Intestinal Barrier Function in Gluten-Related Disorders

 ,
,
Abstract
1. The Intestinal Barrier
1.1. Mucus Barrier
Mucus Composition
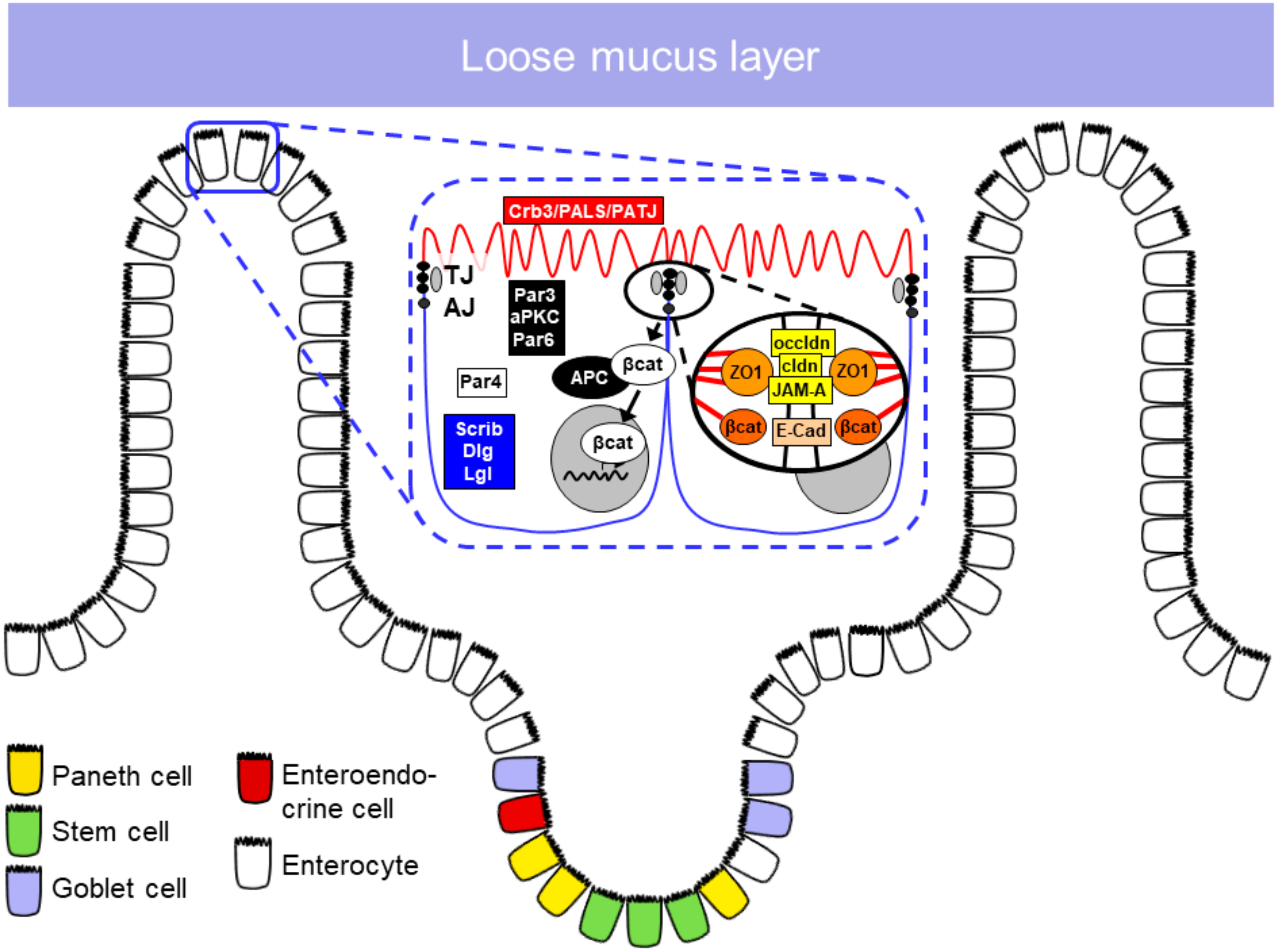
1.2. Epithelial Barrier
1.2.1. Epithelial Cell Types in the Small Intestine
1.2.2. Apical Junctional Complex
Tight Junctions
Adherens Junctions
1.3. The Role of Lamina Propria Cells in Maintaining Barrier Function
1.3.1. Dendritic Cells and Macrophages
1.3.2. Innate Lymphoid Cells
1.4. Impact of Intraepithelial Lymphocytes on Barrier Function
1.5. Role of the Luminal Microbiota on Barrier Function
2. The Role of the Intestinal Barrier in Gluten-Related Disorders
2.1. The Intestinal Barrier in Celiac Disease
Evidence for a Primary Barrier Defect in Celiac Disease
2.2. The Intestinal Barrier in Wheat Allergy
2.3. Barrier Function in Non-Celiac Gluten/Wheat Sensitivity
3. The Role of Barrier Function Tests in Assessing Intestinal Permeability
3.1. In Vivo Tests
3.2. Ex Vivo Analysis of Barrier Function
3.3. Non-Invasive Biomarkers
4. Is the Barrier a Potential Therapeutic Target?
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- France, M.M.; Turner, J.R. The mucosal barrier at a glance. J. Cell Sci. 2017, 130, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Salvo Romero, E.; Alonso Cotoner, C.; Pardo Camacho, C.; Casado Bedmar, M.; Vicario, M. The intestinal barrier function and its involvement in digestive disease. Rev. Esp. Enferm. Dig. 2015, 107, 686–696. [Google Scholar] [CrossRef]
- Johansson, M.E.V.; Gustafsson, J.K.; Holmén-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjövall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Pelaseyed, T.; Bergström, J.H.; Gustafsson, J.K.; Ermund, A.; Birchenough, G.M.H.; Schütte, A.; van der Post, S.; Svensson, F.; Rodríguez-Piñeiro, A.M.; Nyström, E.E.L.; et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014, 260, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Bansil, R.; Turner, B.S. The biology of mucus: Composition, synthesis and organization. Adv. Drug Deliv. Rev. 2018, 124, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.S.D.A.; Melo, E.O. Mucin 2 (MUC2) promoter characterization: An overview. Cell Tissue Res. 2018, 374, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, M.; De Koning, B.A.E.; De Bruijn, A.C.J.M.; Velcich, A.; Meijerink, J.P.P.; Van Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Nakimbugwe, D.; Masschalck, B.; Deckers, D.; Callewaert, L.; Aertsen, A.; Michiels, C.W. Cell wall substrate specificity of six different lysozymes and lysozyme inhibitory activity of bacterial extracts. FEMS Microbiol. Lett. 2006, 259, 41–46. [Google Scholar] [CrossRef]
- Ragland, S.A.; Criss, A.K. From bacterial killing to immune modulation: Recent insights into the functions of lysozyme. PLoS Pathog. 2017, 13, e1006512. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Geuking, M.B.; Slack, E.; Hapfelmeier, S.; McCoy, K.D. The habitat, double life, citizenship, and forgetfulness of IgA. Immunol. Rev. 2012, 245, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Scherf, W.; Burdach, S.; Hansen, G. Reduced expression of transforming growth factor beta 1 exacerbates pathology in an experimental asthma model. Eur. J. Immunol. 2005, 35, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Makinde, T.; Murphy, R.F.; Agrawal, D.K. The regulatory role of TGF-beta in airway remodeling in asthma. Immunol. Cell Biol. 2007, 85, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, e338. [Google Scholar] [CrossRef] [PubMed]
- Martini, E.; Krug, S.M.; Siegmund, B.; Neurath, M.F.; Becker, C. Mend Your Fences: The Epithelial Barrier and its Relationship with Mucosal Immunity in Inflammatory Bowel Disease. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 33–46. [Google Scholar] [CrossRef]
- Miron, N.; Cristea, V. Enterocytes: Active cells in tolerance to food and microbial antigens in the gut. Clin. Exp. Immunol. 2012, 167, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The Intestinal Crypt, A Prototype Stem Cell Compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef]
- Bastide, P.; Darido, C.; Pannequin, J.; Kist, R.; Robine, S.; Marty-Double, C.; Bibeau, F.; Scherer, G.; Joubert, D.; Hollande, F.; et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J. Cell Biol. 2007, 178, 635–648. [Google Scholar] [CrossRef]
- Bevins, C.L.; Salzman, N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 2011, 9, 356–368. [Google Scholar] [CrossRef]
- Armbruster, N.S.; Stange, E.F.; Wehkamp, J. In the Wnt of Paneth Cells: Immune-Epithelial Crosstalk in Small Intestinal Crohn’s Disease. Front. Immunol. 2017, 8, 1204. [Google Scholar] [CrossRef]
- Clevers, H.C.; Bevins, C.L. Paneth cells: Maestros of the small intestinal crypts. Annu. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Kucharzik, T.; Lügering, N.; Rautenberg, K.; Lügering, A.; Schmidt, M.A.; Stoll, R.; Domschke, W. Role of M cells in intestinal barrier function. Ann. N. Y. Acad. Sci. 2000, 915, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Ting, H.-A.; von Moltke, J. The Immune Function of Tuft Cells at Gut Mucosal Surfaces and Beyond. J. Immunol. 2019, 202, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Campbell, H.K.; Maiers, J.L.; DeMali, K.A. Interplay between tight junctions & adherens junctions. Exp. Cell Res. 2017, 358, 39–44. [Google Scholar] [PubMed]
- Balda, M.S.; Flores-Maldonado, C.; Cereijido, M.; Matter, K. Multiple domains of occludin are involved in the regulation of paracellular permeability. J. Cell. Biochem. 2000, 78, 85–96. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Sasaki, H.; Tsukita, S.; Furuse, M.; Tsukita, S. Loss of occludin affects tricellular localization of tricellulin. Mol. Biol. Cell 2008, 19, 4687–4693. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Furuse, M.; Sasaki, H.; Schulzke, J.D.; Fromm, M.; Takano, H.; Noda, T.; Tsukita, S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 2000, 11, 4131–4142. [Google Scholar] [CrossRef]
- Schulzke, J.D.; Gitter, A.H.; Mankertz, J.; Spiegel, S.; Seidler, U.; Amasheh, S.; Saitou, M.; Tsukita, S.; Fromm, M. Epithelial transport and barrier function in occludin-deficient mice. Biochim. Biophys. Acta 2005, 1669, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.F.; Hildner, M.; Schmauder, R.; Turner, J.R.; Schumann, M.; Reiche, J. Occludin knockdown is not sufficient to induce transepithelial macromolecule passage. Tissue Barriers 2019, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and-2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef]
- Günzel, D.; Yu, A.S.L. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Tietgens, A.J.; Krystofiak, E.; Kachar, B.; Anderson, J.M. A complex of ZO-1 and the BAR-domain protein TOCA-1 regulates actin assembly at the tight junction. Mol. Biol. Cell 2015, 26, 2769–2787. [Google Scholar] [CrossRef]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.-D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, S.L.; Argani, P.; Korz, D.; Evron, E.; Raman, V.; Garrett, E.; Rein, A.; Sauter, G.; Kallioniemi, O.-P.; Sukumar, S. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene 2003, 22, 2021–2033. [Google Scholar] [CrossRef]
- Zhu, Y.; Brännström, M.; Janson, P.-O.; Sundfeldt, K. Differences in expression patterns of the tight junction proteins, claudin 1, 3, 4 and 5, in human ovarian surface epithelium as compared to epithelia in inclusion cysts and epithelial ovarian tumours. Int. J. Cancer 2006, 118, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Sasaki, H.; Furuse, M.; Ozaki, H.; Kita, T.; Tsukita, S. Junctional adhesion molecule (JAM) binds to PAR-3. J. Cell Biol. 2001, 154, 491–498. [Google Scholar] [CrossRef]
- Laukoetter, M.G.; Nava, P.; Lee, W.Y.; Severson, E.A.; Capaldo, C.T.; Babbin, B.A.; Williams, I.R.; Koval, M.; Peatman, E.; Campbell, J.A.; et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007, 204, 3067–3076. [Google Scholar] [CrossRef]
- Vetrano, S.; Rescigno, M.; Cera, M.R.; Correale, C.; Rumio, C.; Doni, A.; Fantini, M.; Sturm, A.; Borroni, E.; Repici, A.; et al. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology 2008, 135, 173–184. [Google Scholar] [CrossRef]
- Nelson, W.J. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem. Soc. Trans. 2008, 36, 149–155. [Google Scholar] [CrossRef]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-Cadherin/β-Catenin Complex and the Epithelial Barrier. J. Biomed Biotechnol. 2011. [Google Scholar] [CrossRef]
- Baum, B.; Georgiou, M. Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J. Cell Biol. 2011, 192, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Gehren, A.S.; Rocha, M.R.; de Souza, W.F.; Morgado-Díaz, J.A. Alterations of the apical junctional complex and actin cytoskeleton and their role in colorectal cancer progression. Tissue Barriers 2015, 3, e1017688. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, W.; Nakanishi, H.; Miyoshi, J.; Mandai, K.; Ishizaki, H.; Tanaka, M.; Togawa, A.; Takahashi, K.; Nishioka, H.; Yoshida, H.; et al. Afadin: A key molecule essential for structural organization of cell-cell junctions of polarized epithelia during embryogenesis. J. Cell Biol. 1999, 146, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Fujita, N.; Yamada, A.; Ooshio, T.; Okamoto, R.; Irie, K.; Takai, Y. Regulation of the assembly and adhesion activity of E-cadherin by nectin and afadin for the formation of adherens junctions in Madin-Darby canine kidney cells. J. Biol. Chem. 2006, 281, 5288–5299. [Google Scholar] [CrossRef] [PubMed]
- Moens, E.; Veldhoen, M. Epithelial barrier biology: Good fences make good neighbours. Immunology 2012, 135, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Varol, C.; Zigmond, E.; Jung, S. Securing the immune tightrope: Mononuclear phagocytes in the intestinal lamina propria. Nat. Rev. Immunol. 2010, 10, 415–426. [Google Scholar] [CrossRef]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef]
- Pull, S.L.; Doherty, J.M.; Mills, J.C.; Gordon, J.I.; Stappenbeck, T.S. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc. Natl. Acad. Sci. USA 2005, 102, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Grainger, J.R.; Wohlfert, E.A.; Fuss, I.J.; Bouladoux, N.; Askenase, M.H.; Legrand, F.; Koo, L.Y.; Brenchley, J.M.; Fraser, I.D.C.; Belkaid, Y. Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat. Med. 2013, 19, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Denning, T.L.; Wang, Y.; Patel, S.R.; Williams, I.R.; Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 2007, 8, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Tumanov, A.V.; Koroleva, E.P.; Guo, X.; Wang, Y.; Kruglov, A.; Nedospasov, S.; Fu, Y.-X. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe 2011, 10, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Qiu, J.; Tu, T.; Yang, X.; Deng, L.; Anders, R.A.; Zhou, L.; Fu, Y.-X. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity 2014, 40, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Eken, A.; Singh, A.K.; Treuting, P.M.; Oukka, M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. 2014, 7, 143–154. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Dewi, F.N.; Hisaeda, H.; Smith, A.L.; Jimi, F.; Miyahira, M.; Abdel-Aleem, A.S.F.; Horii, Y.; Nawa, Y. Intestinal intraepithelial lymphocytes sustain the epithelial barrier function against Eimeria vermiformis infection. Infect. Immun. 2006, 74, 5292–5301. [Google Scholar] [CrossRef] [PubMed]
- Saurer, L.; Mueller, C. T cell-mediated immunoregulation in the gastrointestinal tract. Allergy 2009, 64, 505–519. [Google Scholar] [CrossRef]
- Kuhn, K.A.; Schulz, H.M.; Regner, E.H.; Severs, E.L.; Hendrickson, J.D.; Mehta, G.; Whitney, A.K.; Ir, D.; Ohri, N.; Robertson, C.E.; et al. Bacteroidales recruit IL-6-producing intraepithelial lymphocytes in the colon to promote barrier integrity. Mucosal Immunol. 2018, 11, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.; Murray, D. Quantitation of intraepithelial lymphocytes in human jejunum. Gut 1971, 12, 988–994. [Google Scholar] [CrossRef]
- Mayassi, T.; Jabri, B. Human intraepithelial lymphocytes. Mucosal Immunol. 2018, 11, 1281–1289. [Google Scholar] [CrossRef]
- Hüe, S.; Mention, J.-J.; Monteiro, R.C.; Zhang, S.; Cellier, C.; Schmitz, J.; Verkarre, V.; Fodil, N.; Bahram, S.; Cerf-Bensussan, N.; et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 2004, 21, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Meresse, B.; Curran, S.A.; Ciszewski, C.; Orbelyan, G.; Setty, M.; Bhagat, G.; Lee, L.; Tretiakova, M.; Semrad, C.; Kistner, E.; et al. Reprogramming of CTLs into natural killer–like cells in celiac disease. J. Exp. Med. 2006, 203, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Jabri, B.; Abadie, V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol. 2015, 15, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Ostaff, M.J.; Stange, E.F.; Wehkamp, J. Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol. Med. 2013, 5, 1465–1483. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, L.A.G.; Ruigómez, A. Increased risk of irritable bowel syndrome after bacterial gastroenteritis: Cohort study. Bmj 1999, 318, 565–566. [Google Scholar] [CrossRef] [PubMed]
- Al-Toma, A.; Volta, U.; Auricchio, R.; Castillejo, G.; Sanders, D.S.; Cellier, C.; Mulder, C.J.; Lundin, K.E.A. European Society for the Study of Coeliac Disease (ESsCD) guideline for coeliac disease and other gluten-related disorders. United Eur. Gastroenterol. J. 2019, 7, 583–613. [Google Scholar] [CrossRef]
- Malamut, G.; El Machhour, R.; Montcuquet, N.; Martin-Lannerée, S.; Dusanter-Fourt, I.; Verkarre, V.; Mention, J.-J.; Rahmi, G.; Kiyono, H.; Butz, E.A.; et al. IL-15 triggers an antiapoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease-associated inflammation and lymphomagenesis. J. Clin. Investig. 2010, 120, 2131–2143. [Google Scholar] [CrossRef]
- Hamilton, I.; Cobden, I.; Rothwell, J.; Axon, A.T. Intestinal permeability in coeliac disease: The response to gluten withdrawal and single-dose gluten challenge. Gut 1982, 23, 202–210. [Google Scholar] [CrossRef]
- Maiuri, L.; Ciacci, C.; Ricciardelli, I.; Vacca, L.; Raia, V.; Auricchio, S.; Picard, J.; Osman, M.; Quaratino, S.; Londei, M. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 2003, 362, 30–37. [Google Scholar] [CrossRef]
- Sander, G.R.; Cummins, A.G.; Henshall, T.; Powell, B.C. Rapid disruption of intestinal barrier function by gliadin involves altered expression of apical junctional proteins. FEBS Lett. 2005, 579, 4851–4855. [Google Scholar] [CrossRef]
- Drago, S.; El Asmar, R.; Di Pierro, M.; Grazia Clemente, M.; Tripathi, A.; Sapone, A.; Thakar, M.; Iacono, G.; Carroccio, A.; D’Agate, C.; et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand. J. Gastroenterol. 2006, 41, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Lammers, K.M.; Lu, R.; Brownley, J.; Lu, B.; Gerard, C.; Thomas, K.; Rallabhandi, P.; Shea-Donohue, T.; Tamiz, A.; Alkan, S.; et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 2008, 135, 194–204.e3. [Google Scholar] [CrossRef] [PubMed]
- Lindfors, K.; Ciacci, C.; Kurppa, K.; Lundin, K.E.A.; Makharia, G.K.; Mearin, M.L.; Murray, J.A.; Verdu, E.F.; Kaukinen, K. Coeliac disease. Nat. Rev. Dis. Primers 2019, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Stamnaes, J.; Sollid, L.M. Celiac disease: Autoimmunity in response to food antigen. Semin. Immunol. 2015, 27, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Cobden, I.; Dickinson, R.J.; Rothwell, J.; Axon, A.T. Intestinal permeability assessed by excretion ratios of two molecules: Results in coeliac disease. Br. Med. J. 1978, 2, 1060. [Google Scholar] [CrossRef]
- Menzies, I.S.; Laker, M.F.; Pounder, R.; Bull, J.; Heyer, S.; Wheeler, P.G.; Creamer, B. Abnormal intestinal permeability to sugars in villous atrophy. Lancet 1979, 2, 1107–1109. [Google Scholar] [CrossRef]
- Pearson, A.D.; Eastham, E.J.; Laker, M.F.; Craft, A.W.; Nelson, R. Intestinal permeability in children with Crohn’s disease and coeliac disease. Br. Med. J. (Clin. Res. Ed.) 1982, 285, 20–21. [Google Scholar] [CrossRef]
- Schulzke, J.D.; Bentzel, C.J.; Schulzke, I.; Riecken, E.O.; Fromm, M. Epithelial tight junction structure in the jejunum of children with acute and treated celiac sprue. Pediatr. Res. 1998, 43, 435–441. [Google Scholar] [CrossRef]
- Schulzke, J.D.; Schulzke, I.; Fromm, M.; Riecken, E.O. Epithelial barrier and ion transport in coeliac sprue: Electrical measurements on intestinal aspiration biopsy specimens. Gut 1995, 37, 777–782. [Google Scholar] [CrossRef]
- Reims, A.; Strandvik, B.; Sjövall, H. Epithelial electrical resistance as a measure of permeability changes in pediatric duodenal biopsies. J. Pediatr. Gastroenterol. Nutr. 2006, 43, 619–623. [Google Scholar] [CrossRef]
- Schumann, M.; Günzel, D.; Buergel, N.; Richter, J.F.; Troeger, H.; May, C.; Fromm, A.; Sorgenfrei, D.; Daum, S.; Bojarski, C.; et al. Cell polarity-determining proteins Par-3 and PP-1 are involved in epithelial tight junction defects in coeliac disease. Gut 2012, 61, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Schumann, M.; Siegmund, B.; Schulzke, J.D.; Fromm, M. Celiac Disease: Role of the Epithelial Barrier. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Schumann, M.; Kamel, S.; Pahlitzsch, M.-L.; Lebenheim, L.; May, C.; Krauss, M.; Hummel, M.; Daum, S.; Fromm, M.; Schulzke, J.-D. Defective tight junctions in refractory celiac disease. Ann. N. Y. Acad. Sci. 2012, 1258, 43–51. [Google Scholar] [CrossRef]
- Ciccocioppo, R.; Finamore, A.; Ara, C.; Di Sabatino, A.; Mengheri, E.; Corazza, G.R. Altered expression, localization, and phosphorylation of epithelial junctional proteins in celiac disease. Am. J. Clin. Pathol. 2006, 125, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Szakál, D.N.; Gyorffy, H.; Arató, A.; Cseh, A.; Molnár, K.; Papp, M.; Dezsofi, A.; Veres, G. Mucosal expression of claudins 2, 3 and 4 in proximal and distal part of duodenum in children with coeliac disease. Virchows Arch. 2010, 456, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Trynka, G.; Hunt, K.A.; Bockett, N.A.; Romanos, J.; Mistry, V.; Szperl, A.; Bakker, S.F.; Bardella, M.T.; Bhaw-Rosun, L.; Castillejo, G.; et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 2011, 43, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Gutierrez-Achury, J.; Kanduri, K.; Almeida, R.; Hrdlickova, B.; Zhernakova, D.V.; Westra, H.-J.; Karjalainen, J.; Ricaño-Ponce, I.; Li, Y.; et al. Systematic annotation of celiac disease loci refines pathological pathways and suggests a genetic explanation for increased interferon-gamma levels. Hum. Mol. Genet. 2015, 24, 397–409. [Google Scholar] [CrossRef]
- Almeida, R.; Ricaño-Ponce, I.; Kumar, V.; Deelen, P.; Szperl, A.; Trynka, G.; Gutierrez-Achury, J.; Kanterakis, A.; Westra, H.-J.; Franke, L.; et al. Fine mapping of the celiac disease-associated LPP locus reveals a potential functional variant. Hum. Mol. Genet. 2014, 23, 2481–2489. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Tietgens, A.J.; Aponte, A.; Fredriksson, K.; Fanning, A.S.; Gucek, M.; Anderson, J.M. Biotin ligase tagging identifies proteins proximal to E-cadherin, including lipoma preferred partner, a regulator of epithelial cell-cell and cell-substrate adhesion. J. Cell Sci. 2014, 127, 885–895. [Google Scholar] [CrossRef]
- Mohanan, V.; Nakata, T.; Desch, A.N.; Lévesque, C.; Boroughs, A.; Guzman, G.; Cao, Z.; Creasey, E.; Yao, J.; Boucher, G.; et al. C1orf106 is a colitis risk gene that regulates stability of epithelial adherens junctions. Science 2018, 359, 1161–1166. [Google Scholar] [CrossRef]
- Fearnley, G.W.; Young, K.A.; Edgar, J.R.; Antrobus, R.; Hay, I.M.; Liang, W.-C.; Martinez-Martin, N.; Lin, W.; Deane, J.E.; Sharpe, H.J. The homophilic receptor PTPRK selectively dephosphorylates multiple junctional regulators to promote cell-cell adhesion. Elife 2019, 8, e44597. [Google Scholar] [CrossRef]
- Wapenaar, M.C.; Monsuur, A.J.; van Bodegraven, A.A.; Weersma, R.K.; Bevova, M.R.; Linskens, R.K.; Howdle, P.; Holmes, G.; Mulder, C.J.; Dijkstra, G.; et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitis. Gut 2008, 57, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Dubois, P.C.A.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.R.; Adány, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Inomata, N. Wheat allergy. Curr. Opin. Allergy Clin. Immunol. 2009, 9, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Pasha, I.; Saeed, F.; Sultan, M.T.; Batool, R.; Aziz, M.; Ahmed, W. Wheat Allergy and Intolerence; Recent Updates and Perspectives. Crit. Rev. Food Sci. Nutr. 2016, 56, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ruffner, M.A.; Spergel, J.M. Non-IgE Mediated Food Allergy Syndromes. Ann. Allergy Asthma Immunol. 2016, 117, 452–454. [Google Scholar] [CrossRef]
- Cianferoni, A. Wheat allergy: Diagnosis and management. J. Asthma Allergy 2016, 9, 13–25. [Google Scholar] [CrossRef]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat Med 2012, 18, 693–704. [Google Scholar] [CrossRef]
- Catassi, C.; Alaedini, A.; Bojarski, C.; Bonaz, B.; Bouma, G.; Carroccio, A.; Castillejo, G.; De Magistris, L.; Dieterich, W.; Di Liberto, D.; et al. The Overlapping Area of Non-Celiac Gluten Sensitivity (NCGS) and Wheat-Sensitive Irritable Bowel Syndrome (IBS): An Update. Nutrients 2017, 9, 1268. [Google Scholar] [CrossRef]
- Albert-Bayo, M.; Paracuellos, I.; González-Castro, A.M.; Rodríguez-Urrutia, A.; Rodríguez-Lagunas, M.J.; Alonso-Cotoner, C.; Santos, J.; Vicario, M. Intestinal Mucosal Mast Cells: Key Modulators of Barrier Function and Homeostasis. Cells 2019, 8, 135. [Google Scholar] [CrossRef]
- Sehra, S.; Serezani, A.P.M.; Ocaña, J.A.; Travers, J.B.; Kaplan, M.H. Mast Cells Regulate Epidermal Barrier Function and the Development of Allergic Skin Inflammation. J. Investig. Dermatol. 2016, 136, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.R.; Bartram, R.E.; Knight, P.A.; Miller, H.R.P.; Garrod, D.R.; Grencis, R.K. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc. Natl. Acad. Sci. USA 2003, 100, 7761–7766. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Saeki, K.; Liu, M.; Sasaki, F.; Koga, T.; Kitajima, K.; Meno, C.; Okuno, T.; Yokomizo, T. Leukotriene B4 receptor type 2 (BLT2) enhances skin barrier function by regulating tight junction proteins. FASEB J. 2016, 30, 933–947. [Google Scholar] [CrossRef]
- Iizuka, Y.; Okuno, T.; Saeki, K.; Uozaki, H.; Okada, S.; Misaka, T.; Sato, T.; Toh, H.; Fukayama, M.; Takeda, N.; et al. Protective role of the leukotriene B4 receptor BLT2 in murine inflammatory colitis. FASEB J. 2010, 24, 4678–4690. [Google Scholar] [CrossRef] [PubMed]
- Sherrill, J.D.; Kc, K.; Wu, D.; Djukic, Z.; Caldwell, J.M.; Stucke, E.M.; Kemme, K.A.; Costello, M.S.; Mingler, M.K.; Blanchard, C.; et al. Desmoglein-1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal Immunol. 2014, 7, 718–729. [Google Scholar] [CrossRef]
- Roszkowska, A.; Pawlicka, M.; Mroczek, A.; Bałabuszek, K.; Nieradko-Iwanicka, B. Non-Celiac Gluten Sensitivity: A Review. Medicina (Kaunas) 2019, 55, 222. [Google Scholar] [CrossRef] [PubMed]
- Catassi, C.; Bai, J.C.; Bonaz, B.; Bouma, G.; Calabrò, A.; Carroccio, A.; Castillejo, G.; Ciacci, C.; Cristofori, F.; Dolinsek, J.; et al. Non-Celiac Gluten sensitivity: The new frontier of gluten related disorders. Nutrients 2013, 5, 3839–3853. [Google Scholar] [CrossRef]
- Barmeyer, C.; Schumann, M.; Meyer, T.; Zielinski, C.; Zuberbier, T.; Siegmund, B.; Schulzke, J.-D.; Daum, S.; Ullrich, R. Long-term response to gluten-free diet as evidence for non-celiac wheat sensitivity in one third of patients with diarrhea-dominant and mixed-type irritable bowel syndrome. Int. J. Colorectal Dis. 2017, 32, 29–39. [Google Scholar] [CrossRef]
- Schuppan, D.; Pickert, G.; Ashfaq-Khan, M.; Zevallos, V. Non-celiac wheat sensitivity: Differential diagnosis, triggers and implications. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 469–476. [Google Scholar] [CrossRef]
- Biesiekierski, J.R.; Peters, S.L.; Newnham, E.D.; Rosella, O.; Muir, J.G.; Gibson, P.R. No effects of gluten in patients with self-reported non-celiac gluten sensitivity after dietary reduction of fermentable, poorly absorbed, short-chain carbohydrates. Gastroenterology 2013, 145, 320–328. [Google Scholar] [CrossRef]
- Leccioli, V.; Oliveri, M.; Romeo, M.; Berretta, M.; Rossi, P. A New Proposal for the Pathogenic Mechanism of Non-Coeliac/Non-Allergic Gluten/Wheat Sensitivity: Piecing Together the Puzzle of Recent Scientific Evidence. Nutrients 2017, 9, 1203. [Google Scholar] [CrossRef] [PubMed]
- Sapone, A.; Lammers, K.M.; Casolaro, V.; Cammarota, M.; Giuliano, M.T.; De Rosa, M.; Stefanile, R.; Mazzarella, G.; Tolone, C.; Russo, M.I.; et al. Divergence of gut permeability and mucosal immune gene expression in two gluten-associated conditions: Celiac disease and gluten sensitivity. BMC Med. 2011, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Hollon, J.; Puppa, E.L.; Greenwald, B.; Goldberg, E.; Guerrerio, A.; Fasano, A. Effect of gliadin on permeability of intestinal biopsy explants from celiac disease patients and patients with non-celiac gluten sensitivity. Nutrients 2015, 7, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Fritscher-Ravens, A.; Schuppan, D.; Ellrichmann, M.; Schoch, S.; Röcken, C.; Brasch, J.; Bethge, J.; Böttner, M.; Klose, J.; Milla, P.J. Confocal endomicroscopy shows food-associated changes in the intestinal mucosa of patients with irritable bowel syndrome. Gastroenterology 2014, 147, 1012–1020. [Google Scholar] [CrossRef]
- Fritscher-Ravens, A.; Pflaum, T.; Mösinger, M.; Ruchay, Z.; Röcken, C.; Milla, P.J.; Das, M.; Böttner, M.; Wedel, T.; Schuppan, D. Many Patients with Irritable Bowel Syndrome Have Atypical Food Allergies Not Associated with Immunoglobulin E. Gastroenterology 2019, 157, 109–118. [Google Scholar] [CrossRef]
- Uhde, M.; Ajamian, M.; Caio, G.; De Giorgio, R.; Indart, A.; Green, P.H.; Verna, E.C.; Volta, U.; Alaedini, A. Intestinal cell damage and systemic immune activation in individuals reporting sensitivity to wheat in the absence of coeliac disease. Gut 2016, 65, 1930–1937. [Google Scholar] [CrossRef]
- Wang, L.; Llorente, C.; Hartmann, P.; Yang, A.-M.; Chen, P.; Schnabl, B. Methods to determine intestinal permeability and bacterial translocation during liver disease. J. Immunol. Methods 2015, 421, 44–53. [Google Scholar] [CrossRef]
- Galipeau, H.J.; Verdu, E.F. The complex task of measuring intestinal permeability in basic and clinical science. Neurogastroenterol. Motil. 2016, 28, 957–965. [Google Scholar] [CrossRef]
- Bjarnason, I.; MacPherson, A.; Hollander, D. Intestinal permeability: An overview. Gastroenterology 1995, 108, 1566–1581. [Google Scholar] [CrossRef]
- Fasano, A. Intestinal Permeability and its Regulation by Zonulin: Diagnostic and Therapeutic Implications. Clin. Gastroenterol. Hepatol. 2012, 10, 1096–1100. [Google Scholar] [CrossRef]
- Ajamian, M.; Steer, D.; Rosella, G.; Gibson, P.R. Serum zonulin as a marker of intestinal mucosal barrier function: May not be what it seems. PLoS ONE 2019, 14, e0210728. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.; Krug, S.M.; Zeissig, S.; Richter, J.F.; Rosenthal, R.; Schulzke, J.-D.; Günzel, D. High-resolution analysis of barrier function. Ann. N. Y. Acad. Sci. 2009, 1165, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Hegel, U.; Fromm, M.; Kreusel, K.M.; Wiederholt, M. Bovine and porcine large intestine as model epithelia in a student lab course. Am. J. Physiol. 1993, 265, S10–S19. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.F.; Schmauder, R.; Krug, S.M.; Gebert, A.; Schumann, M. A novel method for imaging sites of paracellular passage of macromolecules in epithelial sheets. J. Controll. Release 2016, 229, 70–79. [Google Scholar] [CrossRef]
- Reiche, J.; Schumann, M.; Richter, J.F. The Sandwich Assay: A Method for Subcellular Visualization of Paracellular Macromolecule Passage in Epithelial Sheets. Curr. Protoc. Cell Biol. 2018, 78, 20.10.1–20.10.13. [Google Scholar] [CrossRef] [PubMed]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Fragkos, K.C.; Forbes, A. Citrulline as a marker of intestinal function and absorption in clinical settings: A systematic review and meta-analysis. United Eur. Gastroenterol. J. 2018, 6, 181–191. [Google Scholar] [CrossRef]
- Sturgeon, C.; Fasano, A. Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers 2016, 4, e1251384. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Durai, M.; Kitchens, K.; Tamiz, A.P.; Somerville, R.; Ginski, M.; Paterson, B.M.; Murray, J.A.; Verdu, E.F.; Alkan, S.S.; et al. Larazotide acetate regulates epithelial tight junctions in vitro and in vivo. Peptides 2012, 35, 86–94. [Google Scholar] [CrossRef]
- Paterson, B.M.; Lammers, K.M.; Arrieta, M.C.; Fasano, A.; Meddings, J.B. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: A proof of concept study. Aliment. Pharmacol. Ther. 2007, 26, 757–766. [Google Scholar] [CrossRef]
- Leffler, D.A.; Kelly, C.P.; Abdallah, H.Z.; Colatrella, A.M.; Harris, L.A.; Leon, F.; Arterburn, L.A.; Paterson, B.M.; Lan, Z.H.; Murray, J.A. A randomized, double-blind study of larazotide acetate to prevent the activation of celiac disease during gluten challenge. Am. J. Gastroenterol. 2012, 107, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.P.; Green, P.H.R.; Murray, J.A.; Dimarino, A.; Colatrella, A.; Leffler, D.A.; Alexander, T.; Arsenescu, R.; Leon, F.; Jiang, J.G.; et al. Larazotide acetate in patients with coeliac disease undergoing a gluten challenge: A randomised placebo-controlled study. Aliment. Pharmacol. Ther. 2013, 37, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Leffler, D.A.; Kelly, C.P.; Green, P.H.R.; Fedorak, R.N.; DiMarino, A.; Perrow, W.; Rasmussen, H.; Wang, C.; Bercik, P.; Bachir, N.M.; et al. Larazotide Acetate for Persistent Symptoms of Celiac Disease Despite a Gluten-Free Diet: A Randomized Controlled Trial. Gastroenterology 2015, 148, 1311–1319. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Tight Junction | Functions |
| Occludin | Constitution of TJ strand? |
| Claudins | TJ and epithelial barrier formation Constitution of TJ strands Cytoskeleton organization |
| JAM | TJ maintenance Role in barrier regulation |
| Adherens junction | Functions |
| Nectin-afadin | AJ organization and maturation |
| E-cadherin-β-catenin | Interaction with components of the cytoskeleton Organization and maintenance of AJ |
| Type of Change | Observations | Reference |
|---|---|---|
| Functional | Increased cellobiose/mannitol excretion ratio | [75] |
| Functional | Increased Lactulose/L-rhamnose excretion ratio | [76] |
| Functional | 5-fold increase in Lactulose/mannitol excretion ratio | [77] |
| Functional | 13-fold increase in cellobiose/mannitol ratios in active CeD. 2-fold increase in treated patients and 5-fold increase in non-responders | [68] |
| Cellobiose/mannitol excretion ratio reached normal levels after treatment and increased transiently after a gluten challenge | ||
| Functional | 56% reduction in electrical epithelial resistance in active CeD and 25% reduction in treated CeD | [79] |
| Structural | Decreased number of tight junction strands and depth of TJ meshwork in active CeD. Partial recovery in treated CeD | [78] |
| Discontinued strands and aberrant strands below the main junctional meshwork | ||
| Molecular | Loss of co-immunoprecipitation of occludin and ZO-1 despite no changes in total protein levels. Decrease in the membrane localization of ZO-1 and occludin | [84] |
| Loss of co-immunoprecipitation of E-cadherin and β-Catenin despite normal levels of in total protein. Extensive phosphorylation of β-Catenin. Redistribution of both AJ proteins from the membrane to the cytoplasm | ||
| Functional | 26% reduction in paracellular resistance in CeD patients with partial and subtotal atrophy. 16% reduction in treated CeD | [80] |
| Functional | Electrical resistance decrease after exposure to PT-gliadin in samples from CeD patients | [71] |
| Molecular | Zonulin release and decrease in occludin gene expression after PT-gliadin exposure | |
| Molecular | Increased claudin-2 and -3 in patient samples with villous atrophy | [85] |
| Molecular | Increased claudin-2, -15 and decreased claudin-3, -5, -7 and occludin. No changes in claudin-2 RNA. Intense claudin-2 staining in the TJ of the crypts. Reduced membrane localization of claudin-3 and ZO-1. Partial membrane localization of claudin 5 and 15 and inhomogeneous claudin-7 staining | [81] |
| Functional | 48% reduction of electrical resistance in CeD | |
| Molecular | Increased Claudin-2 in the crypts and decreased Claudin-4 and -5 in Refractory patients | [83] |
| Functional | 40% decrease in epithelial resistance in refractory patients | |
| Functional | Only partial recovery of epithelial resistance in treated CeD | [82] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso-Silva, D.; Delbue, D.; Itzlinger, A.; Moerkens, R.; Withoff, S.; Branchi, F.; Schumann, M. Intestinal Barrier Function in Gluten-Related Disorders. Nutrients 2019, 11, 2325. https://doi.org/10.3390/nu11102325
Cardoso-Silva D, Delbue D, Itzlinger A, Moerkens R, Withoff S, Branchi F, Schumann M. Intestinal Barrier Function in Gluten-Related Disorders. Nutrients. 2019; 11(10):2325. https://doi.org/10.3390/nu11102325
Chicago/Turabian StyleCardoso-Silva, Danielle, Deborah Delbue, Alice Itzlinger, Renée Moerkens, Sebo Withoff, Federica Branchi, and Michael Schumann. 2019. "Intestinal Barrier Function in Gluten-Related Disorders" Nutrients 11, no. 10: 2325. https://doi.org/10.3390/nu11102325
APA StyleCardoso-Silva, D., Delbue, D., Itzlinger, A., Moerkens, R., Withoff, S., Branchi, F., & Schumann, M. (2019). Intestinal Barrier Function in Gluten-Related Disorders. Nutrients, 11(10), 2325. https://doi.org/10.3390/nu11102325