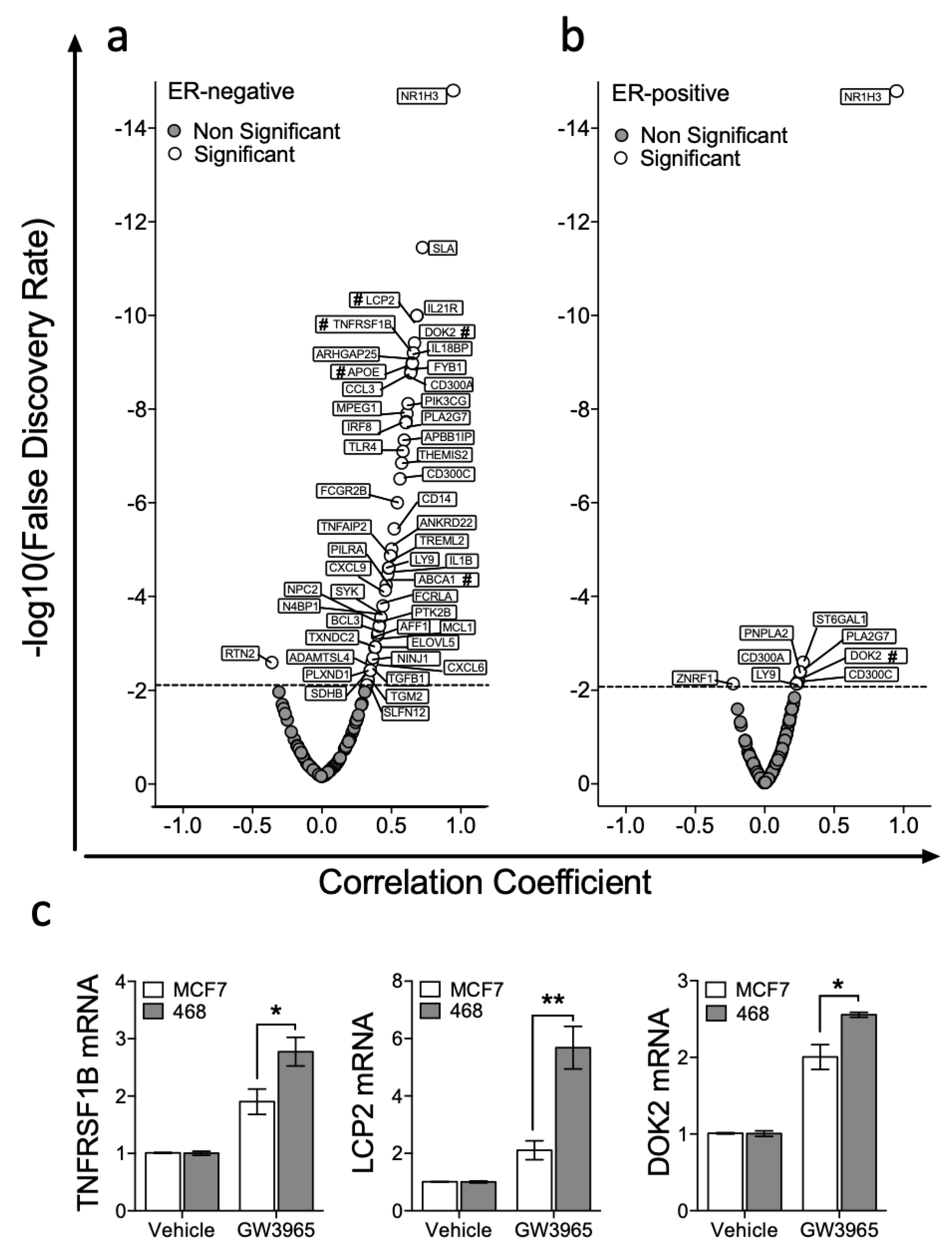
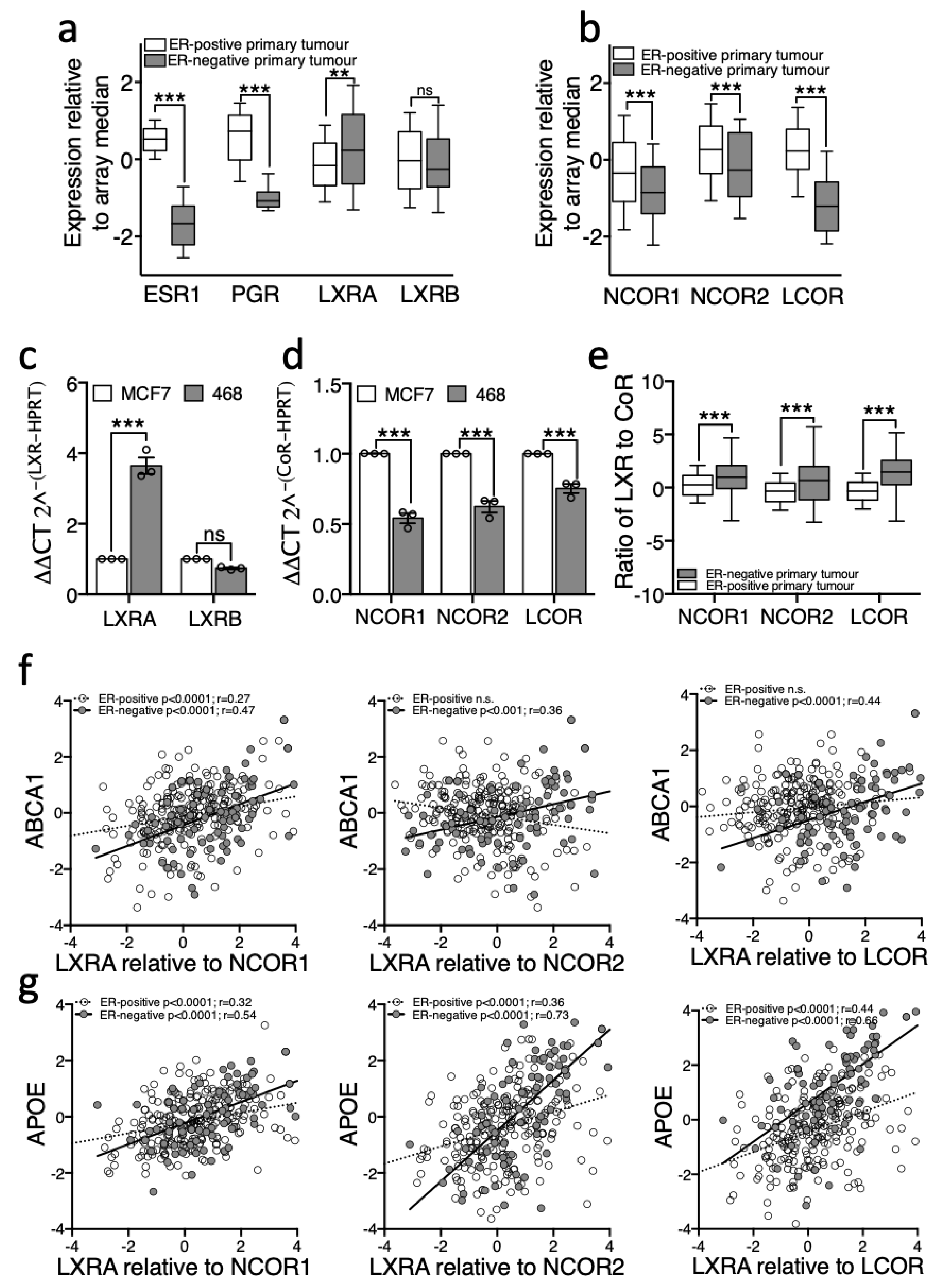
3.2. Expression of LXRA Correlates with Expression of Its Target Genes in Primary ER-Negative Tumours but not in ER-Positive Tumours
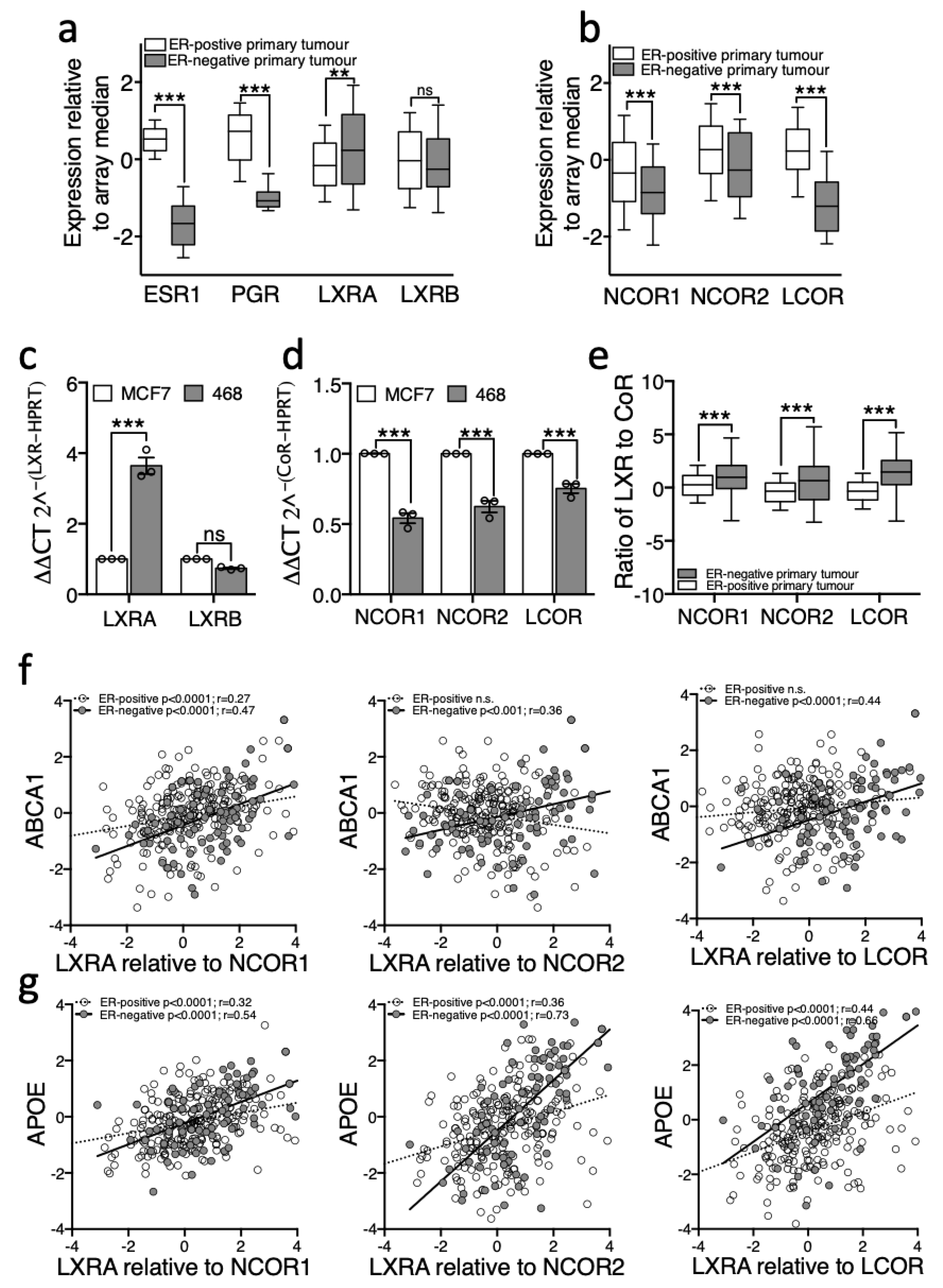
3.3. LXR Is Poised for Transcription in ER-Negative BCa but Repressed in ER-Positive BCa
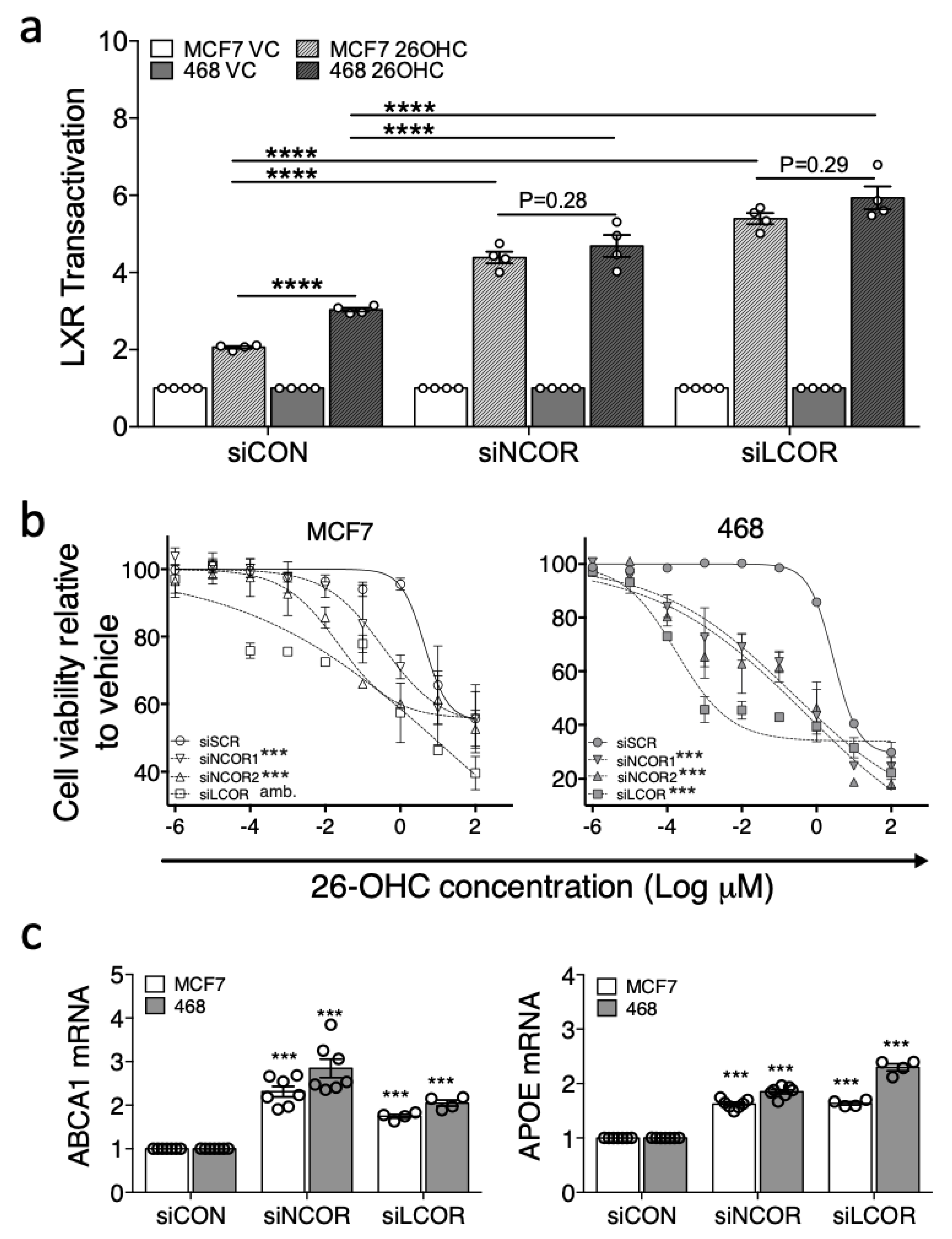
3.4. Removal of Corepressors Equalizes the Response of ER-Negative and ER-Positive Cell Lines to Ligand
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; Kliewer, S.A.; Moore, L.B.; Smith-Oliver, T.A.; Oliver, B.B.; Su, J.L.; Sundseth, S.S.; Winegar, D.A.; Blanchard, D.E.; Spencer, T.A.; et al. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem. 1997, 272, 3137–3140. [Google Scholar] [CrossRef] [PubMed]
- Stiles, A.R.; Kozlitina, J.; Thompson, B.M.; McDonald, J.G.; King, K.S.; Russell, D.W. Genetic, anatomic, and clinical determinants of human serum sterol and vitamin D levels. Proc. Natl. Acad. Sci. USA 2014, 111, E4006–E4014. [Google Scholar] [CrossRef]
- Wu, Q.; Ishikawa, T.; Sirianni, R.; Tang, H.; McDonald, J.G.; Yuhanna, I.S.; Thompson, B.; Girard, L.; Mineo, C.; Brekken, R.A.; et al. 27-Hydroxycholesterol Promotes Cell-Autonomous, ER-Positive Breast Cancer Growth. Cell Rep. 2013, 5, 637–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakheri, R.J.; Javitt, N.B. 27-Hydroxycholesterol, does it exist? On the nomenclature and stereochemistry of 26-hydroxylated sterols. Steroids 2012, 77, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271. [Google Scholar] [CrossRef]
- Solheim, S.; Hutchinson, S.A.; Lundanes, E.; Wilson, S.R.; Thorne, J.L.; Roberg-Larsen, H. Fast liquid chromatography-mass spectrometry reveals side chain oxysterol heterogeneity in breast cancer tumour samples. J. Steroid Biochem. Mol. Biol. 2019, 192, 105309. [Google Scholar] [CrossRef]
- Thorne, J.L.; Campbell, M.J. Nuclear receptors and the Warburg effect in cancer. Int. J. Cancer 2015, 137, 1519–1527. [Google Scholar] [CrossRef]
- Doig, C.L.; Singh, P.K.; Dhiman, V.K.; Thorne, J.L.; Battaglia, S.; Sobolewski, M.; Maguire, O.; O’Neill, L.P.; Turner, B.M.; McCabe, C.J.; et al. Recruitment of NCOR1 to VDR target genes is enhanced in prostate cancer cells and associates with altered DNA methylation patterns. Carcinogenesis 2012, 34, 248–256. [Google Scholar] [CrossRef]
- Battaglia, S.; Maguire, O.; Thorne, J.L.; Hornung, L.B.; Doig, C.L.; Liu, S.; Sucheston, L.E.; Bianchi, A.; Khanim, F.L.; Gommersall, L.M.; et al. Elevated NCOR1 disrupts PPARalpha/gamma signaling in prostate cancer and forms a targetable epigenetic lesion. Carcinogenesis 2010, 31, 1650–1660. [Google Scholar] [CrossRef]
- Abedin, S.A.; Thorne, J.L.; Battaglia, S.; Maguire, O.; Hornung, L.B.; Doherty, A.P.; Mills, I.G.; Campbell, M.J. Elevated NCOR1 disrupts a network of dietary-sensing nuclear receptors in bladder cancer cells. Carcinogenesis 2009, 30, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Li, S.; Wu, J.; Xia, C.; Lala, D.S. Liver X receptors interact with corepressors to regulate gene expression. Mol. Endocrinol. 2003, 17, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Long, M.D.; Thorne, J.L.; Russell, J.; Battaglia, S.; Singh, P.K.; Sucheston-Campbell, L.E.; Campbell, M.J. Cooperative behavior of the nuclear receptor superfamily and its deregulation in prostate cancer. Carcinogenesis 2014, 35, 262–271. [Google Scholar] [CrossRef]
- Pencheva, N.; Buss, C.G.; Posada, J.; Merghoub, T.; Tavazoie, S.F. Broad-Spectrum Therapeutic Suppression of Metastatic Melanoma through Nuclear Hormone Receptor Activation. Cell 2014, 156, 986–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Sun, L.; Yang, X.; Ma, X.; Li, Q.; Chen, Y.; Liu, Y.; Zhang, D.; Li, X.; Xiang, R.; et al. Activation of liver X receptor inhibits the development of pulmonary carcinomas induced by 3-methylcholanthrene and butylated hydroxytoluene in BALB/c mice. Sci. Rep. 2016, 6, 27295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, E.R.; Wardell, S.E.; Jasper, J.S.; Park, S.; Suchindran, S.; Howe, M.K.; Carver, N.J.; Pillai, R.V.; Sullivan, P.M.; Sondhi, V.; et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 2013, 342, 1094–1098. [Google Scholar] [CrossRef]
- Baek, A.E.; Yu, Y.R.A.; He, S.S.; Wardell, S.E.; Chang, C.Y.; Kwon, S.; Pillai, R.V.; McDowell, H.B.; Thompson, J.W.; Dubois, L.G.; et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat. Commun. 2017, 8, 864. [Google Scholar] [CrossRef]
- Dalenc, F.; Iuliano, L.; Filleron, T.; Zerbinati, C.; Voisin, M.; Arellano, C.; Chatelut, E.; Marquet, P.; Samadi, M.; Roche, H.; et al. Circulating oxysterol metabolites as potential new surrogate markers in patients with hormone receptor-positive breast cancer: Results of the OXYTAM study. J. Steroid Biochem. Mol. Biol. 2016, 169, 210–218. [Google Scholar] [CrossRef]
- Hutchinson, S.A.; Lianto, P.; Moore, J.B.; Hughes, T.A.; Thorne, J.L. Phytosterols Inhibit Side-Chain Oxysterol Mediated Activation of LXR in Breast Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3241. [Google Scholar] [CrossRef]
- Kim, B.; Stephen, S.L.; Hanby, A.M.; Horgan, K.; Perry, S.L.; Richardson, J.; Roundhill, E.A.; Valleley, E.M.; Verghese, E.T.; Williams, B.J.; et al. Chemotherapy induces Notch1-dependent MRP1 up-regulation, inhibition of which sensitizes breast cancer cells to chemotherapy. BMC Cancer 2015, 15, 634. [Google Scholar] [CrossRef]
- Thorne, J.L.; Battaglia, S.; Baxter, D.E.; Hayes, J.L.; Hutchinson, S.A.; Jana, S.; Millican-Slater, R.A.; Smith, L.; Teske, M.C.; Wastall, L.M.; et al. MiR-19b non-canonical binding is directed by HuR and confers chemosensitivity through regulation of P-glycoprotein in breast cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Broekema, M.F.; Hollman, D.A.A.; Koppen, A.; van den Ham, H.J.; Melchers, D.; Pijnenburg, D.; Ruijtenbeek, R.; van Mil, S.W.C.; Houtman, R.; Kalkhoven, E. Profiling of 3696 Nuclear Receptor-Coregulator Interactions: A Resource for Biological and Clinical Discovery. Endocrinology 2018, 159, 2397–2407. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.C.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Long, M.D.; Campbell, M.J. Integrative genomic approaches to dissect clinically-significant relationships between the VDR cistrome and gene expression in primary colon cancer. J. Steroid Biochem. Mol. Biol. 2017, 173, 130–138. [Google Scholar] [CrossRef]
- Gross, A.M.; Kreisberg, J.F.; Ideker, T. Analysis of Matched Tumor and Normal Profiles Reveals Common Transcriptional and Epigenetic Signals Shared across Cancer Types. PLoS ONE 2015, 10, e0142618. [Google Scholar] [CrossRef]
- Liu, T.; Ortiz, J.A.; Taing, L.; Meyer, C.A.; Lee, B.; Zhang, Y.; Shin, H.; Wong, S.S.; Ma, J.; Lei, Y.; et al. Cistrome: An integrative platform for transcriptional regulation studies. Genome Biol. 2011, 12, R83. [Google Scholar] [CrossRef]
- Oishi, Y.; Spann, N.J.; Link, V.M.; Muse, E.D.; Strid, T.; Edillor, C.; Kolar, M.J.; Matsuzaka, T.; Hayakawa, S.; Tao, J.; et al. SREBP1 Contributes to Resolution of Pro-inflammatory TLR4 Signaling by Reprogramming Fatty Acid Metabolism. Cell Metab. 2017, 25, 412–427. [Google Scholar] [CrossRef]
- Savic, D.; Ramaker, R.C.; Roberts, B.S.; Dean, E.C.; Burwell, T.C.; Meadows, S.K.; Cooper, S.J.; Garabedian, M.J.; Gertz, J.; Myers, R.M. Distinct gene regulatory programs define the inhibitory effects of liver X receptors and PPARG on cancer cell proliferation. Genome Med. 2016, 8, 74. [Google Scholar] [CrossRef]
- Galhardo, M.; Sinkkonen, L.; Berninger, P.; Lin, J.; Sauter, T.; Heinaniemi, M. Integrated analysis of transcript-level regulation of metabolism reveals disease-relevant nodes of the human metabolic network. Nucleic Acids Res. 2014, 42, 1474–1496. [Google Scholar] [CrossRef] [PubMed]
- Vedin, L.L.; Lewandowski, S.A.; Parini, P.; Gustafsson, J.A.; Steffensen, K.R. The oxysterol receptor LXR inhibits proliferation of human breast cancer cells. Carcinogenesis 2009, 30, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Bełtowski, J. Liver X Receptors (LXR) as Therapeutic Targets in Dyslipidemia. Cardiovascular Therapeutics 2008, 26, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Zuercher, W.J.; Buckholz, R.G.; Campobasso, N.; Collins, J.L.; Galardi, C.M.; Gampe, R.T.; Hyatt, S.M.; Merrihew, S.L.; Moore, J.T.; Oplinger, J.A.; et al. Discovery of Tertiary Sulfonamides as Potent Liver X Receptor Antagonists. J. Med. Chem. 2010, 53, 3412–3416. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Quinn, C.M.; Brown, A.J. SREBP-2 positively regulates transcription of the cholesterol efflux gene, ABCA1, by generating oxysterol ligands for LXR. Biochem. J. 2006, 400, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Laffitte, B.A.; Repa, J.J.; Joseph, S.B.; Wilpitz, D.C.; Kast, H.R.; Mangelsdorf, D.J.; Tontonoz, P. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 507–512. [Google Scholar] [CrossRef]
- Jalaguier, S.; Teyssier, C.; Nait Achour, T.; Lucas, A.; Bonnet, S.; Rodriguez, C.; Elarouci, N.; Lapierre, M.; Cavailles, V. Complex regulation of LCoR signaling in breast cancer cells. Oncogene 2017, 36, 4790–4801. [Google Scholar] [CrossRef]
- Heiser, L.M.; Sadanandam, A.; Kuo, W.L.; Benz, S.C.; Goldstein, T.C.; Ng, S.; Gibb, W.J.; Wang, N.J.; Ziyad, S.; Tong, F.; et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2724–2729. [Google Scholar] [CrossRef]
- Manthravadi, S.; Shrestha, A.; Madhusudhana, S. Impact of statin use on cancer recurrence and mortality in breast cancer: A systematic review and meta-analysis. Int. J. Cancer 2016, 139, 1281–1288. [Google Scholar] [CrossRef]
- Liu, B.; Yi, Z.; Guan, X.; Zeng, Y.X.; Ma, F. The relationship between statins and breast cancer prognosis varies by statin type and exposure time: A meta-analysis. Breast Cancer Res. Treat. 2017, 164, 1–11. [Google Scholar] [CrossRef]
- Link, L.B.; Canchola, A.J.; Bernstein, L.; Clarke, C.A.; Stram, D.O.; Ursin, G.; Horn-Ross, P.L. Dietary patterns and breast cancer risk in the California Teachers Study cohort. Am. J. Clin. Nutr. 2013, 98, 1524–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, E.; Hill, A.D.K.; Kelly, G.; McDermott, E.W.; O’Higgins, N.J.; Buggy, Y.; Young, L.S. Associations and interactions between Ets-1 and Ets-2 and coregulatory proteins, SRC-1, AIB1, and NCoR in breast cancer. Clin. Cancer Res. 2005, 11, 2111–2122. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.Q.; Hu, X.B.; Zhou, J.M.; Sun, J.J.; Zhu, A.Z.; Xu, X.F.; Zheng, H.; Gao, X.; Wang, X.; Jin, H.C.; et al. COPS5 amplification and overexpression confers tamoxifen-resistance in ER alpha-positive breast cancer by degradation of NCoR. Nat. Commun. 2016, 7, 12044. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Bin Hafeez, B.; Siddiqui, I.A.; Gerlach, C.; Patz, M.; Mukhtar, H.; Baniahmad, A. Ligand-dependent Corepressor Acts as a Novel Androgen Receptor Corepressor, Inhibits Prostate Cancer Growth, and Is Functionally Inactivated by the Src Protein Kinase. J. Biol. Chem. 2011, 286, 37108–37117. [Google Scholar] [CrossRef] [PubMed]
- Sixou, S.; Muller, K.; Jalaguier, S.; Kuhn, C.; Harbeck, N.; Mayr, D.; Engel, J.; Jeschke, U.; Ditsch, N.; Cavailles, V. Importance of RIP140 and LCoR Sub-Cellular Localization for Their Association With Breast Cancer Aggressiveness and Patient Survival. Transl. Oncol. 2018, 11, 1090–1096. [Google Scholar] [CrossRef]
- Chu, S.; Wang, H.; Yu, M. A putative molecular network associated with colon cancer metastasis constructed from microarray data. World J. Surg. Oncol. 2017, 15, 115. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Dong, L.; Gong, F.; Gu, Y.; Zhang, H.; Fan, D.; Sun, Z. Inflammatory genes are novel prognostic biomarkers for colorectal cancer. Int. J. Mol. Med. 2018, 42, 368–380. [Google Scholar] [CrossRef]
- Xu, F.; Zhou, G.; Han, S.; Yuan, W.; Chen, S.; Fu, Z.; Li, D.; Zhang, H.; Li, D.; Pang, D. Association of TNF-alpha, TNFRSF1A and TNFRSF1B gene polymorphisms with the risk of sporadic breast cancer in northeast Chinese Han women. PLoS ONE 2014, 9, e101138. [Google Scholar] [CrossRef]
- Han, W.; Kang, S.Y.; Kang, D.; Park, S.K.; Lee, J.Y.; Kim, H.; Park, A.K.; Noh, D.Y. Multiplex genotyping of 1107 SNPs from 232 candidate genes identified an association between IL1A polymorphism and breast cancer risk. Oncol. Rep. 2010, 23, 763–769. [Google Scholar]
- Yang, F.; Zhao, N.; Wu, N. TNFR2 promotes Adriamycin resistance in breast cancer cells by repairing DNA damage. Mol. Med. Rep. 2017, 16, 2962–2968. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, J.; Berger, A.H.; Diolombi, M.S.; Ng, C.; Fung, J.; Bronson, R.T.; Castillo-Martin, M.; Thin, T.H.; Cordon-Cardo, C.; et al. Compound haploinsufficiency of Dok2 and Dusp4 promotes lung tumorigenesis. J. Clin. Investig. 2019, 129, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Peng, X.; Zhang, K.; Li, C.; Su, B.; Zhang, Y.; Yu, W. Co-expression and significance of Dok2 and Ras p21 protein activator 1 in breast cancer. Oncol. Lett. 2017, 14, 5386–5392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WCRF/AICR. Continuous Update Project Report: Diet, Nutrition, Physical Actvity and Cancer; World Cancer Research Fund International/American Institute for Cancer Research: London UK, 2017. [Google Scholar]
- Dos Santos, C.R.; Fonseca, I.; Dias, S.; de Almeida, J.C.M. Plasma level of LDL-cholesterol at diagnosis is a predictor factor of breast tumor progression. BMC Cancer 2014, 14, 132. [Google Scholar] [CrossRef]
- Dos Santos, C.R.; Domingues, G.; Matias, I.; Matos, J.; Fonseca, I.; de Almeida, J.M.; Dias, S. LDL-cholesterol signaling induces breast cancer proliferation and invasion. Lipids Health Dis. 2014, 13, 16. [Google Scholar] [CrossRef]
- Chlebowski, R.T.; Blackburn, G.L.; Thomson, C.A.; Nixon, D.W.; Shapiro, A.; Hoy, M.K.; Goodman, M.T.; Giuliano, A.E.; Karanja, N.; McAndrew, P.; et al. Dietary fat reduction and breast cancer outcome: Interim efficacy results from the Women’s Intervention Nutrition Study. J. Natl. Cancer Inst. 2006, 98, 1767–1776. [Google Scholar] [CrossRef]
- Toledo, E.; Salas-Salvado, J.; Donat-Vargas, C.; Buil-Cosiales, P.; Estruch, R.; Ros, E.; Corella, D.; Fito, M.; Hu, F.B.; Aros, F.; et al. Mediterranean Diet and Invasive Breast Cancer Risk Among Women at High Cardiovascular Risk in the PREDIMED Trial: A Randomized Clinical Trial. JAMA Intern. Med. 2015, 175, 1752–1760. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutchinson, S.A.; Lianto, P.; Roberg-Larsen, H.; Battaglia, S.; Hughes, T.A.; Thorne, J.L. ER-Negative Breast Cancer Is Highly Responsive to Cholesterol Metabolite Signalling. Nutrients 2019, 11, 2618. https://doi.org/10.3390/nu11112618
Hutchinson SA, Lianto P, Roberg-Larsen H, Battaglia S, Hughes TA, Thorne JL. ER-Negative Breast Cancer Is Highly Responsive to Cholesterol Metabolite Signalling. Nutrients. 2019; 11(11):2618. https://doi.org/10.3390/nu11112618
Chicago/Turabian StyleHutchinson, Samantha A, Priscilia Lianto, Hanne Roberg-Larsen, Sebastiano Battaglia, Thomas A Hughes, and James L Thorne. 2019. "ER-Negative Breast Cancer Is Highly Responsive to Cholesterol Metabolite Signalling" Nutrients 11, no. 11: 2618. https://doi.org/10.3390/nu11112618
APA StyleHutchinson, S. A., Lianto, P., Roberg-Larsen, H., Battaglia, S., Hughes, T. A., & Thorne, J. L. (2019). ER-Negative Breast Cancer Is Highly Responsive to Cholesterol Metabolite Signalling. Nutrients, 11(11), 2618. https://doi.org/10.3390/nu11112618