Heterozygotes Are a Potential New Entity among Homozygotes and Compound Heterozygotes in Congenital Sucrase-Isomaltase Deficiency



Abstract
1. Introduction
2. Molecular and Cellular Basis of Genetically-Determined Carbohydrate Malabsorption
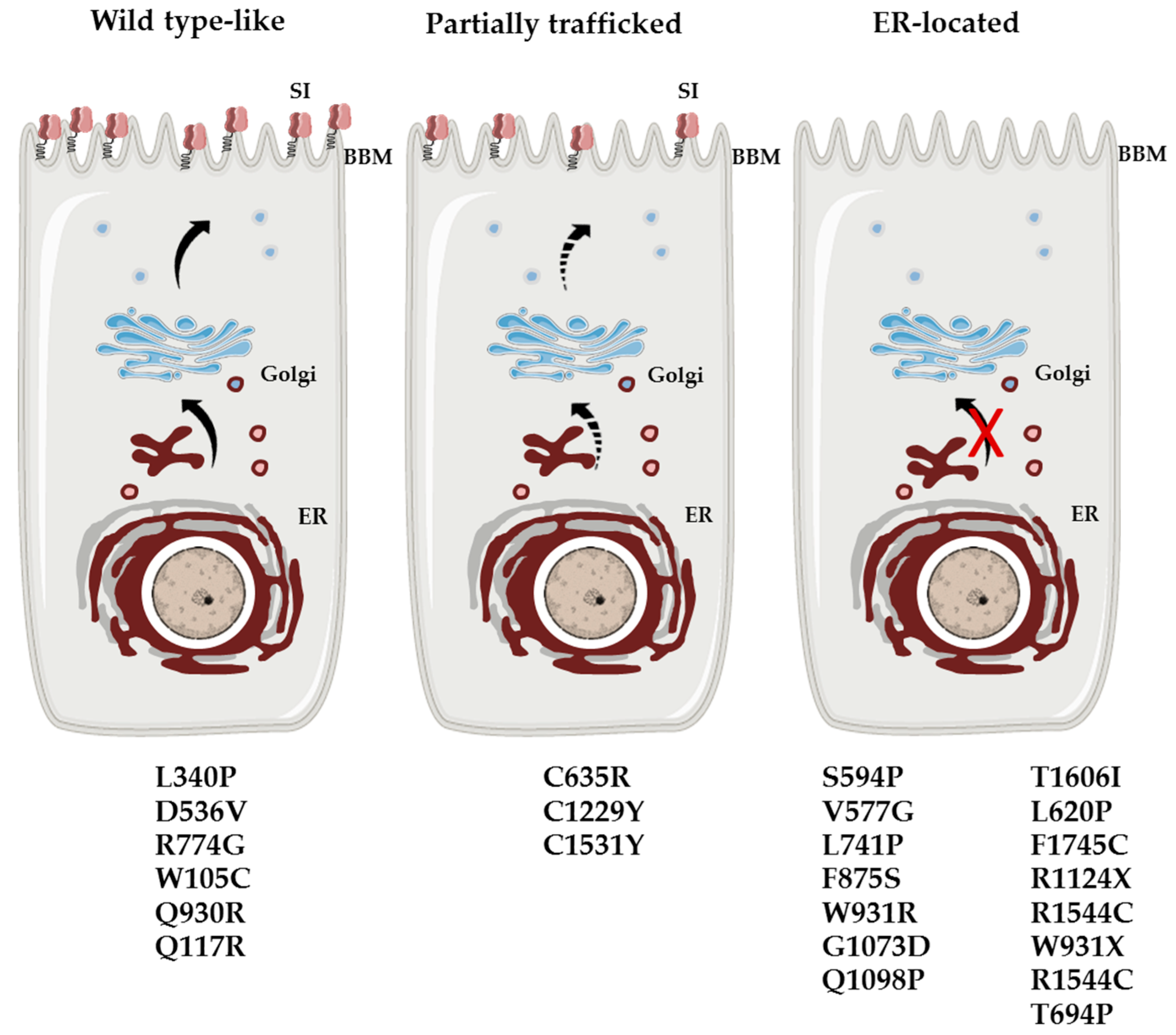
3. Homozygous and Compound Heterozygous Inheritance in CSID
4. Heterozygotes in CSID
4.1. The Quaternary Structure of SI
4.2. The SI Biosynthetic Phenotypes in Intestinal Biopsy Specimens Form CSID Heterozygotes
4.3. The Mosaic Structure of SI in the Enterocytes
5. Future Perspectives
5.1. What Is the Level of the Contribution of the Partner Glycosidase, MGAM, to the Overall Starch-Digestive Capacity and Other Carbohydrates in CSID?
5.2. Do Mutations in Heterozygotes Elicit CSID-Like Symptoms?
5.3. Further Clinical and Nutritional Implications of SI Gene Variants
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Conklin, K.A.; Yamashiro, K.M.; Gray, M.G. Human intestinal sucrase-isomaltase. Identification of free sucrase and isomaltase and cleavage of the hybrid into active distinct subunits. J. Biol. Chem. 1975, 250, 5735–5741. [Google Scholar] [PubMed]
- Ao, Z.; Quezada-Calvillo, R.; Sim, L.; Nichols, B.L.; Rose, D.R.; Sterchi, E.E.; Hamaker, B.R. Evidence of native starch degradation with human small intestinal maltase-glucoamylase (recombinant). FEBS Lett. 2007, 581, 2381–2388. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.H.-M.; Hamaker, B.R.; Nichols, B.L.J. Direct starch digestion by sucrase-isomaltase and maltase-glucoamylase. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-H.; Eskandari, R.; Jones, K.R.; Kongara, R.; Quezada-Calvillo, R.; Nichols, B.L.; Rose, D.R.; Hamaker, B.R.; Pinto, B.M. Modulation of starch digestion for slow glucose release through ‘toggling’ of activities of mucosal alpha-glucosidases. J. Biol. Chem. 2012, 287, 31929–31938. [Google Scholar] [CrossRef] [PubMed]
- Treem, W.R. Clinical aspects and treatment of congenital sucrase-isomaltase deficiency. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Sander, P.; Alfalah, M.; Keiser, M.; Korponay-Szabo, I.; Kovacs, J.B.; Leeb, T.; Naim, H.Y. Novel mutations in the human sucrase-isomaltase gene (SI) that cause congenital carbohydrate malabsorption. Hum. Mutat. 2006, 27, 119. [Google Scholar] [CrossRef] [PubMed]
- Alfalah, M.; Keiser, M.; Leeb, T.; Zimmer, K.-P.; Naim, H.Y. Compound heterozygous mutations affect protein folding and function in patients with congenital sucrase-isomaltase deficiency. Gastroenterology 2009, 136, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Moolenaar, C.E.; Ouwendijk, J.; Wittpoth, M.W.; Heleen, A.; Hauri, H.-P.; Ginsel, L.A.; Naim, H.Y.; Fransen, J.A. A mutation in a highly conserved region in brush-border sucrase-isomaltase and lysosomal alpha-glucosidase results in Golgi retention. J. Cell Sci. 1997, 110, 557–567. [Google Scholar] [PubMed]
- Naim, H.Y.; Heine, M.; Zimmer, K.-P. Congenital sucrase-isomaltase deficiency: Heterogeneity of inheritance, trafficking, and function of an intestinal enzyme complex. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 13–20. [Google Scholar] [CrossRef]
- Amiri, M.; Naim, H.Y. Characterization of Mucosal Disaccharidases from Human Intestine. Nutrients 2017, 9, 1106. [Google Scholar] [CrossRef]
- Robayo-Torres, C.C.; Quezada-Calvillo, R.; Nichols, B.L. Disaccharide digestion: Clinical and molecular aspects. Clin. Gastroenterol. Hepatol. 2006, 4, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Ritz, V.; Alfalah, M.; Zimmer, K.-P.; Schmitz, J.; Jacob, R.; Naim, H.Y. Congenital sucrase-isomaltase deficiency because of an accumulation of the mutant enzyme in the endoplasmic reticulum. Gastroenterology 2003, 125, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Spodsberg, N.; Jacob, R.; Alfalah, M.; Zimmer, K.P.; Naim, H.Y. Molecular basis of aberrant apical protein transport in an intestinal enzyme disorder. J. Biol. Chem. 2001, 276, 23506–23510. [Google Scholar] [CrossRef] [PubMed]
- Fransen, J.A.; Hauri, H.P.; Ginsel, L.A.; Naim, H.Y. Naturally occurring mutations in intestinal sucrase-isomaltase provide evidence for the existence of an intracellular sorting signal in the isomaltase subunit. J. Cell Biol. 1991, 115, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.; Zimmer, K.P.; Schmitz, J.; Naim, H.Y. Congenital sucrase-isomaltase deficiency arising from cleavage and secretion of a mutant form of the enzyme. J. Clin. Investig. 2000, 106, 281–287. [Google Scholar] [CrossRef]
- Gericke, B.; Amiri, M.; Scott, C.R.; Naim, H.Y. Molecular pathogenicity of novel sucrase-isomaltase mutations found in congenital sucrase-isomaltase deficiency patients. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 817–826. [Google Scholar] [CrossRef]
- Keiser, M.; Alfalah, M.; Propsting, M.J.; Castelletti, D.; Naim, H.Y. Altered folding, turnover, and polarized sorting act in concert to define a novel pathomechanism of congenital sucrase-isomaltase deficiency. J. Biol. Chem. 2006, 281, 14393–14399. [Google Scholar] [CrossRef] [PubMed]
- Husein, D.M.; Naim, H. Impaired cell surface expression and digestive function of sucrase-isomaltase gene variants are associated with reduced efficacy of low FODMAPs diet in patients with IBS-D. Gut 2019, 2019, 319411. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, J.; Moolenaar, C.E.; Peters, W.J.; Hollenberg, C.P.; Ginsel, L.A.; Fransen, J.A.; Naim, H.Y. Congenital sucrase-isomaltase deficiency. Identification of a glutamine to proline substitution that leads to a transport block of sucrase-isomaltase in a pre-Golgi compartment. J. Clin. Investig. 1996, 97, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Henstrom, M.; Diekmann, L.; Bonfiglio, F.; Hadizadeh, F.; Kuech, E.; von Kockritz-Blickwede, M.; Thingholm, L.B.; Zheng, T.; Assadi, G.; Dierks, C.; et al. Functional variants in the sucrase-isomaltase gene associate with increased risk of irritable bowel syndrome. Gut 2018, 67, 263–270. [Google Scholar] [CrossRef]
- Zheng, T.; Eswaran, S.; Photenhauer, A.L.; Merchant, J.L.; Chey, W.D.; D’Amato, M. Reduced efficacy of low FODMAPs diet in patients with IBS-D carrying sucrase-isomaltase (SI) hypomorphic variants. Gut 2019, 2018, 318036. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Etxebarria, K.; Zheng, T.; Bonfiglio, F.; Bujanda, L.; Dlugosz, A.; Lindberg, G.; Schmidt, P.T.; Karling, P.; Ohlsson, B.; Simren, M.; et al. Increased Prevalence of Rare Sucrase-isomaltase Pathogenic Variants in Irritable Bowel Syndrome Patients. Clin. Gastroenterol. Hepatol. 2018, 16, 1673–1676. [Google Scholar] [CrossRef] [PubMed]
- Uhrich, S.; Wu, Z.; Huang, J.-Y.; Scott, C.R. SI have been found associated with CSID and were found in a heterozygote background. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 34–35. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, T.; Slusarewicz, P.; Hoe, M.H.; Warren, G. Kin recognition. A model for the retention of Golgi enzymes. FEBS Lett. 1993, 330, 1–4. [Google Scholar] [CrossRef]
- Alfalah, M.; Jacob, R.; Preuss, U.; Zimmer, K.P.; Naim, H.; Naim, H.Y. O-linked glycans mediate apical sorting of human intestinal sucrase-isomaltase through association with lipid rafts. Curr. Biol. 1999, 9, 593–596. [Google Scholar] [CrossRef]
- Reinshagen, K.; Keller, K.M.; Haase, B.; Leeb, T.; Naim, H.Y.; Zimmer, K.P. Mosaic pattern of sucrase isomaltase deficiency in two brothers. Pediatr. Res. 2008, 63, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.L.; Carrazza, F.; Nichols, V.N.; Putman, M.; Johnston, P.; Rodrigues, M.; Quaroni, A.; Shiner, M. Mosaic expression of brush-border enzymes in infants with chronic diarrhea and malnutrition. J. Pediatr. Gastroenterol. Nutr. 1992, 14, 371–379. [Google Scholar] [CrossRef]
- Dahlqvist, A. Assay of intestinal disaccharidases. Scand. J. Clin. Lab. Investig. 1984, 44, 169–172. [Google Scholar] [CrossRef]
- Deng, Y.; Misselwitz, B.; Dai, N.; Fox, M. Lactose Intolerance in Adults: Biological Mechanism and Dietary Management. Nutrients 2015, 7, 8020–8035. [Google Scholar] [CrossRef]
- Lovell, R.M.; Ford, A.C. Global prevalence of and risk factors for irritable bowel syndrome: A meta-analysis. Clin. Gastroenterol. Hepatology 2012, 10, 712–721. [Google Scholar]
- Chumpitazi, B.P.; Shulman, R.J. Dietary Carbohydrates and Childhood Functional Abdominal Pain. Ann. Nutr. Metab. 2016, 68, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Makharia, A.; Catassi, C.; Makharia, G.K. The Overlap between Irritable Bowel Syndrome and Non-Celiac Gluten Sensitivity: A Clinical Dilemma. Nutrients 2015, 7, 10417–10426. [Google Scholar] [CrossRef] [PubMed]
- Dionne, J.; Ford, A.C.; Yuan, Y.; Chey, W.D.; Lacy, B.E.; Saito, Y.A.; Quigley, E.M.M.; Moayyedi, P. A Systematic Review and Meta-Analysis Evaluating the Efficacy of a Gluten-Free Diet and a Low FODMAPs Diet in Treating Symptoms of Irritable Bowel Syndrome. Am. J. Gastroenterol. 2018, 113, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.F.; Burt, E.; Dowling, K.; Davidson, G.; Brooks, D.A.; Butler, R.N. Effect of age on fructose malabsorption in children presenting with gastrointestinal symptoms. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 581–584. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| SI | MGAM | LPH | ||||
|---|---|---|---|---|---|---|
| Content (% total BBM protein) | 8.2 ± 0.7 | 2.7 ± 1.4 | 1.4 ± 0.5 | |||
| Substrate | Sucrose | Isomaltose | Maltose | Maltose | Lactose | |
| Specific activity | Immunopr. | 9.5 ± 1.9 | 5.2 ± 1.4 | 9.5 ± 1.9 | 28.1 ± 12.4 | 1.8 ± 0.3 |
| U·mg−1 | BBM | 27.8 ± 0.5 | 16.5 ± 0.8 | 20.2 ± 0.4 | 5.6 ± 0.3 | |
| Amino Acid Change | SNP | Allele Frequency | SI Domain | Biosynthetic Pattern | Activity | CSID Genotype | References | |
|---|---|---|---|---|---|---|---|---|
| Sucrase | Isomaltase | |||||||
| W105C | rs138564183 | 0.00007054 | Isomaltase trefoil 1 | Wild type-like | reduced | reduced | Compound heterozygote with W931X | [16] |
| Q117R | rs121912612 | n.d. | Isomaltase | Wild type-like | reduced | reduced | Homozygote | [13] |
| L340P | rs267607049 | n.d. | Isomaltase | Wild type-like | normal | normal | Homozygote | [15] |
| D536V | rs376816463 | 0.0002731 | Isomaltase | Wild type-like | reduced | inactive | Compound heterozygote with V577G | [16] |
| V577G | rs121912615 | 0.002710 | Isomaltase | ER-located | inactive | inactive | Compound heterozygote with D536V or G1073D | [6,7,16] |
| S594P | rs765433197 | 0.0001091 | Isomaltase | ER-located | inactive | inactive | Compound heterozygote with splice site (c.26887 + 1G > C) | [6,16] |
| L620P | rs121912613 | n.d. | Isomaltase | ER-located | inactive | inactive | Homozygote | [12] |
| C635R | n.d. | n.d. | Isomaltase | Partial trafficked | reduced | reduced | Homozygote | [17] |
| T694P | n.d. | n.d. | Isomaltase | n.d | n.d | n.d | Heterozygote | [6] |
| L741P | rs1167931116 | 0.00006486 | Isomaltase | ER-located | inactive | inactive | Compound heterozygote with F1745C | [16] |
| R774G | rs147207752 | 0.001401 | Isomaltase | Wild type-like | reduced | reduced | Heterozygote | [18] |
| F875S | n.d. | n.d. | Isomaltase | ER-located | inactive | inactive | Heterozygote | Unpublished |
| Q930R | rs150927256 | 0.0004030 | Isomaltase | Wild type-like | normal | normal | Compound heterozygote with R1544C | [16] |
| W931R | rs914403158 | 0.000008899 | Isomaltase | ER-located | reduced | reduced | Compound heterozygote with T1606I | [16] |
| W931X | rs1314243578 | n.d. | Isomaltase | ER-located | inactive | inactive | Compound heterozygote with W105C | [16] |
| G1073D | rs121912616 | 0.002313 | Sucrase | ER-located | inactive | inactive | Compound heterozygote with R1544C, D577 or R1124X/Heterozygote | [6,7,16] |
| Q1098P | rs121912611 | 0.00003882 | Sucrase | ER-located/cis-Golgi | inactive | inactive | Homozygote | [19] |
| R1124X | rs200451408 | 0.0001629 | Sucrase | ER-located | inactive | inactive | Compound heterozygote with G1073D | [16] |
| C1229Y | rs121912614 | 0.000008825 | Sucrase | Partial trafficked | inactive | reduced | Compound heterozygote/Heterozygote | [6,7] |
| R1367G | rs143388292 | 0.0005431 | Sucrase | n.d | n.d | n.d | Compound heterozygote with frame shift (.1648delC) | [6] |
| C1531Y | n.d. | n.d. | Sucrase | Partial trafficked | inactive | reduced | Compound heterozygote with G1073D | [16] |
| R1544C | rs1340078396 | 0.00001776 | Sucrase | ER-located | reduced | reduced | Compound heterozygote with Q930R | [16] |
| T1606I | rs376062850 | 0.00003900 | Sucrase | ER-located | reduced | reduced | Compound heterozygote with W931R | [16] |
| F1745C | rs79717168 | 0.001581 | Sucrase | ER-located | inactive | inactive | Compound heterozygote with L741P or C1229Y/Heterozygote | [6,7,16] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Husein, D.M.; Wanes, D.; Marten, L.M.; Zimmer, K.-P.; Naim, H.Y. Heterozygotes Are a Potential New Entity among Homozygotes and Compound Heterozygotes in Congenital Sucrase-Isomaltase Deficiency. Nutrients 2019, 11, 2290. https://doi.org/10.3390/nu11102290
Husein DM, Wanes D, Marten LM, Zimmer K-P, Naim HY. Heterozygotes Are a Potential New Entity among Homozygotes and Compound Heterozygotes in Congenital Sucrase-Isomaltase Deficiency. Nutrients. 2019; 11(10):2290. https://doi.org/10.3390/nu11102290
Chicago/Turabian StyleHusein, Diab M., Dalanda Wanes, Lara M. Marten, Klaus-Peter Zimmer, and Hassan Y. Naim. 2019. "Heterozygotes Are a Potential New Entity among Homozygotes and Compound Heterozygotes in Congenital Sucrase-Isomaltase Deficiency" Nutrients 11, no. 10: 2290. https://doi.org/10.3390/nu11102290
APA StyleHusein, D. M., Wanes, D., Marten, L. M., Zimmer, K.-P., & Naim, H. Y. (2019). Heterozygotes Are a Potential New Entity among Homozygotes and Compound Heterozygotes in Congenital Sucrase-Isomaltase Deficiency. Nutrients, 11(10), 2290. https://doi.org/10.3390/nu11102290