Role of Zinc in Immune System and Anti-Cancer Defense Mechanisms


{kind=link}
Abstract
1. Introduction
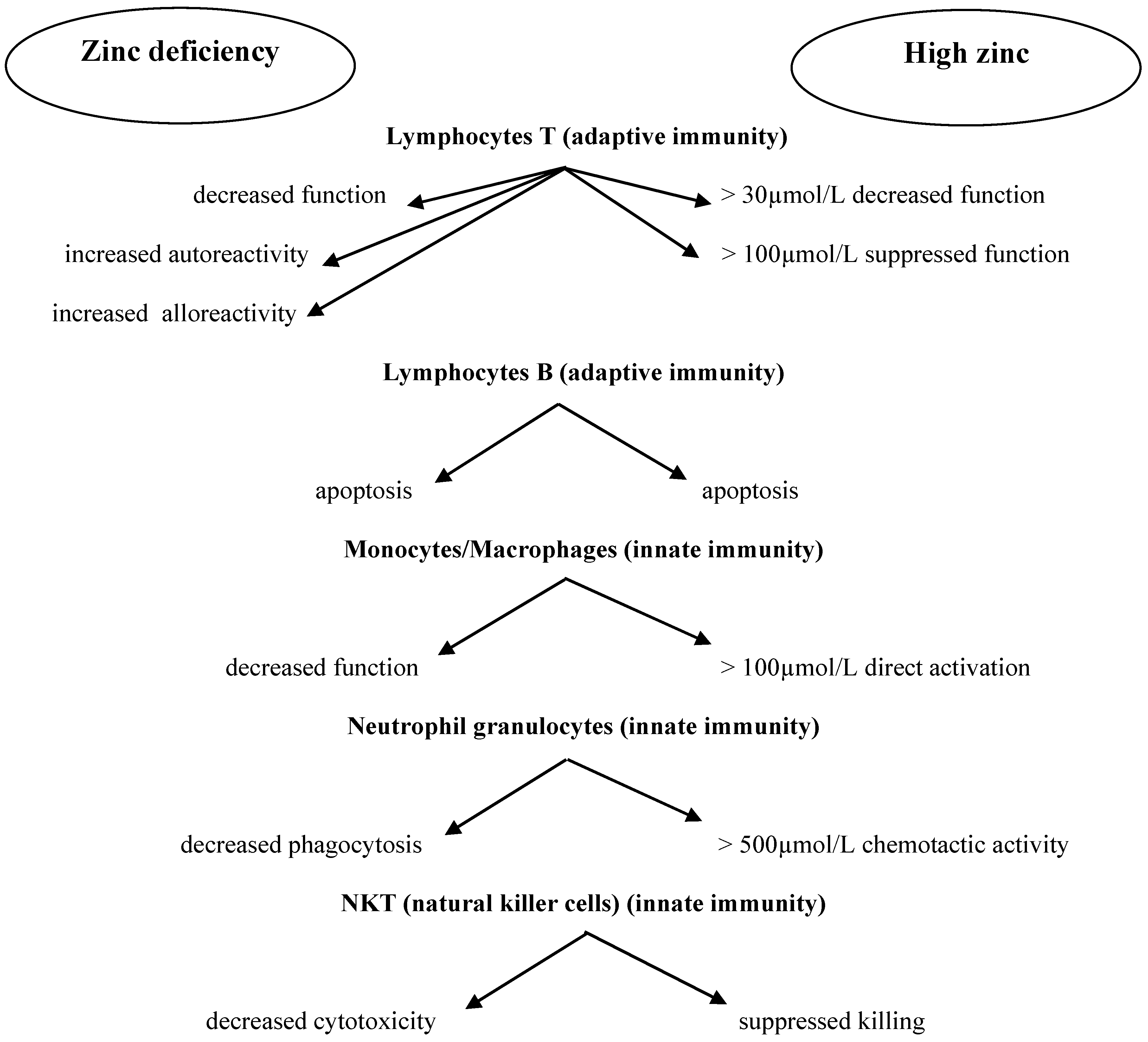
2. Zinc and the Immune System
Human Intestinal Microbiome and Immune System
3. Zinc and Nuclear Transcription Factor Nf Kappa B (Nf-κB)
3.1. Zinc and Cyclic Nucleotide Phosphodiesterases (PDEs)
3.2. Zinc and Zinc Transporters
3.3. Zinc and Expression of Protein A20
3.4. Zinc and Peroxisome Proliferator-Activated Receptor α (PPAR-α)
4. Zinc and Antioxidant Processes
4.1. Zinc and Antioxidant Enzymes
4.2. Zinc as an Iron and Copper Antagonist
4.3. Zinc and Protein Keap1 and Transcription Factor Nrf2
4.4. Zinc and Metallothioneins (MTs)
5. Zinc and Zinc Fingers
5.1. The Effect of Zinc on Genes
5.2. Zinc and Histone Deacetylases (HDACs)
6. Zinc and Apoptosis and Autophagy
7. Zinc and the Psyche (NDMA Receptors)
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Garazd, Y.; Garazd, M.; Lesyk, R. Synthesis and evaluation of anticancer activity of 6-pyrazolinylcoumarin derivatives. Saudi Pharm. J. 2017, 25, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Amaral, R.G. Natural Products as Treatment against Cancer: A Historical and Current Vision. Clin. Oncol. 2019, 4, 5. [Google Scholar]
- Spaczyńska, E.; Mrozek-Wilczkiewicz, A.; Malarz, K.; Kos, J.; Gonec, T.; Oravec, M.; Gawecki, R.; Bak, A.; Dohanosova, J.; Kapustikova, I.; et al. Design and synthesis of anticancer 1-hydroxynaphthalene-2-carboxanilides with a p53 independent mechanism of action. Sci. Rep. 2019, 9, 6387. [Google Scholar] [CrossRef] [PubMed]
- Dhammaraj, T.; Phintha, A.; Pinthong, C.; Medhanavyn, D.; Tinikul, R.; Chenprakhon, P.; Sucharitakul, J.; Vardhanabhuti, N.; Jiarpinitnun, C.; Chaiyen, P. p-Hydroxyphenylacetate 3-Hydroxylase as a Biocatalyst for the Synthesis of Trihydroxyphenolic Acids. ACS Catal. 2015, 5, 4492–4502. [Google Scholar] [CrossRef]
- Faa, G.; Nurchi, V.M.; Ravarino, A.; Fanni, D.; Nemolato, S.; Gerosa, C.; Van Eyken, P.; Geboes, K. Zinc in gastrointestinal and liver disease. Coord. Chem. Rev. 2008, 252, 1257–1269. [Google Scholar] [CrossRef]
- Fukada, T. Zinc biology and zinc signaling. Biomed. Res. Trace Elem. 2015, 26, 1–6. [Google Scholar]
- Zastrow, M.L.; Pecoraro, V.L. Designing Hydrolytic Zinc Metalloenzymes. Biochemistry 2014, 53, 957–978. [Google Scholar] [CrossRef]
- Krishna, S.S.; Majumdar, I.; Grishin, N.V. Structural classification of zinc fingers: Survey and summary. Nucleic Acids Res. 2003, 31, 532–550. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636. [Google Scholar] [CrossRef]
- Wessells, K.R.; Singh, G.M.; Brown, K.H. Estimating the Global Prevalence of Inadequate Zinc Intake from National Food Balance Sheets: Effects of Methodological Assumptions. PLoS ONE 2012, 7, e50565. [Google Scholar] [CrossRef]
- Brown, K.H.; Hambidge, K.M.; Ranum, P. Zinc Fortification Working Group Zinc fortification of cereal flours: Current recommendations and research needs. Food Nutr. Bull. 2010, 31, S62–S74. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Zinc in human health: Effect of zinc on immune cells. Mol. Med. Camb. Mass 2008, 14, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Impact of the discovery of human zinc deficiency on health. J. Trace Elem. Med. Biol. 2014, 28, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Stepien, M.; Hughes, D.J.; Hybsier, S.; Bamia, C.; Tjønneland, A.; Overvad, K.; Affret, A.; His, M.; Boutron-Ruault, M.-C.; Katzke, V.; et al. Circulating copper and zinc levels and risk of hepatobiliary cancers in Europeans. Br. J. Cancer 2017, 116, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Razab, S.; Prabhu, K.; Ray, S.; Prakash, B. Serum butyrylcholinesterase and zinc in breast cancer. J. Cancer Res. Ther. 2017, 13, 367–370. [Google Scholar] [PubMed]
- Khoshdel, Z.; Naghibalhossaini, F.; Abdollahi, K.; Shojaei, S.; Moradi, M.S.; Malekzadeh, M. Serum Copper and Zinc Levels Among Iranian Colorectal Cancer Patients. Biol. Trace Elem. Res. 2015, 170, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Okunade, K.S.; Dawodu, O.O.; Salako, O.; Osanyin, G.E.; Okunowo, A.A.; Anorlu, R.I. Comparative analysis of serum trace element levels in women with invasive cervical cancer in Lagos, Nigeria. Pan Afr. Med. J. 2018, 31, 194. [Google Scholar] [CrossRef] [PubMed]
- Zabłocka-Słowińska, K.; Płaczkowska, S.; Prescha, A.; Pawełczyk, K.; Porębska, I.; Kosacka, M.; Pawlik-Sobecka, L.A.; Grajeta, H. Serum and whole blood Zn, Cu and Mn profiles and their relation to redox status in lung cancer patients. J. Trace Elem. Med. Biol. 2018, 45, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z.; Li, A.; Zhang, Y. Association between serum zinc levels and lung cancer: A meta-analysis of observational studies. World J. Surg. Oncol. 2019, 17, 78. [Google Scholar] [CrossRef]
- Emami, A.; Nazem, M.R.; Shekarriz, R.; Hedayati, M. Micronutrient status (calcium, zinc, vitamins D and E) in patients with medullary thyroid carcinoma: A cross-sectional study. Nutrition 2017, 41, 86–89. [Google Scholar] [CrossRef]
- Lu, H.; Cai, L.; Mu, L.-N.; Lu, Q.-Y.; Zhao, J.; Cui, Y.; Sul, J.H.; Zhou, X.-F.; Ding, B.-G.; Elashoff, R.M.; et al. Dietary mineral and trace element intake and squamous cell carcinoma of the esophagus in a Chinese population. Nutr. Cancer 2006, 55, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Bidoli, E.; Bosetti, C.; La Vecchia, C.; Levi, F.; Parpinel, M.; Talamini, R.; Negri, E.; Maso, L.D.; Franceschi, S. Micronutrients and laryngeal cancer risk in Italy and Switzerland: A case-control study. Cancer Causes Control CCC 2003, 14, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, D.K.; Chadha, V.D. Zinc: A promising agent in dietary chemoprevention of cancer. Indian J. Med. Res. 2010, 132, 676–682. [Google Scholar] [PubMed]
- Chasapis, C.T.; Loutsidou, A.C.; Spiliopoulou, C.A.; Stefanidou, M.E. Zinc and human health: An update. Arch. Toxicol. 2012, 86, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Fraker, P.J.; King, L.E.; Laakko, T.; Vollmer, T.L. The dynamic link between the integrity of the immune system and zinc status. J. Nutr. 2000, 130, 1399S–1406S. [Google Scholar] [CrossRef] [PubMed]
- Sangthawan, D.; Phungrassami, T.; Sinkitjarurnchai, W. Effects of zinc sulfate supplementation on cell-mediated immune response in head and neck cancer patients treated with radiation therapy. Nutr. Cancer 2015, 67, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Bae, S.-C. TGF-beta-dependent cell growth arrest and apoptosis. J. Biochem. Mol. Biol. 2002, 35, 47–53. [Google Scholar]
- Mungunsukh, O.; Day, R.M. Transforming growth factor-β1 selectively inhibits hepatocyte growth factor expression via a micro-RNA-199–dependent posttranscriptional mechanism. Mol. Biol. Cell 2013, 24, 2088–2097. [Google Scholar] [CrossRef]
- Smyth, G.P.; Stapleton, P.P.; Barden, C.B.; Mestre, J.R.; Freeman, T.A.; Duff, M.D.; Maddali, S.; Yan, Z.; Daly, J.M. Renal cell carcinoma induces prostaglandin E2 and T-helper type 2 cytokine production in peripheral blood mononuclear cells. Ann. Surg. Oncol. 2003, 10, 455–462. [Google Scholar] [CrossRef]
- Snijdewint, F.G.; Kaliński, P.; Wierenga, E.A.; Bos, J.D.; Kapsenberg, M.L. Prostaglandin E2 differentially modulates cytokine secretion profiles of human T helper lymphocytes. J. Immunol. 1993, 150, 5321–5329. [Google Scholar]
- McComb, S.; Thiriot, A.; Akache, B.; Krishnan, L.; Stark, F. Introduction to the Immune System. Methods Mol. Biol. 2019, 2024, 1–24. [Google Scholar] [PubMed]
- Bienenstock, J.; McDermott, M.R. Bronchus- and nasal-associated lymphoid tissues. Immunol. Rev. 2005, 206, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-L.; Hung, T.-C.; Hsieh, B.-S.; Chen, Y.-H.; Chen, T.-F.; Cheng, H.-L. Zinc at pharmacologic concentrations affects cytokine expression and induces apoptosis of human peripheral blood mononuclear cells. Nutrition 2006, 22, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Baltaci, S.B.; Mogulkoc, R.; Baltaci, A.K.; Emsen, A.; Artac, H. The effect of zinc and melatonin supplementation on immunity parameters in breast cancer induced by DMBA in rats. Arch. Physiol. Biochem. 2018, 124, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.-C.; et al. Interleukin 4 inhibits TGF-β-induced-Foxp3+T cells and generates, in combination with TGF-β, Foxp3− effector T cells that produce interleukins 9 and 10. Nat. Immunol. 2008, 9, 1347. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.Z.; Rink, L. Zinc in Infection and Inflammation. Nutrients 2017, 9, 624. [Google Scholar] [CrossRef]
- Ibs, K.-H.; Rink, L. Zinc-altered immune function. J. Nutr. 2003, 133, 1452S–1456S. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Rink, L. Zinc signals and immune function. BioFactors 2014, 40, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Di Santo, J. The expanding family of innate lymphoid cells: Regulators and effectors of immunity and tissue remodeling. Nat. Immunol. 2011, 12, 21. [Google Scholar] [CrossRef]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367. [Google Scholar] [CrossRef]
- Zamarron, B.F.; Chen, W. Dual Roles of Immune Cells and Their Factors in Cancer Development and Progression. Int. J. Biol. Sci. 2011, 7, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Baltaci, A.K.; Mogulkoc, R. Leptin and zinc relation: In regulation of food intake and immunity. Indian J. Endocrinol. Metab. 2012, 16, S611–S616. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Rink, L.; Haase, H. Chelation of Free Zn2+ Impairs Chemotaxis, Phagocytosis, Oxidative Burst, Degranulation, and Cytokine Production by Neutrophil Granulocytes. Biol. Trace Elem. Res. 2016, 171, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Oteiza, P.I. Zinc and the modulation of redox homeostasis. Free Radic. Biol. Med. 2012, 53, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- DeCoursey, T.E.; Morgan, D.; Cherny, V.V. The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature 2003, 422, 531. [Google Scholar] [CrossRef]
- Hasegawa, H.; Suzuki, K.; Suzuki, K.; Nakaji, S.; Sugawara, K. Effects of zinc on the reactive oxygen species generating capacity of human neutrophils and on the serum opsonic activity in vitro. Lumin. J. Biol. Chem. Lumin. 2000, 15, 321–327. [Google Scholar] [CrossRef]
- Lee, S.; Eskin, S.G.; Shah, A.K.; Schildmeyer, L.A.; McIntire, L.V. Effect of Zinc and Nitric Oxide on Monocyte Adhesion to Endothelial Cells under Shear Stress. Ann. Biomed. Eng. 2012, 40, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Hujanen, E.S.; Seppä, S.T.; Virtanen, K. Polymorphonuclear leukocyte chemotaxis induced by zinc, copper and nickel in vitro. Biochim. Biophys. Acta 1995, 1245, 145–152. [Google Scholar] [CrossRef]
- Gao, H.; Dai, W.; Zhao, L.; Min, J.; Wang, F. The Role of Zinc and Zinc Homeostasis in Macrophage Function. Available online: https://www.hindawi.com/journals/jir/2018/6872621/ (accessed on 13 September 2019).
- Wessels, I.; Haase, H.; Engelhardt, G.; Rink, L.; Uciechowski, P. Zinc deficiency induces production of the proinflammatory cytokines IL-1β and TNFα in promyeloid cells via epigenetic and redox-dependent mechanisms. J. Nutr. Biochem. 2013, 24, 289–297. [Google Scholar] [CrossRef]
- Krzysik, M.; Biernat, J.; Grajeta, H. the influence of chojen nutrients on immune system functioning. Part II. Immunomodulatory effects of vitamins and trace elements on the human body. Adv. Clin. Exp. Med. 2006, 15, 1055–1062. [Google Scholar]
- Koyasu, S.; Moro, K. Type 2 innate immune responses and the natural helper cell. Immunology 2011, 132, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Van Stry, M.; Bix, M. Explaining discordant coordination. Nat. Immunol. 2011, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Sheu, B.C.; Lin, R.H.; Lien, H.C.; Ho, H.N.; Hsu, S.M.; Huang, S.C. Predominant Th2/Tc2 polarity of tumor-infiltrating lymphocytes in human cervical cancer. J. Immunol. 2001, 167, 2972–2978. [Google Scholar] [CrossRef] [PubMed]
- Rink, L.; Kirchner, H. Zinc-Altered Immune Function and Cytokine Production. J. Nutr. 2000, 130, 1407S–1411S. [Google Scholar] [CrossRef] [PubMed]
- Fraker, P.J. Roles for cell death in zinc deficiency. J. Nutr. 2005, 135, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Fraker, P.J.; King, L.E. Reprogramming of the immune system during zinc deficiency. Annu. Rev. Nutr. 2004, 24, 277–298. [Google Scholar] [CrossRef]
- Wellinghausen, N. Immunobiology of gestational zinc deficiency. Br. J. Nutr. 2001, 85, S81–S86. [Google Scholar] [CrossRef]
- Chavakis, T.; May, A.E.; Preissner, K.T.; Kanse, S.M. Molecular mechanisms of zinc-dependent leukocyte adhesion involving the urokinase receptor and beta2-integrins. Blood 1999, 93, 2976–2983. [Google Scholar]
- Fernandes, G.; Nair, M.; Onoe, K.; Tanaka, T.; Floyd, R.; Good, R.A. Impairment of cell-mediated immunity functions by dietary zinc deficiency in mice. Proc. Natl. Acad. Sci. USA 1979, 76, 457–461. [Google Scholar] [CrossRef]
- Chávez-Galán, L.; Arenas-Del Angel, M.C.; Zenteno, E.; Chávez, R.; Lascurain, R. Cell death mechanisms induced by cytotoxic lymphocytes. Cell. Mol. Immunol. 2009, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Driessen, C.; Hirv, K.; Rink, L.; Kirchner, H. Induction of cytokines by zinc ions in human peripheral blood mononuclear cells and separated monocytes. Lymphokine Cytokine Res. 1994, 13, 15–20. [Google Scholar] [PubMed]
- Truong-Tran, A.Q.; Ho, L.H.; Chai, F.; Zalewski, P.D. Cellular zinc fluxes and the regulation of apoptosis/gene-directed cell death. J. Nutr. 2000, 130, 1459S–1466S. [Google Scholar] [CrossRef] [PubMed]
- Ranjbar, E.; Shams, J.; Sabetkasaei, M.; M-Shirazi, M.; Rashidkhani, B.; Mostafavi, A.; Bornak, E.; Nasrollahzadeh, J. Effects of zinc supplementation on efficacy of antidepressant therapy, inflammatory cytokines, and brain-derived neurotrophic factor in patients with major depression. Nutr. Neurosci. 2014, 17, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, M.; Nagata, M.; Nakamura, K.; Kawai, M.; Baba, T.; Yamaki, K.; Yoshino, S. Adjuvant effect of zinc oxide on Th2 but not Th1 immune responses in mice. Immunopharmacol. Immunotoxicol. 2010, 32, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Sandstead, H.H.; Prasad, A.S.; Penland, J.G.; Beck, F.W.J.; Kaplan, J.; Egger, N.G.; Alcock, N.W.; Carroll, R.M.; Ramanujam, V.M.S.; Dayal, H.H.; et al. Zinc deficiency in Mexican American children: Influence of zinc and other micronutrients on T cells, cytokines, and antiinflammatory plasma proteins. Am. J. Clin. Nutr. 2008, 88, 1067–1073. [Google Scholar] [CrossRef]
- Bryant, V.L.; Ma, C.S.; Avery, D.T.; Li, Y.; Good, K.L.; Corcoran, L.M.; de Waal Malefyt, R.; Tangye, S.G. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: Predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J. Immunol. 2007, 179, 8180–8190. [Google Scholar] [CrossRef]
- Benchetrit, F.; Ciree, A.; Vives, V.; Warnier, G.; Gey, A.; Sautès-Fridman, C.; Fossiez, F.; Haicheur, N.; Fridman, W.H.; Tartour, E. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood 2002, 99, 2114–2121. [Google Scholar] [CrossRef]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef]
- Kitabayashi, C.; Fukada, T.; Kanamoto, M.; Ohashi, W.; Hojyo, S.; Atsumi, T.; Ueda, N.; Azuma, I.; Hirota, H.; Murakami, M.; et al. Zinc suppresses Th17 development via inhibition of STAT3 activation. Int. Immunol. 2010, 22, 375–386. [Google Scholar] [CrossRef]
- Cardenas, A.E.A.; Tapia, M.T.S.; Chacon, B.M.; Palacios, R.P. Impact of zinc supplementation on the production of IL-17 in perinatal stages. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef]
- King, L.E.; Osati-Ashtiani, F.; Fraker, P.J. Apoptosis plays a distinct role in the loss of precursor lymphocytes during zinc deficiency in mice. J. Nutr. 2002, 132, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Besold, A.N.; Gilston, B.A.; Radin, J.N.; Ramsoomair, C.; Culbertson, E.M.; Li, C.X.; Cormack, B.P.; Chazin, W.J.; Kehl-Fie, T.E.; Culotta, V.C. Role of Calprotectin in Withholding Zinc and Copper from Candida albicans. Infect. Immun. 2018, 86, e00779-17. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.R.; McKenzie, R.C.; Beckett, G.J. Selenium in the immune system. J. Nutr. 2003, 133, 1457S–1459S. [Google Scholar] [CrossRef]
- Rink, L.; Gabriel, P. Zinc and the immune system. Proc. Nutr. Soc. 2000, 59, 541–552. [Google Scholar] [CrossRef]
- Wiggins, H.L.; Wymant, J.M.; Solfa, F.; Hiscox, S.E.; Taylor, K.M.; Westwell, A.D.; Jones, A.T. Disulfiram-induced cytotoxicity and endo-lysosomal sequestration of zinc in breast cancer cells. Biochem. Pharmacol. 2015, 93, 332–342. [Google Scholar] [CrossRef]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-κB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. [Google Scholar] [CrossRef]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Lyte, M. Microbial endocrinology: Host-microbiota neuroendocrine interactions influencing brain and behavior. Gut Microbes 2014, 5, 381–389. [Google Scholar] [CrossRef]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef]
- Tsilimigras, M.C.B.; Fodor, A.; Jobin, C. Carcinogenesis and therapeutics: The microbiota perspective. Nat. Microbiol. 2017, 2, 17008. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Daillère, R.; Roberti, M.P.; Routy, B.; Kroemer, G. Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol. 2017, 15, 465. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lécuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; De Paepe, M.; Brandi, G.; et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 2009, 31, 677–689. [Google Scholar] [CrossRef]
- Geuking, M.B.; Cahenzli, J.; Lawson, M.A.E.; Ng, D.C.K.; Slack, E.; Hapfelmeier, S.; McCoy, K.D.; Macpherson, A.J. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity 2011, 34, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Zarour, H.M. Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin. Cancer Res. 2016, 22, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Sturniolo, G.C.; Di Leo, V.; Ferronato, A.; D’Odorico, A.; D’Incà, R. Zinc supplementation tightens “leaky gut” in Crohn’s disease. Inflamm. Bowel Dis. 2001, 7, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Usama, U.; Khan, M.J.; Fatima, S. Role of Zinc in Shaping the Gut Microbiome; Proposed Mechanisms and Evidence from the Literature. J. Gastrointest. Dig. Syst. 2018, 8, 1. [Google Scholar] [CrossRef]
- Platts-Mills, J.A.; Babji, S.; Bodhidatta, L.; Gratz, J.; Haque, R.; Havt, A.; McCormick, B.J.; McGrath, M.; Olortegui, M.P.; Samie, A.; et al. Pathogen-specific burdens of community diarrhoea in developing countries: A multisite birth cohort study (MAL-ED). Lancet Glob. Health 2015, 3, e564–e575. [Google Scholar] [CrossRef]
- Faiz, U.; Butt, T.; Satti, L.; Hussain, W.; Hanif, F. Efficacy of zinc as an antibacterial agent against enteric bacterial pathogens. J. Ayub Med. Coll. Abbottabad JAMC 2011, 23, 18–21. [Google Scholar]
- Sarker, S.A.; Brüssow, H. From bench to bed and back again: Phage therapy of childhood Escherichia coli diarrhea. Ann. N. Y. Acad. Sci. 2016, 1372, 42–52. [Google Scholar] [CrossRef]
- Zackular, J.P.; Moore, J.L.; Jordan, A.T.; Juttukonda, L.J.; Noto, M.J.; Nicholson, M.R.; Crews, J.D.; Semler, M.W.; Zhang, Y.; Ware, L.B.; et al. Dietary Zinc Alters the Microbiota and Decreases Resistance to Clostridium difficile Infection. Nat. Med. 2016, 22, 1330. [Google Scholar] [CrossRef]
- Tergaonkar, V. NF-κB pathway: A good signaling paradigm and therapeutic target. Int. J. Biochem. Cell Biol. 2006, 38, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Zingarelli, B. Nuclear factor-κB. Crit. Care Med. 2005, 33, S414–S416. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the NF-κB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Jarosz, M.; Olbert, M.; Wyszogrodzka, G.; Młyniec, K.; Librowski, T. Antioxidant and anti-inflammatory effects of zinc. Zinc-dependent NF-κB signaling. Inflammopharmacology 2017, 25, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; DeGuzman, A.; Bucana, C.D.; Fidler, I.J. Level of interleukin-8 expression by metastatic human melanoma cells directly correlates with constitutive NF-κB activity. Cytokines Cell. Mol. Ther. 2000, 6, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Riehemann, K.; Behnke, B.; Schulze-Osthoff, K. Plant extracts from stinging nettle (Urtica dioica), an antirheumatic remedy, inhibit the proinflammatory transcription factor NF-κB. FEBS Lett. 1999, 442, 89–94. [Google Scholar] [CrossRef]
- Waddick, K.G.; Uckun, F.M. Innovative treatment programs against cancer: II. Nuclear factor-κB (NF-κB) as a molecular target. Biochem. Pharmacol. 1999, 57, 9–17. [Google Scholar] [CrossRef]
- Garg, A.; Aggarwal, B.B. Nuclear transcription factor-κB as a target for cancer drug development. Leukemia 2002, 16, 1053–1068. [Google Scholar] [CrossRef]
- Ma, M.H.; Yang, H.H.; Parker, K.; Manyak, S.; Friedman, J.M.; Altamirano, C.; Wu, Z.; Borad, M.J.; Frantzen, M.; Roussos, E.; et al. The proteasome inhibitor PS-341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents. Clin. Cancer Res. 2003, 9, 1136–1144. [Google Scholar]
- Qureshi, N.; Vogel, S.N.; Van Way, C.; Papasian, C.J.; Qureshi, A.A.; Morrison, D.C. The proteasome: A central regulator of inflammation and macrophage function. Immunol. Res. 2005, 31, 243–260. [Google Scholar] [CrossRef]
- Baldwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J. Clin. Investig. 2001, 107, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Bentires-Alj, M.; Hellin, A.C.; Ameyar, M.; Chouaib, S.; Merville, M.P.; Bours, V. Stable inhibition of nuclear factor κB in cancer cells does not increase sensitivity to cytotoxic drugs. Cancer Res. 1999, 59, 811–815. [Google Scholar] [PubMed]
- Chen, L.F.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-κB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Ober-Blöbaum, J.L.; Engelhardt, G.; Hebel, S.; Heit, A.; Heine, H.; Rink, L. Zinc Signals Are Essential for Lipopolysaccharide-Induced Signal Transduction in Monocytes. J. Immunol. 2008, 181, 6491–6502. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.; Samman, S. Zinc and Regulation of Inflammatory Cytokines: Implications for Cardiometabolic Disease. Nutrients 2012, 4, 676–694. [Google Scholar] [CrossRef] [PubMed]
- Ke, H. Implications of PDE4 structure on inhibitor selectivity across PDE families. Int. J. Impot. Res. 2004, 16, S24. [Google Scholar] [CrossRef][Green Version]
- Dodge-Kafka, K.L.; Soughayer, J.; Pare, G.C.; Carlisle Michel, J.J.; Langeberg, L.K.; Kapiloff, M.S.; Scott, J.D. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 2005, 437, 574. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef] [PubMed]
- Horbinski, C.; Chu, C.T. Kinase signaling cascades in the mitochondrion: A matter of life or death. Free Radic. Biol. Med. 2005, 38, 2–11. [Google Scholar] [CrossRef]
- Razzaque, M.A.; Nishizawa, T.; Komoike, Y.; Yagi, H.; Furutani, M.; Amo, R.; Kamisago, M.; Momma, K.; Katayama, H.; Nakagawa, M.; et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat. Genet. 2007, 39, 1013. [Google Scholar] [CrossRef]
- Roskoski, R. RAF protein-serine/threonine kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2010, 399, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.G.; Reed, J.C. Bc1-2, Raf-1 and mitochondrial regulation of apoptosis. BioFactors 1998, 8, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, K.-W.; Murray, F.; Nakajima, T.; Zhao, Y.; Perkins, D.L.; Finn, P.W. Regulation of cytotoxic T lymphocyte antigen 4 by cyclic AMP. Am. J. Respir. Cell Mol. Biol. 2013, 48, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Semmler, J.; Gebert, U.; Eisenhut, T.; Moeller, J.; Schönharting, M.M.; Alléra, A.; Endres, S. Xanthine derivatives: Comparison between suppression of tumour necrosis factor-alpha production and inhibition of cAMP phosphodiesterase activity. Immunology 1993, 78, 520–525. [Google Scholar] [PubMed]
- Nishida, K.; Hasegawa, A.; Nakae, S.; Oboki, K.; Saito, H.; Yamasaki, S.; Hirano, T. Zinc transporter Znt5/Slc30a5 is required for the mast cell-mediated delayed-type allergic reaction but not the immediate-type reaction. J. Exp. Med. 2009, 206, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Von Bülow, V.; Dubben, S.; Engelhardt, G.; Hebel, S.; Plümäkers, B.; Heine, H.; Rink, L.; Haase, H. Zinc-dependent suppression of TNF-alpha production is mediated by protein kinase A-induced inhibition of Raf-1, I kappa B kinase beta, and NF-kappa B. J. Immunol. 2007, 179, 4180–4186. [Google Scholar] [CrossRef]
- Takahashi, N.; Tetsuka, T.; Uranishi, H.; Okamoto, T. Inhibition of the NF-κB transcriptional activity by protein kinase A. Eur. J. Biochem. 2002, 269, 4559–4565. [Google Scholar] [CrossRef]
- Brieger, A.; Rink, L.; Haase, H. Differential regulation of TLR-dependent MyD88 and TRIF signaling pathways by free zinc ions. J. Immunol. 2013, 191, 1808–1817. [Google Scholar] [CrossRef]
- Gálvez-Peralta, M.; Wang, Z.; Bao, S.; Knoell, D.L.; Nebert, D.W. Tissue-Specific Induction of Mouse ZIP8 and ZIP14 Divalent Cation/Bicarbonate Symporters by, and Cytokine Response to, Inflammatory Signals. Int. J. Toxicol. 2014, 33, 246–258. [Google Scholar] [CrossRef]
- Liu, M.-J.; Bao, S.; Gálvez-Peralta, M.; Pyle, C.J.; Rudawsky, A.C.; Pavlovicz, R.E.; Killilea, D.W.; Li, C.; Nebert, D.W.; Wewers, M.D.; et al. ZIP8 regulates host defense through zinc-mediated inhibition of NF-κB. Cell Rep. 2013, 3, 386–400. [Google Scholar] [CrossRef]
- Kagara, N.; Tanaka, N.; Noguchi, S.; Hirano, T. Zinc and its transporter ZIP10 are involved in invasive behavior of breast cancer cells. Cancer Sci. 2007, 98, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Vichova, P.; Jordan, N.; Hiscox, S.; Hendley, R.; Nicholson, R.I. ZIP7-mediated intracellular zinc transport contributes to aberrant growth factor signaling in antihormone-resistant breast cancer Cells. Endocrinology 2008, 149, 4912–4920. [Google Scholar] [CrossRef] [PubMed]
- Lopez, V.; Kelleher, S.L. Zip6-attenuation promotes epithelial-to-mesenchymal transition in ductal breast tumor (T47D) cells. Exp. Cell Res. 2010, 316, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, L.; Verhelst, K.; van Loo, G.; Carpentier, I.; Ley, S.C.; Beyaert, R. Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochem. Pharmacol. 2010, 80, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694. [Google Scholar] [CrossRef] [PubMed]
- Balakirev, M.Y.; Tcherniuk, S.O.; Jaquinod, M.; Chroboczek, J. Otubains: A new family of cysteine proteases in the ubiquitin pathway. EMBO Rep. 2003, 4, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-C.; Chung, J.Y.; Lamothe, B.; Rajashankar, K.; Lu, M.; Lo, Y.-C.; Lam, A.Y.; Darnay, B.G.; Wu, H. Molecular basis for the unique deubiquitinating activity of the NF-κB inhibitor A20. J. Mol. Biol. 2008, 376, 526–540. [Google Scholar] [CrossRef]
- Li, L.; Soetandyo, N.; Wang, Q.; Ye, Y. The zinc finger protein A20 targets TRAF2 to the lysosomes for degradation. Biochim. Biophys. Acta 2009, 1793, 346–353. [Google Scholar] [CrossRef]
- Prasad, A.S.; Bao, B.; Beck, F.W.J.; Sarkar, F.H. Zinc-suppressed inflammatory cytokines by induction of A20-mediated inhibition of nuclear factor-κB. Nutrients 2011, 27, 816–823. [Google Scholar] [CrossRef]
- Mabilleau, G.; Chappard, D.; Sabokbar, A. Role of the A20-TRAF6 Axis in Lipopolysaccharide-mediated Osteoclastogenesis. J. Biol. Chem. 2011, 286, 3242–3249. [Google Scholar] [CrossRef]
- Heyninck, K.; De Valck, D.; Vanden Berghe, W.; Van Criekinge, W.; Contreras, R.; Fiers, W.; Haegeman, G.; Beyaert, R. The zinc finger protein A20 inhibits TNF-induced NF-κB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-κB-inhibiting protein ABIN. J. Cell Biol. 1999, 145, 1471–1482. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Bao, B.; Beck, F.W.J.; Kucuk, O.; Sarkar, F.H. Antioxidant effect of zinc in humans. Free Radic. Biol. Med. 2004, 37, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Lemberger, T.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: A nuclear receptor signaling pathway in lipid physiology. Annu. Rev. Cell Dev. Biol. 1996, 12, 335–363. [Google Scholar] [CrossRef] [PubMed]
- Das, K.C.; White, C.W. Redox systems of the cell: Possible links and implications. Proc. Natl. Acad. Sci. USA 2002, 99, 9617–9618. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cochemé, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef]
- Lau, A.T.Y.; Wang, Y.; Chiu, J.-F. Reactive oxygen species: Current knowledge and applications in cancer research and therapeutic. J. Cell. Biochem. 2008, 104, 657–667. [Google Scholar] [CrossRef]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.-X.; Mayo, J.C.; Sainz, R.M.; Leon, J.; Czarnocki, Z. Melatonin as an antioxidant: Biochemical mechanisms and pathophysiological implications in humans. Acta Biochim. Pol. 2003, 50, 1129–1146. [Google Scholar] [PubMed]
- Sliwinski, T.; Czechowska, A.; Kolodziejczak, M.; Jajte, J.; Wisniewska-Jarosinska, M.; Blasiak, J. Zinc salts differentially modulate DNA damage in normal and cancer cells. Cell Biol. Int. 2009, 33, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Strange, R.W.; Antonyuk, S.; Hough, M.A.; Doucette, P.A.; Rodriguez, J.A.; Hart, P.J.; Hayward, L.J.; Valentine, J.S.; Hasnain, S.S. The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J. Mol. Biol. 2003, 328, 877–891. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Eapen, C.E.; Madesh, M.; Balasubramanian, K.A.; Pulimood, A.; Mathan, M.; Ramakrishna, B.S. Mucosal mitochondrial function and antioxidant defences in patients with gastric carcinoma. Scand. J. Gastroenterol. 1998, 33, 975–981. [Google Scholar]
- Liaw, K.Y.; Lee, P.H.; Wu, F.C.; Tsai, J.S.; Lin-Shiau, S.Y. Zinc, copper, and superoxide dismutase in hepatocellular carcinoma. Am. J. Gastroenterol. 1997, 92, 2260–2263. [Google Scholar]
- Branco, R.J.F.; Fernandes, P.A.; Ramos, M.J. Cu, Zn Superoxide dismutase: Distorted active site binds substrate without significant energetic cost. Theor. Chem. Acc. 2006, 115, 27–31. [Google Scholar] [CrossRef]
- Wong, P.C.; Waggoner, D.; Subramaniam, J.R.; Tessarollo, L.; Bartnikas, T.B.; Culotta, V.C.; Price, D.L.; Rothstein, J.; Gitlin, J.D. Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. Proc. Natl. Acad. Sci. USA 2000, 97, 2886–2891. [Google Scholar] [CrossRef]
- Faraci, F.M.; Didion, S.P. Vascular protection: Superoxide dismutase isoforms in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1367–1373. [Google Scholar] [CrossRef]
- Wang, T.; Liu, B.; Qin, L.; Wilson, B.; Hong, J.S. Protective effect of the SOD/catalase mimetic MnTMPyP on inflammation-mediated dopaminergic neurodegeneration in mesencephalic neuronal-glial cultures. J. Neuroimmunol. 2004, 147, 68–72. [Google Scholar] [CrossRef]
- Frank, S.; Kämpfer, H.; Podda, M.; Kaufmann, R.; Pfeilschifter, J. Identification of copper/zinc superoxide dismutase as a nitric oxide-regulated gene in human (HaCaT) keratinocytes: Implications for keratinocyte proliferation. Biochem. J. 2000, 346, 719–728. [Google Scholar] [CrossRef]
- Lin, Y.; Kikuchi, S.; Obata, Y.; Yagyu, K. Serum Copper/Zinc Superoxide Dismutase (Cu/Zn SOD) and Gastric Cancer Risk: A Case-Control Study. Jpn. J. Cancer Res. Gann 2002, 93, 1071–1075. [Google Scholar] [CrossRef]
- Skrzydlewska, E.; Kozuszko, B.; Sulkowska, M.; Bogdan, Z.; Kozlowski, M.; Snarska, J.; Puchalski, Z.; Sulkowski, S.; Skrzydlewski, Z. Antioxidant potential in esophageal, stomach and colorectal cancers. Hepatogastroenterology. 2003, 50, 126–131. [Google Scholar]
- Janssen, A.M.; Bosman, C.B.; van Duijn, W.; Oostendorp-van de Ruit, M.M.; Kubben, F.J.; Griffioen, G.; Lamers, C.B.; van Krieken, J.H.; van de Velde, C.J.; Verspaget, H.W. Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clin. Cancer Res. 2000, 6, 3183–3192. [Google Scholar]
- Bóka, B.; Myari, A.; Sóvágó, I.; Hadjiliadis, N. Copper(II) and zinc(II) complexes of the peptides Ac-HisValHis-NH2 and Ac-HisValGlyAsp-NH2 related to the active site of the enzyme CuZnSOD. J. Inorg. Biochem. 2004, 98, 113–122. [Google Scholar] [CrossRef]
- Kong, Q.; Beel, J.A.; Lillehei, K.O. A threshold concept for cancer therapy. Med. Hypotheses 2000, 55, 29–35. [Google Scholar] [CrossRef]
- Golab, J.; Nowis, D.; Skrzycki, M.; Czeczot, H.; Baranczyk-Kuzma, A.; Wilczynski, G.M.; Makowski, M.; Mroz, P.; Kozar, K.; Kaminski, R.; et al. Antitumor effects of photodynamic therapy are potentiated by 2-methoxyestradiol. A superoxide dismutase inhibitor. J. Biol. Chem. 2003, 278, 407–414. [Google Scholar]
- Higuchi, Y. Chromosomal DNA fragmentation in apoptosis and necrosis induced by oxidative stress. Biochem. Pharmacol. 2003, 66, 1527–1535. [Google Scholar] [CrossRef]
- Huang, P.; Feng, L.; Oldham, E.A.; Keating, M.J.; Plunkett, W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature 2000, 407, 390. [Google Scholar] [CrossRef]
- Mooberry, S.L. Mechanism of action of 2-methoxyestradiol: New developments. Drug Resist. Updates 2003, 6, 355–361. [Google Scholar] [CrossRef]
- Zhou, Y.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003, 101, 4098–4104. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide anion radical (O-2), superoxide dismutases, and related matters. J. Biol. Chem. 1997, 272, 18515–18517. [Google Scholar] [CrossRef]
- Pláteník, J.; Stopka, P.; Vejrazka, M.; Stípek, S. Quinolinic acid-iron(ii) complexes: Slow autoxidation, but enhanced hydroxyl radical production in the Fenton reaction. Free Radic. Res. 2001, 34, 445–459. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Wakabayashi, N. Keap1, the Sensor for Electrophiles and Oxidants that Regulates the Phase 2 Response, Is a Zinc Metalloprotein. Biochemistry 2005, 44, 6889–6899. [Google Scholar] [CrossRef]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef]
- Kröncke, K.-D. Cellular stress and intracellular zinc dyshomeostasis. Arch. Biochem. Biophys. 2007, 463, 183–187. [Google Scholar] [CrossRef]
- Królicka, A.; Kobierzycki, C.; Puła, B.; Podhorska-Okołów, M.; Piotrowska, A.; Rzeszutko, M.; Rzeszutko, W.; Rabczyński, J.; Domosławski, P.; Wojtczak, B.; et al. Comparison of metallothionein (MT) and Ki-67 antigen expression in benign and malignant thyroid tumours. Anticancer Res. 2010, 30, 4945–4949. [Google Scholar]
- Lansdown, A.B. Metallothioneins: Potential therapeutic aids for wound healing in the skin. Wound Repair Regen. 2002, 10, 130–132. [Google Scholar] [CrossRef]
- Sakulsak, N. Metallothionein: An Overview on its Metal Homeostatic Regulation in Mammals. Int. J. Morphol. 2012, 30, 1007–1012. [Google Scholar] [CrossRef]
- Carpenè, E.; Andreani, G.; Isani, G. Metallothionein functions and structural characteristics. J. Trace Elem. Med. Biol. 2007, 21, 35–39. [Google Scholar] [CrossRef]
- Vasák, M. Advances in metallothionein structure and functions. J. Trace Elem. Med. Biol. 2005, 19, 13–17. [Google Scholar] [CrossRef]
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. The role of metallothionein in oxidative stress. Int. J. Mol. Sci. 2013, 14, 6044–6066. [Google Scholar] [CrossRef]
- Fukada, T.; Yamasaki, S.; Nishida, K.; Murakami, M.; Hirano, T. Zinc homeostasis and signaling in health and diseases: Zinc signaling. JBIC J. Biol. Inorg. Chem. 2011, 16, 1123–1134. [Google Scholar] [CrossRef]
- Stefanidou, M.; Maravelias, C.; Dona, A.; Spiliopoulou, C. Zinc: A multipurpose trace element. Arch. Toxicol. 2006, 80, 1. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Arriaga, J.M.; Bravo, I.A.; Bruno, L.; Morales Bayo, S.; Hannois, A.; Sanchez Loria, F.; Pairola, F.; Huertas, E.; Roberti, M.P.; Rocca, Y.S.; et al. Combined metallothioneins and p53 proteins expression as a prognostic marker in patients with Dukes stage B and C colorectal cancer. Hum. Pathol. 2012, 43, 1695–1703. [Google Scholar] [CrossRef]
- Chimienti, F.; Seve, M.; Richard, S.; Mathieu, J.; Favier, A. Role of cellular zinc in programmed cell death: Temporal relationship between zinc depletion, activation of caspases, and cleavage of Sp family transcription factors. Biochem. Pharmacol. 2001, 62, 51–62. [Google Scholar] [CrossRef]
- Dutsch-Wicherek, M.; Sikora, J.; Tomaszewska, R. The possible biological role of metallothionein in apoptosis. Front Biosci 2008, 13, 4029–4038. [Google Scholar] [CrossRef]
- Coyle, P.; Philcox, J.C.; Carey, L.C.; Rofe, A.M. Metallothionein: The multipurpose protein. Cell. Mol. Life Sci. CMLS 2002, 59, 627–647. [Google Scholar] [CrossRef]
- Dziegiel, P. Expression of metallothioneins in tumor cells. Pol. J. Pathol. 2004, 55, 3–12. [Google Scholar]
- Cherian, M.G.; Jayasurya, A.; Bay, B.-H. Metallothioneins in human tumors and potential roles in carcinogenesis. Mutat. Res. 2003, 533, 201–209. [Google Scholar] [CrossRef]
- Lynes, M.A.; Zaffuto, K.; Unfricht, D.W.; Marusov, G.; Samson, J.S.; Yin, X. The physiological roles of extracellular metallothionein. Exp. Biol. Med. 2006, 231, 1548–1554. [Google Scholar] [CrossRef]
- Manso, Y.; Adlard, P.A.; Carrasco, J.; Vašák, M.; Hidalgo, J. Metallothionein and brain inflammation. JBIC J. Biol. Inorg. Chem. 2011, 16, 1103. [Google Scholar] [CrossRef]
- Milnerowicz, H.; Jabłonowska, M.; Bizoń, A. Change of zinc, copper, and metallothionein concentrations and the copper-zinc superoxide dismutase activity in patients with pancreatitis. Pancreas 2009, 38, 681–688. [Google Scholar] [CrossRef]
- Suzuki, J.S.; Nishimura, N.; Zhang, B.; Nakatsuru, Y.; Kobayashi, S.; Satoh, M.; Tohyama, C. Metallothionein deficiency enhances skin carcinogenesis induced by 7,12-dimethylbenz[a]anthracene and 12-O-tetradecanoylphorbol-13-acetate in metallothionein-null mice. Carcinogenesis 2003, 24, 1123–1132. [Google Scholar] [CrossRef]
- Nielsen, A.E.; Bohr, A.; Penkowa, M. The Balance between Life and Death of Cells: Roles of Metallothioneins. Biomark. Insights 2006, 1, 117727190600100016. [Google Scholar] [CrossRef]
- Miyashita, H.; Sato, Y. Metallothionein 1 is a downstream target of vascular endothelial zinc finger 1 (VEZF1) in endothelial cells and participates in the regulation of angiogenesis. Endothelium 2005, 12, 163–170. [Google Scholar] [CrossRef]
- Balleza, E.; López-Bojorquez, L.N.; Martínez-Antonio, A.; Resendis-Antonio, O.; Lozada-Chávez, I.; Balderas-Martínez, Y.I.; Encarnación, S.; Collado-Vides, J. Regulation by transcription factors in bacteria: Beyond description. FEMS Microbiol. Rev. 2009, 33, 133–151. [Google Scholar] [CrossRef]
- Amarasinghe, G.K.; De Guzman, R.N.; Turner, R.B.; Chancellor, K.J.; Wu, Z.R.; Summers, M.F. NMR structure of the HIV-1 nucleocapsid protein bound to stem-loop SL2 of the psi-RNA packaging signal. Implications for genome recognition. J. Mol. Biol. 2000, 301, 491–511. [Google Scholar] [CrossRef]
- Berg, J.M.; Shi, Y. The galvanization of biology: A growing appreciation for the roles of zinc. Science 1996, 271, 1081–1085. [Google Scholar] [CrossRef]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Elrod-Erickson, M.; Benson, T.E.; Pabo, C.O. High-resolution structures of variant Zif268-DNA complexes: Implications for understanding zinc finger-DNA recognition. Structure 1998, 6, 451–464. [Google Scholar] [CrossRef]
- Maret, W. Zinc biochemistry: From a single zinc enzyme to a key element of life. Adv. Nutr. 2013, 4, 82–91. [Google Scholar] [CrossRef]
- Carroll, D. Progress and prospects: Zinc-finger nucleases as gene therapy agents. Gene Ther. 2008, 15, 1463–1468. [Google Scholar] [CrossRef]
- Cornu, T.I.; Thibodeau-Beganny, S.; Guhl, E.; Alwin, S.; Eichtinger, M.; Joung, J.K.; Joung, J.K.; Cathomen, T. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol. Ther. 2008, 16, 352–358. [Google Scholar] [CrossRef]
- Wyvekens, N.; Topkar, V.V.; Khayter, C.; Joung, J.K.; Tsai, S.Q. Dimeric CRISPR RNA-Guided FokI-dCas9 Nucleases Directed by Truncated gRNAs for Highly Specific Genome Editing. Hum. Gene Ther. 2015, 26, 425–431. [Google Scholar] [CrossRef]
- Björndahl, L.; Kvist, U. A model for the importance of zinc in the dynamics of human sperm chromatin stabilization after ejaculation in relation to sperm DNA vulnerability. Syst. Biol. Reprod. Med. 2011, 57, 86–92. [Google Scholar] [CrossRef]
- Fabris, N.; Mocchegiani, E. Zinc, human diseases and aging. Aging Clin. Exp. Res. 1995, 7, 77–93. [Google Scholar] [CrossRef]
- Ho, E. Zinc deficiency, DNA damage and cancer risk. J. Nutr. Biochem. 2004, 15, 572–578. [Google Scholar] [CrossRef]
- Casadevall, M.; Sarkar, B. Effect of redox conditions on the DNA-binding efficiency of the retinoic acid receptor zinc-finger. J. Inorg. Biochem. 1998, 71, 147–152. [Google Scholar] [CrossRef]
- Lafon-Hughes, L.; Di Tomaso, M.V.; Méndez-Acuña, L.; Martínez-López, W. Chromatin-remodelling mechanisms in cancer. Mutat. Res. 2008, 658, 191–214. [Google Scholar] [CrossRef]
- Mercurio, C.; Minucci, S.; Pelicci, P.G. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol. Res. 2010, 62, 18–34. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Schneider-Stock, R.; Ocker, M. Epigenetic therapy in cancer: Molecular background and clinical development of histone deacetylase and DNA methyltransferase inhibitors. IDrugs Investig. Drugs J. 2007, 10, 557–561. [Google Scholar]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391. [Google Scholar] [CrossRef]
- Kim, H.-J.; Bae, S.-C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- John, E.; Laskow, T.C.; Buchser, W.J.; Pitt, B.R.; Basse, P.H.; Butterfield, L.H.; Kalinski, P.; Lotze, M.T. Zinc in innate and adaptive tumor immunity. J. Transl. Med. 2010, 8, 118. [Google Scholar] [CrossRef]
- Ahn, Y.H.; Kim, Y.H.; Hong, S.H.; Koh, J.Y. Depletion of intracellular zinc induces protein synthesis-dependent neuronal apoptosis in mouse cortical culture. Exp. Neurol. 1998, 154, 47–56. [Google Scholar] [CrossRef]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Carter, J.E.; Truong-Tran, A.Q.; Grosser, D.; Ho, L.; Ruffin, R.E.; Zalewski, P.D. Involvement of redox events in caspase activation in zinc-depleted airway epithelial cells. Biochem. Biophys. Res. Commun. 2002, 297, 1062–1070. [Google Scholar] [CrossRef]
- Clegg, M.S.; Hanna, L.A.; Niles, B.J.; Momma, T.Y.; Keen, C.L. Zinc deficiency-induced cell death. IUBMB Life 2005, 57, 661–669. [Google Scholar] [CrossRef]
- Freedman, D.A.; Levine, A.J. Regulation of the p53 protein by the MDM2 oncoprotein--thirty-eighth G.H.A. Clowes Memorial Award Lecture. Cancer Res. 1999, 59, 1–7. [Google Scholar]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194. [Google Scholar] [CrossRef]
- Lymburner, S.; McLeod, S.; Purtzki, M.; Roskelley, C.; Xu, Z. Zinc inhibits magnesium-dependent migration of human breast cancer MDA-MB-231 cells on fibronectin. J. Nutr. Biochem. 2013, 24, 1034–1040. [Google Scholar] [CrossRef]
- Marleau, A.M.; Sarvetnick, N. T cell homeostasis in tolerance and immunity. J. Leukoc. Biol. 2005, 78, 575–584. [Google Scholar] [CrossRef]
- Barry, M.; Bleackley, R.C. Cytotoxic T lymphocytes: All roads lead to death. Nat. Rev. Immunol. 2002, 2, 401. [Google Scholar] [CrossRef]
- Sepiashvili, R.I.; Shubich, M.G.; Kolesnikova, N.V.; Slavyanskaya, T.A.; Lomtatidze, L.V. Apoptosis in immunologic processes. Clin. Appl. Immunol. Rev. 2001, 1, 163–172. [Google Scholar] [CrossRef]
- Garg, A.D.; Maes, H.; Romano, E.; Agostinis, P. Autophagy, a major adaptation pathway shaping cancer cell death and anticancer immunity responses following photodynamic therapy. Photochem. Photobiol. Sci. 2015, 14, 1410–1424. [Google Scholar] [CrossRef]
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035. [Google Scholar] [CrossRef]
- Fagundes, C.; LeRoy, A.; Karuga, M. Behavioral Symptoms after Breast Cancer Treatment: A Biobehavioral Approach. J. Pers. Med. 2015, 5, 280–295. [Google Scholar] [CrossRef]
- Harbottle, L.; Schonfelder, N. Nutrition and depression: A review of the evidence. J. Ment Health 2008, 17, 576–587. [Google Scholar] [CrossRef]
- Bartosz, G. Reactive oxygen species: Destroyers or messengers? Biochem. Pharmacol. 2009, 77, 1303–1315. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef]
- Nowak, G.; Szewczyk, B.; Pilc, A. Zinc and depression. An update. Pharmacol. Rep. 2005, 57, 713–718. [Google Scholar]
- Siwek, M.; Szewczyk, B.; Dudek, D.; Styczeń, K.; Sowa-Kućma, M.; Młyniec, K.; Siwek, A.; Witkowski, L.; Pochwat, B.; Nowak, G. Zinc as a marker of affective disorders. Pharmacol. Rep. 2013, 65, 1512–1518. [Google Scholar] [CrossRef]
- Maeng, S.; Zarate, C.A. The role of glutamate in mood disorders: Results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr. Psychiatry Rep. 2007, 9, 467–474. [Google Scholar] [CrossRef]
- Swardfager, W.; Herrmann, N.; Mazereeuw, G.; Goldberger, K.; Harimoto, T.; Lanctôt, K.L. Zinc in depression: A meta-analysis. Biol. Psychiatry 2013, 74, 872–878. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skrajnowska, D.; Bobrowska-Korczak, B. Role of Zinc in Immune System and Anti-Cancer Defense Mechanisms. Nutrients 2019, 11, 2273. https://doi.org/10.3390/nu11102273
Skrajnowska D, Bobrowska-Korczak B. Role of Zinc in Immune System and Anti-Cancer Defense Mechanisms. Nutrients. 2019; 11(10):2273. https://doi.org/10.3390/nu11102273
Chicago/Turabian StyleSkrajnowska, Dorota, and Barbara Bobrowska-Korczak. 2019. "Role of Zinc in Immune System and Anti-Cancer Defense Mechanisms" Nutrients 11, no. 10: 2273. https://doi.org/10.3390/nu11102273
APA StyleSkrajnowska, D., & Bobrowska-Korczak, B. (2019). Role of Zinc in Immune System and Anti-Cancer Defense Mechanisms. Nutrients, 11(10), 2273. https://doi.org/10.3390/nu11102273