Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry

 and
and 
{kind=link}
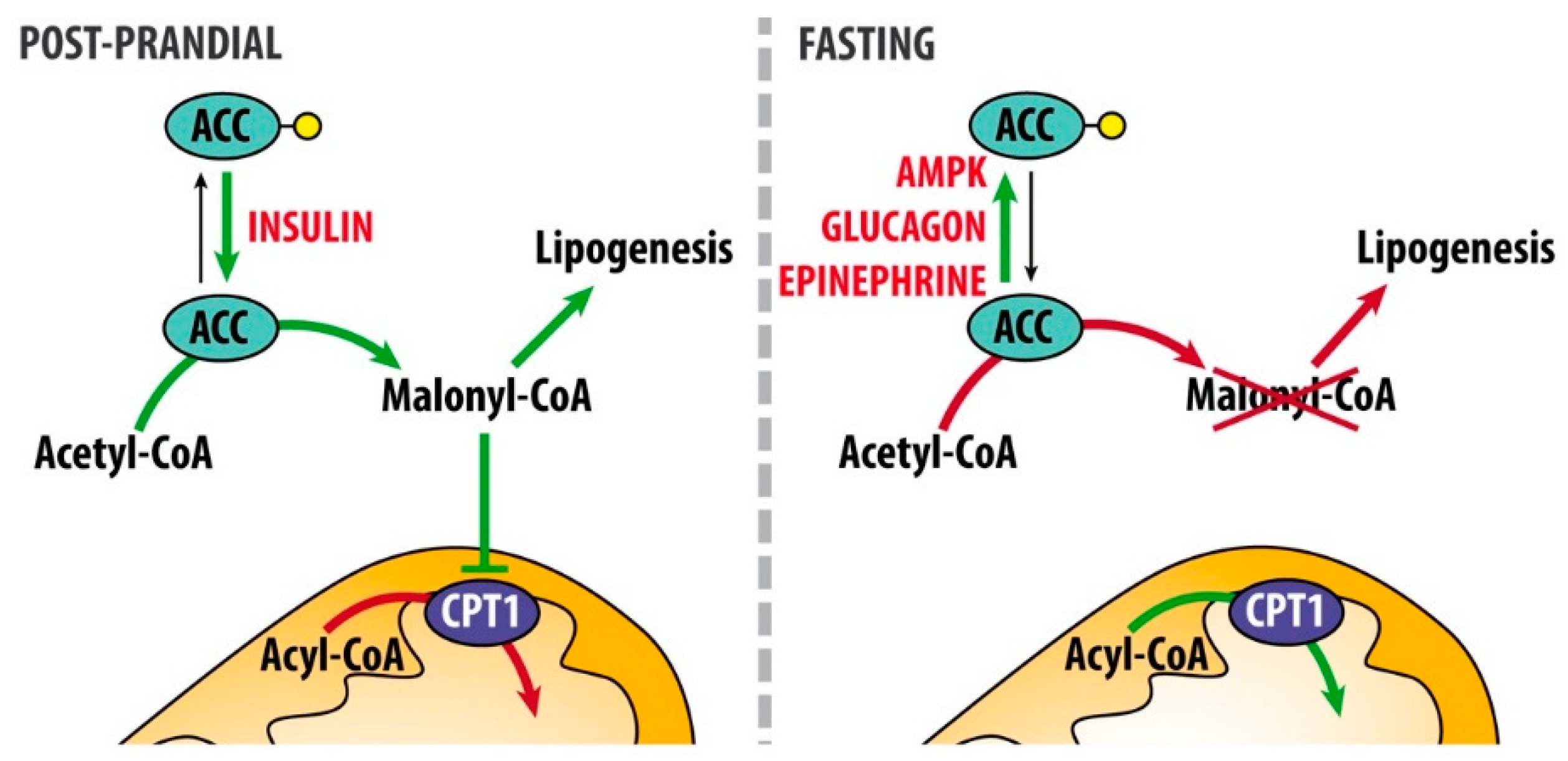{kind=link}
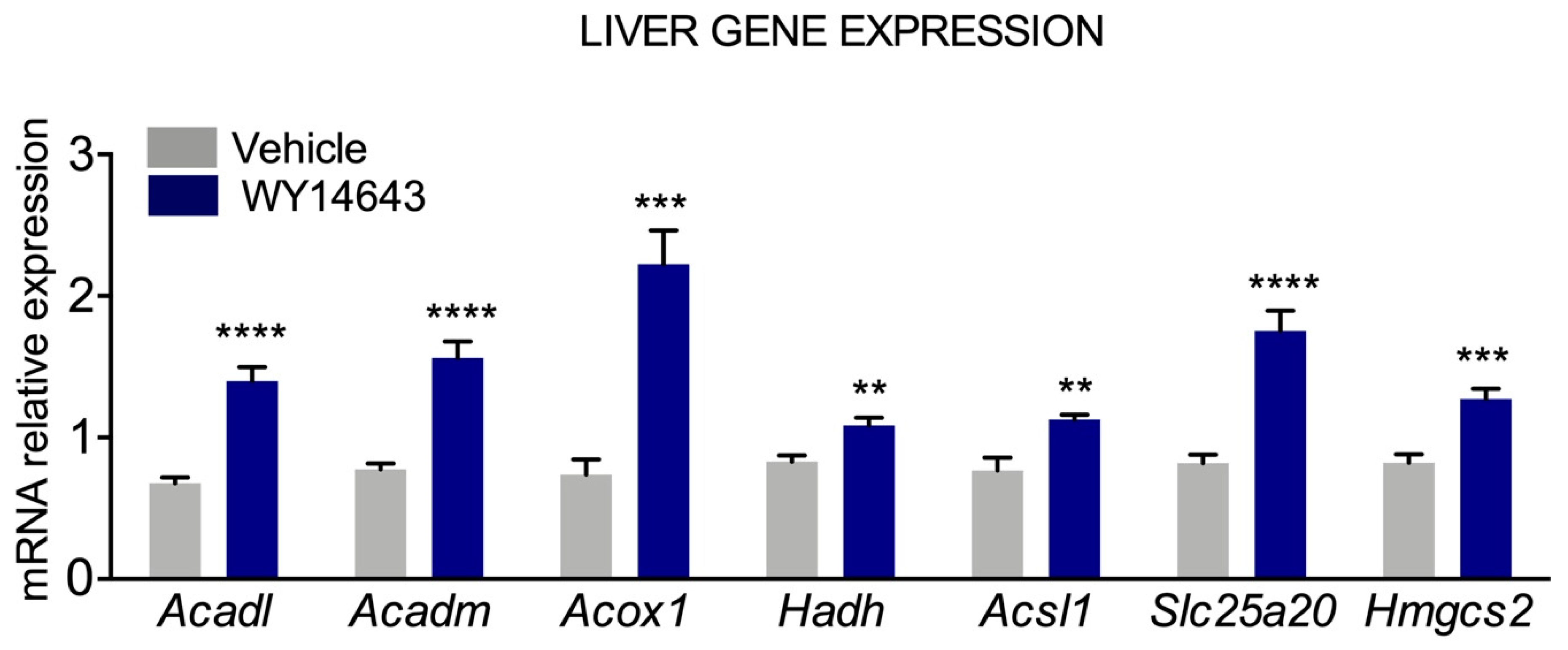{kind=link}
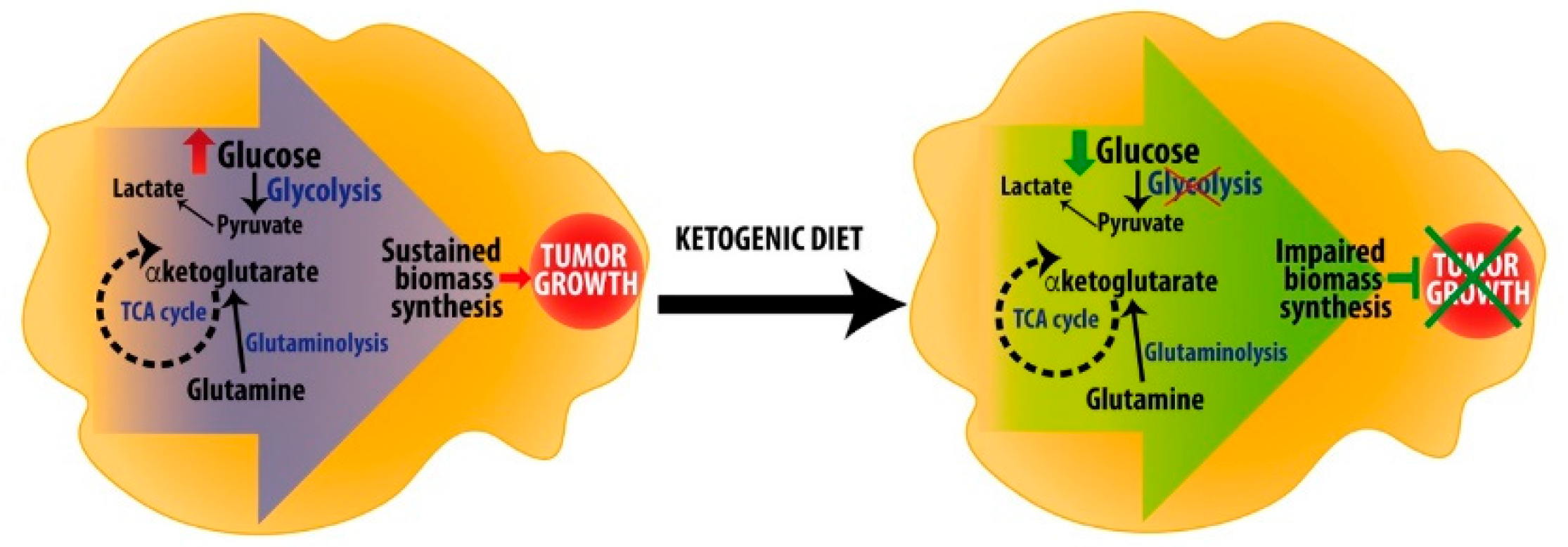{kind=link}
Abstract
1. Introduction
2. Overview of the Biochemical Reactions of KB Synthesis and Utilization
2.1. Ketone Body Production
2.2. Ketone Body Utilization (Ketolysis)
3. Regulation of Ketogenesis
3.1. Hormonal Regulation
3.2. Transcriptional Regulation
3.3. FGF21 is an Endocrine Regulator of the Ketotic State
3.4. The Ketogenic Factor FGF21 as Pharmacological Target
3.5. Epigenetic Regulation by Ketone Bodies
4. Clinical Relevance of Ketogenic Diet
5. Biochemistry of KD Action
5.1. Inhibition of Glycolysis
5.2. Mitochondrial Metabolism
5.3. Modulation of Neuronal Transmission
5.4. Regulation of Polyunsaturated Fatty Acid Levels
6. Ketone Bodies in Inflammation and Immune System
7. Ketogenic Diets as Anti-Cancer Adjuvant Therapy to Target Metabolic Rewiring
8. Could Ketogenic Diet Help in Treating Neurodegenerative Diseases?
9. Ketogenic Diet as a Strategy to Cope Obesity
10. Adverse Effects of Ketogenic Diet
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- d’Avignon, D.A.; Puchalska, P.; Ercal, B.; Chang, Y.; Martin, S.E.; Graham, M.J.; Patti, G.J.; Han, X.; Crawford, P.A. Hepatic ketogenic insufficiency reprograms hepatic glycogen metabolism and the lipidome. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of ketone body metabolism and the role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.L.; Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry; Macmillan: London, UK, 2005. [Google Scholar]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Lopaschuk, G.D.; Mitchell, G.A. Pathways and control of ketone body metabolism: On the fringe of lipid biochemistry. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 243–251. [Google Scholar] [CrossRef]
- Hegardt, F.G. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: A control enzyme in ketogenesis. Biochem. J. 1999, 338 Pt 3, 569–582. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Carson, J.J.; Hebert, A.S.; Hubler, S.L.; Niemi, N.M.; Bailey, D.J.; Jochem, A.; Stapleton, D.S.; Keller, M.P.; Westphall, M.S.; et al. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 2012, 16, 672–683. [Google Scholar] [CrossRef]
- Desvergne, B. Peroxisome Proliferator-Activated Receptors: Nuclear Control of Metabolism. Endocr. Rev. 2004, 20, 649–688. [Google Scholar]
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 1–10. [Google Scholar] [CrossRef]
- Kersten, S.; Seydoux, J.; Peters, J. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef]
- Misto, A.; Provensi, G.; Vozella, V.; Passani, M.B.; Piomelli, D. Mast Cell-Derived Histamine Regulates Liver Ketogenesis via Oleoylethanolamide Signaling. Cell Metab. 2019, 29, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Tognini, P.; Murakami, M.; Liu, Y.; Eckel-Mahan, K.L.; Newman, J.C.; Verdin, E.; Baldi, P.; Sassone-Corsi, P. Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metab. 2017, 26, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Chavan, R.; Feillet, C.; Costa, S.S.F.; Delorme, J.E.; Okabe, T.; Ripperger, J.A.; Albrecht, U. Liver-derived ketone bodies are necessary for food anticipation. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic Fibroblast Growth Factor 21 Is Regulated by PPARα and Is a Key Mediator of Hepatic Lipid Metabolism in Ketotic States. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine Regulation of the Fasting Response by PPARα-Mediated Induction of Fibroblast Growth Factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef]
- Véniant, M.M.; Komorowski, R.; Chen, P.; Stanislaus, S.; Winters, K.; Hager, T.; Zhou, L.; Wada, R.; Hecht, R.; Xu, J. Long-acting FGF21 has enhanced efficacy in diet-induced obese mice and in obese rhesus monkeys. Endocrinology 2012, 153, 4192–4203. [Google Scholar] [CrossRef]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The effects of LY2405319, an FGF21 Analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef]
- De Ruijter, A.J.M.; Van Gennip, A.H.; Caron, H.N.; Kemp, S.; Van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370 Pt 3, 737–749. [Google Scholar] [CrossRef]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfi, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef]
- Finkel, T.; Deng, C.-X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 185, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.C.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of Oxidative Stress by b-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.P.L.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.A.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M.T. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef]
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. The role of metallothionein in oxidative stress. Int. J. Mol. Sci. 2013, 14, 6044–6066. [Google Scholar] [CrossRef]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. Elife 2016, 5, 1–21. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef]
- Hildebrand, M.S.; Damiano, J.A.; Mullen, S.A.; Bellows, S.T.; Oliver, K.L.; Dahl, H.H.M.; Scheffer, I.E.; Berkovic, S.F. Glucose metabolism transporters and epilepsy: Only GLUT1 has an established role. Epilepsia 2014, 55, 18–21. [Google Scholar] [CrossRef]
- Wang, D.; Kranz-Eble, P.; De Vivo, D.C. Mutational analysis of GLUT1 (SLC2A1) in Glut-1 deficiency syndrome. Hum. Mutat. 2000, 16, 224–231. [Google Scholar] [CrossRef]
- Klepper, J. Autosomal dominant transmission of GLUT1 deficiency. Hum. Mol. Genet. 2001, 10, 63–68. [Google Scholar] [CrossRef]
- Wang, D.; Pascual, J.M.; Yang, H.; Engelstad, K.; Jhung, S.; Sun, R.P.; De Vivo, D.C. Glut-1 deficiency syndrome: Clinical, genetic, and therapeutic aspects. Ann. Neurol. 2005, 57, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Klepper, J.; Diefenbach, S.; Kohlschütter, A.; Voit, T. Effects of the ketogenic diet in the glucose transporter 1 deficiency syndrome. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Klepper, J.; Leiendecker, B.; Bredahl, R.; Athanassopoulos, S.; Heinen, F.; Gertsen, E.; Flörcken, A.; Metz, A.; Voit, T. Introduction of a ketogenic diet in young infants. J. Inherit. Metab. Dis. 2002, 25, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Pascual, J.M.; Yang, H.; Engelstad, K.; Mao, X.; Cheng, J.; Yoo, J.; Noebels, J.L.; De Vivo, D.C. A mouse model for Glut-1 haploinsufficiency. Hum. Mol. Genet. 2006, 15, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Chida, R.; Shimura, M.; Nishimata, S.; Kashiwagi, Y.; Kawashima, H. Efficacy of ketogenic diet for pyruvate dehydrogenase complex deficiency. Pediatr. Int. 2018, 60, 1041–1042. [Google Scholar] [CrossRef]
- Lissens, W.; De Meirleir, L.; Seneca, S.; Liebaers, I.; Brown, G.K.; Ruth, M.; Ito, M.; Naito, E.; Kuroda, Y.; Kerr, D.S.; et al. Mutations in the X-Linked Pyruvate Dehydrogenase (E1) α Subunit Gene (PDHA1) in Patients With a Pyruvate Dehydrogenase Complex Deficiency. Hum. Mutat. 2000, 15, 209–219. [Google Scholar] [CrossRef]
- Sato, S.; Ioi, H.; Kashiwagi, Y.; Kawashima, H.; Miyajima, T.; Naito, E.; Hoshika, A. Novel mutation (R263X) of the E1 subunit in pyruvate dehydrogenase complex deficiency. Pediatr. Int. 2010, 52, 181–183. [Google Scholar] [CrossRef]
- Stowe, R.C.; Sun, Q.; Elsea, S.H.; Scaglia, F. LIPT1 deficiency presenting as early infantile epileptic encephalopathy, Leigh disease, and secondary pyruvate dehydrogenase complex deficiency. Am. J. Med. Genet. Part A 2018, 176, 1184–1189. [Google Scholar] [CrossRef]
- Nimmo, G.A.M.; Venkatesh, S.; Pandey, A.K.; Marshall, C.R.; Hazrati, L.; Blaser, S.; Ahmed, S.; Cameron, J.; Singh, K.; Ray, P.N.; et al. Bi-allelic mutations of LONP1 encoding the mitochondrial LonP1 protease cause pyruvate dehydrogenase deficiency and profound neurodegeneration with progressive cerebellar atrophy. Hum. Mol. Genet. 2018, 28, 290–306. [Google Scholar] [CrossRef]
- Jakkamsetti, V.; Marin-Valencia, I.; Ma, Q.; Good, L.B.; Terrill, T.; Rajasekaran, K.; Pichumani, K.; Khemtong, C.; Hooshyar, M.A.; Sundarrajan, C.; et al. Brain metabolism modulates neuronal excitability in a mouse model of pyruvate dehydrogenase deficiency. Sci. Transl. Med. 2019, 11, eaan0457. [Google Scholar] [CrossRef]
- Kerr, D.S.; Hemalatha, S.G.; Wexler, I.D.; Buist, N.R.M.; Berry, S.A.; McConnell, J.; Patel, M.S.; Dahl, H.M.; Cederbaum, S.D. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets: Studies in patients with identical mutations. Neurology 2012, 49, 1655–1661. [Google Scholar]
- Murray, C.J.L.; Vos, T.; Lozano, R.; Naghavi, M.; Flaxman, A.D.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2197–2223. [Google Scholar] [CrossRef]
- Fiest, K.M.; Sauro, K.M.; Wiebe, S.; Patten, S.B.; Kwon, C.-S.; Dykeman, J.; Pringsheim, T.; Lorenzetti, D.L.; Jette, N. Prevalence and incidence of epilepsy: A systematic review and meta-analysis of international studies. Neurology 2017, 89, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Martin-McGill, K.J.; Jackson, C.F.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Roehl, K.; Falco-Walter, J.; Ouyang, B.; Balabanov, A. Modified ketogenic diets in adults with refractory epilepsy: Efficacious improvements in seizure frequency, seizure severity, and quality of life. Epilepsy Behav. 2019, 93, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Shah, L.M.; Turner, Z.; Bessone, S.K.; Winesett, S.P.; Stanfield, A.; Kossoff, E.H. How often is antiseizure drug-free ketogenic diet therapy achieved? Epilepsy Behav. 2019, 93, 29–31. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 3, 175–192. [Google Scholar] [CrossRef]
- Roehl, K.; Sewak, S.L. Practice Paper of the Academy of Nutrition and Dietetics: Classic and Modified Ketogenic Diets for Treatment of Epilepsy. J. Acad. Nutr. Diet. 2017, 117, 1279–1292. [Google Scholar] [CrossRef]
- Wasterlain, C.G.; Thompson, K.W.; Suchomelova, L.; Niquet, J. Brain energy metabolism during experimental neonatal seizures. Neurochem. Res. 2010, 35, 2193–2198. [Google Scholar] [CrossRef][Green Version]
- Stafstrom, C.E.; Ockuly, J.C.; Murphree, L.; Valley, M.T.; Roopra, A.; Sutula, T.P. Anticonvulsant and antiepileptic actions of 2-deoxy-D-glucose in epilepsy models. Ann. Neurol. 2009, 65, 435–448. [Google Scholar] [CrossRef]
- Shao, L.; Rho, J.M.; Stafstrom, C.E. Glycolytic inhibition: A novel approach toward controlling neuronal excitability and seizures. Epilepsia Open 2018, 3, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Sutula, T.P.; Rutecki, P.A. 2-D-deoxyglucose Reduces Epileptiform Activity by Presynaptic Mechanisms. J. Neurophysiol. 2019, 121, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Garriga-Canut, M.; Schoenike, B.; Qazi, R.; Bergendahl, K.; Daley, T.J.; Pfender, R.M.; Morrison, J.F.; Ockuly, J.; Stafstrom, C.; Sutula, T.; et al. 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat. Neurosci. 2006, 9, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovács, R.; Kann, O. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, 641–657. [Google Scholar] [CrossRef]
- Giménez-Cassina, A.; Martínez-François, J.R.; Fisher, J.K.; Szlyk, B.; Polak, K.; Wiwczar, J.; Tanner, G.R.; Lutas, A.; Yellen, G.; Danial, N.N. BAD-Dependent Regulation of Fuel Metabolism and KATP Channel Activity Confers Resistance to Epileptic Seizures. Neuron 2012, 74, 719–730. [Google Scholar] [CrossRef]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. ; A.D. Mechanisms of Action of Bcl-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, 104–116. [Google Scholar]
- Giménez-cassina, A.; Garcia-haro, L.; Choi, C.S.; Mayowa, A.; Lane, E.; Huang, H.; Yildirim, M.A.; Szlyk, B.; Jill, K.; Polak, K.; et al. Regulation of hepatic energy metabolism and gluconeogenesis by BAD. Cell Metab. 2014, 19, 272–284. [Google Scholar] [CrossRef]
- Danial, N.N.; Walensky, L.D.; Zhang, C.Y.; Choi, C.S.; Fisher, J.K.; Molina, A.J.A.; Datta, S.R.; Pitter, K.L.; Bird, G.H.; Wikstrom, J.D.; et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat. Med. 2008, 14, 144–153. [Google Scholar] [CrossRef]
- Milder, J.; Patel, M. Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. 2012, 100, 295–303. [Google Scholar] [CrossRef]
- Shin, E.J.; Jeong, J.H.; Chung, Y.H.; Kim, W.K.; Ko, K.H.; Bach, J.H.; Hong, J.S.; Yoneda, Y.; Kim, H.C. Role of oxidative stress in epileptic seizures. Neurochem. Int. 2011, 59, 122–137. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Milder, J.B.; Liang, L.P.; Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 2008, 106, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Milder, J.B.; Liang, L.P.; Patel, M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol. Dis. 2010, 40, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Masino, A. ketogenic diet suppresses seizures in mice through adenosine A (1) receptors. J. Clin. Investig. 2011, 121, 2679–2683. [Google Scholar] [CrossRef] [PubMed]
- Keith, H.M. Factors influencing experimentally produced convulsions. Arch. Neurol. Psychiatry 1933, 29, 148–154. [Google Scholar] [CrossRef]
- Erecińska, M.; Nelson, D.; Daikhin, Y.; Yudkoff, M. Regulation of GABA level in rat brain synaptosomes: Fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. J. Neurochem. 1996, 67, 2325–2334. [Google Scholar] [CrossRef]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Pleasure, D.; Stern, J.; Nissim, I. Inhibition of Astrocyte Glutamine Production by α-Ketoisocaproic Acid. J. Neurochem. 2002, 63, 1508–1515. [Google Scholar] [CrossRef]
- Juge, N.; Gray, J.A.; Omote, H.; Miyaji, T.; Inoue, T.; Hara, C.; Uneyama, H.; Edwards, R.H.; Nicoll, R.A.; Moriyama, Y. Metabolic Control of Vesicular Glutamate Transport and Release. Neuron 2010, 68, 99–112. [Google Scholar] [CrossRef]
- Thio, L.L.; Wong, M.; Yamada, K.A. Ketone bodies do not directly alter excitatory or inhibitory hippocampal synaptic transmission. Neurology 2012, 54, 325. [Google Scholar] [CrossRef]
- Cheng, C.M.; Hicks, K.; Wang, J.; Eagles, D.A.; Bondy, C.A. Caloric restriction augments brain glutamic acid decarboxylase-65 and -67 expression. J. Neurosci. Res. 2004, 77, 270–276. [Google Scholar] [CrossRef]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Horyn, O.; Lazarow, A.; Luhovyy, B.; Wehrli, S.; Nissim, I. Response of brain amino acid metabolism to ketosis. Neurochem. Int. 2005, 47, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.D.; Whiting, S.; Andrew, R.D.; McDonald, E.A.; Veloso, K.M.; Cunnane, S.C. Elevated polyunsaturated fatty acids in blood serum obtained from children on the ketogenic diet. Neurology 2003, 60, 1026–1029. [Google Scholar] [CrossRef] [PubMed]
- Taha, A.Y.; Filo, E.; Ma, D.W.L.; McIntyre Burnham, W. Dose-dependent anticonvulsant effects of linoleic and α-linolenic polyunsaturated fatty acids on pentylenetetrazol induced seizures in rats. Epilepsia 2009, 50, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Porta, N.; Vallée, L.; Lecointe, C.; Bouchaert, E.; Staels, B.; Bordet, R.; Auvin, S. Fenofibrate, a peroxisome proliferator-activated receptor-α agonist, exerts anticonvulsive properties. Epilepsia 2009, 50, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.A.; Jeon, B.T.; Shin, H.J.; Kim, N.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 2011, 232, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Gotlinger, K.; Hong, S.; Lu, Y.; Siegelman, J.; Baer, T.; Yang, R.; Colgan, S.P.; Petasis, N.A. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: Assignments of dihydroxy-containing docosatrienes. J. Immunol. 2006, 176, 1848–1859. [Google Scholar]
- Moro, K.; Nagahashi, M.; Ramanathan, R.; Takabe, K.; Wakai, T. Resolvins and omega three polyunsaturated fatty acids: Clinical implications in inflammatory diseases and cancer. World J. Clin. Cases 2016, 4, 155. [Google Scholar] [CrossRef]
- Schlanger, S.; Shinitzky, M.; Yam, D. Diet enriched with omega-3 fatty acids alleviates convulsion symptoms in epilepsy patients. Epilepsia 2002, 43, 103–104. [Google Scholar] [CrossRef]
- Yuen, A.W.C.; Sander, J.W.; Fluegel, D.; Patsalos, P.N.; Bell, G.S.; Johnson, T.; Koepp, M.J. Omega-3 fatty acid supplementation in patients with chronic epilepsy: A randomized trial. Epilepsy Behav. 2005, 7, 253–258. [Google Scholar] [CrossRef]
- Bromfield, E.; Dworetzky, B.; Hurwitz, S.; Eluri, Z.; Lane, L.; Replansky, S.; Mostofsky, D. A randomized trial of polyunsaturated fatty acids for refractory epilepsy. Epilepsy Behav. 2008, 12, 187–190. [Google Scholar] [CrossRef]
- Sarmento Vasconcelos, V.; de Souza Pedrosa, A.; Pereira Gomes Morais, E.; Macedo, C.R.; Torloni, M.R.; Porfírio, G.J.M. Polyunsaturated fatty acid supplementation for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.L.; Asher, J.L.; Molony, R.D.; Shaw, A.C.; Zeiss, C.J.; Wang, C.; Morozova-Roche, L.A.; Herzog, R.I.; Iwasaki, A.; Dixit, V.D. β-Hydroxybutyrate Deactivates Neutrophil NLRP3 Inflammasome to Relieve Gout Flares. Cell Rep. 2017, 18, 2077–2087. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; De Nigris, V.; Micheloni, S.; La Sala, L.; Ceriello, A. Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: Is low-grade inflammation the neglected component? Diabetes Obes. Metab. 2018, 20, 2515–2522. [Google Scholar] [CrossRef] [PubMed]
- Tajima, T.; Yoshifuji, A.; Matsui, A.; Itoh, T.; Uchiyama, K.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. β-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney Int. 2019, 95, 1120–1137. [Google Scholar] [CrossRef] [PubMed]
- Woolf, E.C.; Syed, N.; Scheck, A.C. Tumor Metabolism, the Ketogenic Diet and β-Hydroxybutyrate: Novel Approaches to Adjuvant Brain Tumor Therapy. Front. Mol. Neurosci. 2016, 9, 122. [Google Scholar] [CrossRef]
- Woolf, E.C.; Scheck, A.C. The ketogenic diet for the treatment of malignant glioma. J. Lipid Res. 2015, 56, 5–10. [Google Scholar] [CrossRef]
- Noorlag, L.; De Vos, F.Y.; Kok, A.; Broekman, M.L.D.; Seute, T.; Robe, P.A.; Snijders, T.J. Treatment of malignant gliomas with ketogenic or caloric restricted diets: A systematic review of preclinical and early clinical studies. Clin. Nutr. 2018. [Google Scholar] [CrossRef]
- De Feyter, H.M.; Behar, K.L.; Rao, J.U.; Madden-Hennessey, K.; Ip, K.L.; Hyder, F.; Drewes, L.R.; Geschwind, J.-F.; de Graaf, R.A.; Rothman, D.L. A ketogenic diet increases transport and oxidation of ketone bodies in RG2 and 9L gliomas without affecting tumor growth. Neuro Oncol. 2016, 18, 1079–1087. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef]
- Byrne, F.L.; Hargett, S.R.; Lahiri, S.; Roy, R.J.; Berr, S.S.; Caldwell, S.H.; Hoehn, K.L. Serial MRI imaging reveals minimal impact of ketogenic diet on established liver tumor growth. Cancers 2018, 10, 312. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J.; Champ, C.E.; Otto, C.; Kämmerer, U. Anti-tumor effects of ketogenic diets in mice: A meta-analysis. PLoS ONE 2016, 11, e0155050. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J. The emerging role of ketogenic diets in cancer treatment. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J. Beneficial effects of ketogenic diets for cancer patients: A realist review with focus on evidence and confirmation. Med. Oncol. 2017, 34, 132. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.W.; Fontaine, K.R.; Arend, R.C.; Alvarez, R.D.; Leath, C.A.; Huh, W.K.; Bevis, K.S.; Kim, K.H.; Straughn, J.M.; Gower, B.A. A ketogenic diet reduces central obesity and serum insulin in women with ovarian or endometrial cancer. J. Nutr. 2018, 148, 1253–1260. [Google Scholar] [CrossRef]
- Cohen, C.W.; Fontaine, K.R.; Arend, R.C.; Soleymani, T.; Gower, B.A. Favorable effects of a ketogenic diet on physical function, perceived energy, and food cravings in women with ovarian or endometrial cancer: A randomized, controlled trial. Nutrients 2018, 10, 1187. [Google Scholar] [CrossRef]
- OK, J.H.; Lee, H.; Chung, H.-Y.; Lee, S.H.; Choi, E.J.; Kang, C.M.; Lee, S.M. The Potential Use of a Ketogenic Diet in Pancreatobiliary Cancer Patients After Pancreatectomy. Anticancer Res. 2018, 38, 6519–6527. [Google Scholar] [CrossRef]
- Włodarek, D. Role of ketogenic diets in neurodegenerative diseases (Alzheimer’s disease and parkinson’s disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 28–36. [Google Scholar] [CrossRef]
- Torosyan, N.; Sethanandha, C.; Grill, J.D.; Dilley, M.L.; Lee, J.; Cummings, J.L.; Ossinalde, C.; Silverman, D.H. Changes in regional cerebral blood flow associated with a 45day course of the ketogenic agent, caprylidene, in patients with mild to moderate Alzheimer’s disease: Results of a randomized, double-blinded, pilot study. Exp. Gerontol. 2018, 111, 118–121. [Google Scholar] [CrossRef]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 1–25. [Google Scholar] [CrossRef]
- Broom, G.M.; Shaw, I.C.; Rucklidge, J.J. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition 2019, 60, 118–121. [Google Scholar] [CrossRef]
- VanItallie, T.B.; Nonas, C.; Di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef]
- Phillips, M.C.L.; Murtagh, D.K.J.; Gilbertson, L.J.; Asztely, F.J.S.; Lynch, C.D.P. Low-fat versus ketogenic diet in Parkinson’s disease: A pilot randomized controlled trial. Mov. Disord. 2018, 33, 1306–1314. [Google Scholar] [CrossRef]
- Veyrat-Durebex, C.; Andres, C.R.; Reynier, P.; Hergesheimer, R.; Blasco, H.; Corcia, P.; Procaccio, V. How Can a Ketogenic Diet Improve Motor Function? Front. Mol. Neurosci. 2018, 11, 15. [Google Scholar] [CrossRef]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546. [Google Scholar] [CrossRef]
- Newman, J.C.; Covarrubias, A.J.; Zhao, M.; Yu, X.; Gut, P.; Ng, C.P.; Huang, Y.; Haldar, S.; Verdin, E. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab. 2017, 26, 547–557. [Google Scholar] [CrossRef]
- Mansoor, N.; Vinknes, K.J.; Veierod, M.B.; Retterstol, K. Effects of low-carbohydrate diets v. low-fat diets on body weight and cardiovascular risk factors a meta-analysis of randomised controlled trials. Br. J. Nutr. 2015, 115, 466–479. [Google Scholar] [CrossRef]
- Bueno, N.B.; De Melo, I.S.V.; De Oliveira, S.L.; Da Rocha Ataide, T. Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: A meta-analysis of Randomised controlled trials. Br. J. Nutr. 2013, 110, 1178–1187. [Google Scholar] [CrossRef]
- Harvey, C.J.D.C.; Schofield, G.M.; Zinn, C.; Thornley, S.J.; Crofts, C.; Merien, F.L.R. Low-carbohydrate diets differing in carbohydrate restriction improve cardiometabolic and anthropometric markers in healthy adults: A randomised clinical trial. PeerJ 2019, 7, e6273. [Google Scholar] [CrossRef]
- Sackner-Bernstein, J.; Kanter, D.; Kaul, S. Dietary intervention for overweight and obese adults: Comparison of low-carbohydrate and low-fat diets. a meta-analysis. PLoS ONE 2015, 10, e0139817. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Cotter, D.G.; Patti, G.J.; Crawford, P.A.; Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; Avignon, D.A.; Graham, M.J.; Dietzen, D.J. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia Find the latest version: Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef]
- Browning, J.D.; Baker, J.A.; Rogers, T.; Davis, J.; Satapati, S.; Burgess, S.C. Short-term weight loss and hepatic triglyceride reduction: Evidence of a metabolic advantage with dietary carbohydrate restriction 1–3. Am. J. Clin. Nutr. 2011, 93, 1048–1052. [Google Scholar] [CrossRef]
- Cai, Q.Y.; Zhou, Z.J.; Luo, R.; Gan, J.; Li, S.P.; Mu, D.Z.; Wan, C.M. Safety and tolerability of the ketogenic diet used for the treatment of refractory childhood epilepsy: A systematic review of published prospective studies. World J. Pediatr. 2017, 13, 528–536. [Google Scholar] [CrossRef]
- McDonald, T.J.W.; Cervenka, M.C. Ketogenic Diets for Neurological Disorders. Homeost. Control Brain Funct. 2016, 248–270. [Google Scholar] [CrossRef]
- Klein, P.; Janousek, J.; Barber, A.; Weissberger, R. Ketogenic Diet Treatment in Adults with Refractory Epilepsy. Epilepsy Behav. 2010, 19, 575–579. [Google Scholar] [CrossRef]
- Lin, A.; Turner, Z.; Doerrer, S.C.; Stanfield, A.; Kossoff, E.H. Complications During Ketogenic Diet Initiation: Prevalence, Treatment, and Influence on Seizure Outcomes. Pediatr. Neurol. 2017, 68, 35–39. [Google Scholar] [CrossRef]
- Ferraris, C.; Guglielmetti, M.; Pasca, L.; De Giorgis, V.; Ferraro, O.E.; Brambilla, I.; Leone, A.; De Amicis, R.; Bertoli, S.; Veggiotti, P.; et al. Impact of the ketogenic diet on linear growth in children: A single-center retrospective analysis of 34 cases. Nutrients 2019, 11, 1442. [Google Scholar] [CrossRef]
- Wibisono, C.; Rowe, N.; Beavis, E.; Kepreotes, H.; Mackie, F.E.; Lawson, J.A.; Cardamone, M. Ten-year single-center experience of the ketogenic diet: Factors influencing efficacy, tolerability, and compliance. J. Pediatr. 2015, 166, 1030–1036. [Google Scholar] [CrossRef]
- Lambrechts, D.A.J.E.; de Kinderen, R.J.A.; Vles, H.S.H.; de Louw, A.J.; Aldenkamp, A.P.; Majoie, M.J.M. The MCT-ketogenic diet as a treatment option in refractory childhood epilepsy: A prospective study with 2-year follow-up. Epilepsy Behav. 2015, 51, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Dressler, A.; Stöcklin, B.; Reithofer, E.; Benninger, F.; Freilinger, M.; Hauser, E.; Reiter-Fink, E.; Seidl, R.; Trimmel-Schwahofer, P.; Feucht, M. Long-term outcome and tolerability of the ketogenic diet in drug-resistant childhood epilepsy-the austrian experience. Seizure 2010, 19, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Simm, P.J.; Bicknell-Royle, J.; Lawrie, J.; Nation, J.; Draffin, K.; Stewart, K.G.; Cameron, F.J.; Scheffer, I.E.; Mackay, M.T. The effect of the ketogenic diet on the developing skeleton. Epilepsy Res. 2017, 136, 62–66. [Google Scholar] [CrossRef] [PubMed]
- McNally, M.A.; Pyzik, P.L.; Rubenstein, J.E.; Hamdy, R.F.; Kossoff, E.H. Empiric Use of Potassium Citrate Reduces Kidney-Stone Incidence With the Ketogenic Diet. Pediatrics 2009, 124, e300–e304. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longo, R.; Peri, C.; Cricrì, D.; Coppi, L.; Caruso, D.; Mitro, N.; De Fabiani, E.; Crestani, M. Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry. Nutrients 2019, 11, 2497. https://doi.org/10.3390/nu11102497
Longo R, Peri C, Cricrì D, Coppi L, Caruso D, Mitro N, De Fabiani E, Crestani M. Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry. Nutrients. 2019; 11(10):2497. https://doi.org/10.3390/nu11102497
Chicago/Turabian StyleLongo, Raffaella, Carolina Peri, Dalma Cricrì, Lara Coppi, Donatella Caruso, Nico Mitro, Emma De Fabiani, and Maurizio Crestani. 2019. "Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry" Nutrients 11, no. 10: 2497. https://doi.org/10.3390/nu11102497
APA StyleLongo, R., Peri, C., Cricrì, D., Coppi, L., Caruso, D., Mitro, N., De Fabiani, E., & Crestani, M. (2019). Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry. Nutrients, 11(10), 2497. https://doi.org/10.3390/nu11102497