The Gut–Brain Axis in the Neuropsychological Disease Model of Obesity: A Classical Movie Revised by the Emerging Director “Microbiome”




Abstract
1. Introduction
2. Why a Neuropsychological View of Obesity?
2.1. Neurobiological Aspects of Obesity’s Pathogenesis
2.2. Obesity and Neurological Comorbidities
3. The Microbiota–Gut–Brain Axis
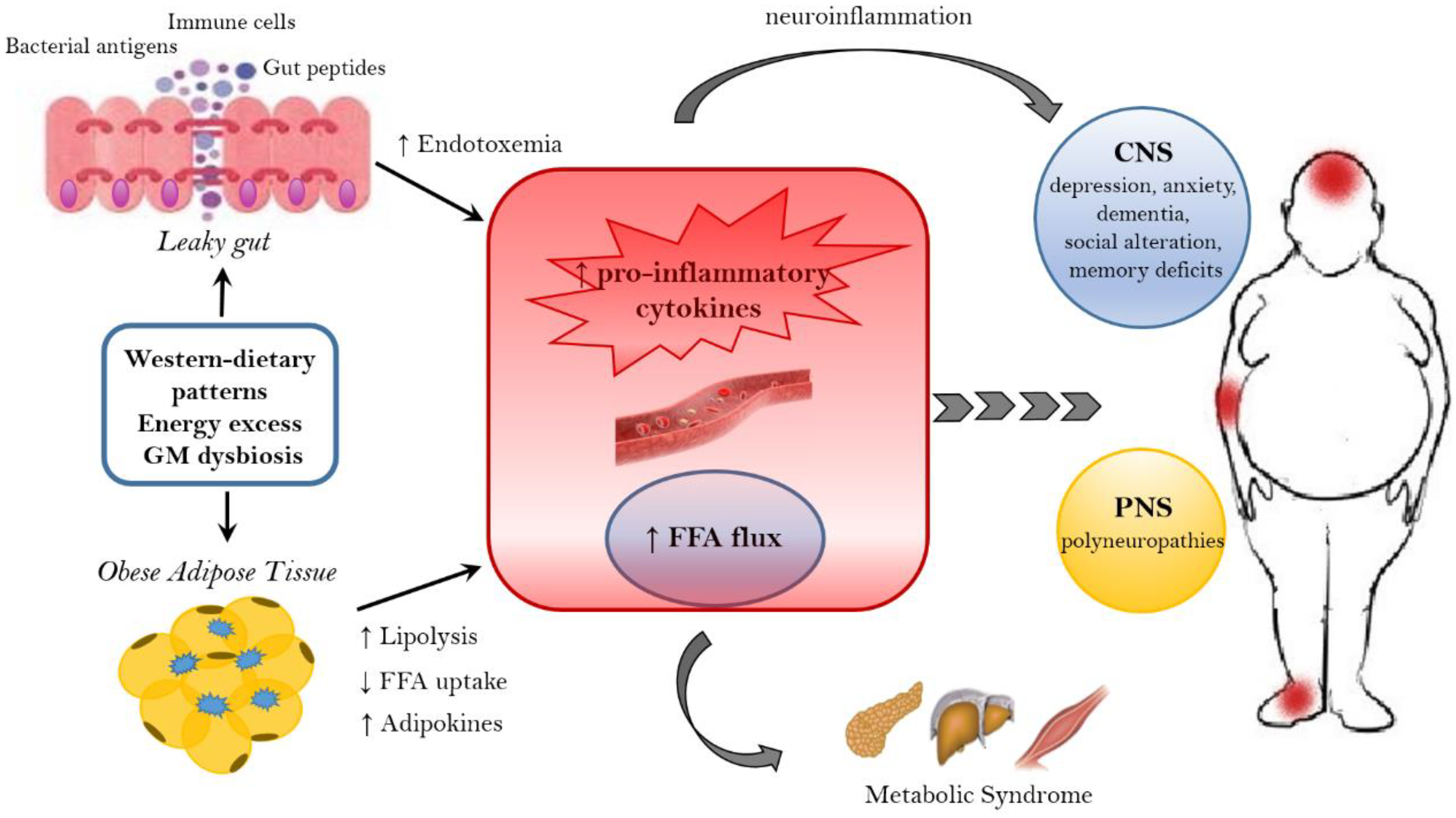
3.1. Microbiome and Energy Harvest
- GM influences energy homeostasis by regulating gene expression via complex mechanisms started by SCFAs and monosaccharides [156]. In particular, the commensal microorganisms stimulate monosaccharide cellular uptake [157] and induce lipogenesis by activating the transcription factors carbohydrate response element binding protein (ChREBP) and sterol response element binding protein (SREBP) [155]. Triacylglycerols, produced trought hepatic lipogenesis, are thus sent from the liver to the blood in the form of very low-density lipoprotein and chylomicrons.
- HF diet triggers an increased absorption of bacterial lipopolysaccharide (LPS) (an endotoxin in the cell wall of Gram-negative bacteria) from the gut lumen to the bloodstream inducing low-grade inflammation, by activating B cells or dendritic cells activating and cytokine production [158]. The inflammation could also be stimulated by endotoxemia condition [157]; moreover, also the damaged gut barrier might contribute to this metabolic endotoxaemia [159].
- GM induces a suppression of angiopoietin-like protein 4 (ANGPTL4) in the intestinal epithelium. ANGPTL4 is a circulating enzyme produced by liver, gut and adipose tissue that inhibits LPL; its suppression provokes an increase of triacylglycerol storage in the adipose tissue [127].
3.2. Microbiome and the Brain
3.3. The Role of Microbiome-Driven Inflammation
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organisation Website. Available online: http://www.who.int/news-room/factsheets/detail/obesity-and-overweight (accessed on 16 February 2018).
- Bessesen, D.H. Update on obesity. J. Clin. Endocrinol. Metab. 2008, 93, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Loos, R.J.F.; Bouchard, C. FTO: The first gene contributing to common forms of human obesity. Obes. Rev. 2008, 9, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Rennie, K.L.; Jebb, S.A. Prevalence of obesity in Great Britain. Obes. Rev. 2005, 6, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Rennie, K.L.; Johnson, L.; Jebb, S.A. Behavioural determinants of obesity. Best Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Jauch-Chara, K.; Oltmanns, K.M. Obesity—A neuropsychological disease? Systematic review and neuropsychological model. Prog. Neurobiol. 2014, 114, 84–101. [Google Scholar] [CrossRef] [PubMed]
- Lyte, M. Microbial Endocrinology in the Microbiome-Gut-Brain Axis: How Bacterial Production and Utilization of Neurochemicals Influence Behavior. PLoS Pathog. 2013, 9, e1003726. [Google Scholar] [CrossRef] [PubMed]
- Mulders, R.J.; de Git, K.C.G.; Schéle, E.; Dickson, S.L.; Sanz, Y.; Adan, R.A.H. Microbiota in obesity: Interactions with enteroendocrine, immune and central nervous systems. Obes. Rev. 2018, 19, 435–451. [Google Scholar] [CrossRef]
- Scriven, M.; Dinan, T.G.; Cryan, J.F.; Wall, M. Neuropsychiatric Disorders: Influence of Gut Microbe to Brain Signalling. Diseases 2018, 6, 78. [Google Scholar] [CrossRef]
- Foster, J.A.; Rinaman, L.; Cryan, J.F. Neurobiology of Stress Stress & the gut-brain axis: Regulation by the microbiome. Neurobiol. Stress 2017, 7, 124–136. [Google Scholar]
- Hamilton, M.K.; Raybould, H.E. Bugs, guts and brains, and the regulation of food intake and body weight. Int. J. Obes. Suppl. 2016, 6, S8–S14. [Google Scholar] [CrossRef]
- Agustí, A.; García-Pardo, M.P.; López-Almela, I.; Campillo, I.; Maes, M.; Romaní-Pérez, M.; Sanz, Y. Interplay between the gut-brain axis, obesity and cognitive function. Front. Neurosci. 2018, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.; Herzog, C.; Pacheco, J.A.; Fujisaka, S.; Bullock, K.; Clish, C.B.; Kahn, C.R. Gut microbiota modulate neurobehavior through changes in brain insulin sensitivity and metabolism. Mol. Psychiatry 2018, 23, 2287–2301. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Noble, E.E.; Hsu, T.M.; Kanoski, S.E. Gut to Brain Dysbiosis: Mechanisms Linking Western Diet Consumption, the Microbiome, and Cognitive Impairment. Front. Beav. Neurosci. 2017, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Bean, M.K.; Stewart, K.; Olbrisch, M.E. Obesity in America: Implications for clinical and health psychologists. J. Clin. Psychol. Med. Settings 2008, 15, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Luppino, F.S.; de Wit, L.M.; Bouvy, P.F.; Stijnen, T.; Cuijpers, P.; Penninx, B.W.; Zitman, F.G. Overweight, obesity, and depression: A systematic review and meta-analysis of longitudinal studies. Arch. Gen. Psychiatry 2010, 67, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Beall, C.; Hanna, L.; Ellacott, K.L.J. CNS Targets of Adipokines. Compr. Physiol. 2017, 7, 1359–1406. [Google Scholar] [PubMed]
- Woods, S.C.; D’Alessio, D.A. Central control of body weight and appetite. J. Clin. Endocrinol. Metab. 2008, 93, S37–S50. [Google Scholar] [CrossRef]
- Benarroch, E.E. Neural control of feeding behavior: Overview and clinical correlations. Neurology 2010, 74, 1643–1650. [Google Scholar] [CrossRef]
- Müller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Woods, S.C.; Porte, D., Jr.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef]
- Pocai, A.; Lam, T.K.; Gutierrez-Juarez, R.; Obici, S.; Schwartz, G.J.; Bryan, J.; Aguilar-Bryan, L.; Rossetti, L. Hypothalamic K(ATP) channels control hepatic glucose production. Nature 2005, 434, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.; Lawson, M.; Zhu, W.; Beverly, J.L.; Sherwin, R.S. ATP-sensitive K(+) channels regulate the release of GABA in the ventromedial hypothalamus during hypoglycemia. Diabetes 2007, 56, 1120–1126. [Google Scholar] [CrossRef]
- Campfield, L.A.; Smith, F.J.; Guisez, Y.; Devos, R.; Burn, P. Recombinant mouse OB protein: Evidence for a peripheral signal linking adiposity and central neural networks. Science 1995, 269, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.; Pellerin, L.; Dallman, M.F.; Oltmanns, K.M.; Schweiger, U.; Born, J.; Fehm, H.L. Causes of obesity: Looking beyond the hypothalamus. Prog. Neurobiol. 2007, 81, 61–88. [Google Scholar] [CrossRef]
- Peters, A.; Schweiger, U.; Pellerin, L.; Hubold, C.; Oltmanns, K.M.; Conrad, M.; Schultes, B.; Born, J.; Fehm, H.L. The selfish brain: Competition for energy resources. Neurosci. Biobehav. Rev. 2004, 28, 143–180. [Google Scholar] [CrossRef] [PubMed]
- Schmoller, A.; Hass, T.; Strugovshchikova, O.; Melchert, U.H.; Scholand-Engler, H.G.; Peters, A.; Schweiger, U.; Hohagen, F.; Oltmanns, K.M. Evidence for a relationship between body mass and energy metabolism in the human brain. J. Cereb. Blood Flow Metab. 2010, 30, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Jauch-Chara, K.; Friedrich, A.; Rezmer, M.; Melchert, U.H.; Scholand-Engler, H.G.; Hallschmid, M.; Oltmanns, K.M. Intranasal insulin suppresses food intake via enhancement of brain energy levels in humans. Diabetes 2012, 61, 2261–2268. [Google Scholar] [CrossRef]
- Jauch-Chara, K.; Binkofski, F.; Loebig, M.; Reetz, K.; Jahn, G.; Melchert, U.H.; Schweiger, U.; Oltmanns, K.M. Blunted brain energy consumption relates to insula atrophy and impaired glucose tolerance in obesity. Diabetes 2015, 64, 2082–2091. [Google Scholar] [CrossRef]
- Wardzinski, E.K.; Kistenmacher, A.; Melchert, U.H.; Jauch-Chara, K.; Oltmanns, K.M. Impaired brain energy gain upon a glucose load in obesity. Metabolism 2018, 85, 90–96. [Google Scholar] [CrossRef]
- Kelley, A.E.; Berridge, K.C. The neuroscience of natural rewards: Relevance to addictive drugs. J. Neurosci. 2002, 22, 3306–3311. [Google Scholar] [CrossRef] [PubMed]
- Adam, T.C.; Epel, E.S. Stress, eating and the reward system. Physiol. Behav. 2007, 91, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Lovallo, W.R.; Thomas, T.R. Stress hormones in psychophysiological research: Emotional, behavioral, and cognitive implications. In Handbook of Psychophysiology, 1st ed.; Cacioppo, J.T., Tassinary, L.G., Berntson, G.G., Eds.; Cambridge University Press: New York, NY, USA, 2000; pp. 342–367. [Google Scholar]
- Johnson, P.M.; Kenny, P.J. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat. Neurosci. 2010, 13, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wise, R.A. How can drug addiction help us understand obesity? Nat. Neurosci. 2005, 8, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Pruessner, J.C.; Champagne, F.; Meaney, M.J.; Dagher, A. Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: A positron emission tomography study using [11C]raclopride. J. Neurosci. 2004, 24, 2825–2831. [Google Scholar] [CrossRef]
- Wand, G.S.; Oswald, L.M.; McCaul, M.E.; Wong, D.F.; Johnson, E.; Zhou, Y.; Kuwabara, H.; Kumar, A. Association of amphetamineinduced striatal dopamine release and cortisol responses to psychological stress. Neuropsychopharmacology 2007, 32, 2310–2320. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R. Chronic stress, drug use, and vulnerability to addiction. Ann. N. Y. Acad. Sci. 2008, 1141, 105–130. [Google Scholar] [CrossRef] [PubMed]
- Jéquier, E. Leptin signaling, adiposity, and energy balance. Ann. N. Y. Acad. Sci. 2002, 967, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Cottone, P.; Sabino, V.; Roberto, M.; Bajo, M.; Pockros, L.; Frihauf, J.B.; Fekete, E.M.; Steardo, L.; Rice, K.C.; Grigoriadis, D.E.; et al. CRF system recruitment mediates dark side of compulsive eating. Proc. Natl. Acad. Sci. USA 2009, 106, 20016–20020. [Google Scholar] [CrossRef]
- Dallman, M.F.; Pecoraro, N.; Akana, S.F.; La Fleur, S.E.; Gomez, F.; Houshyar, H.; Bell, M.E.; Bhatnagar, S.; Laugero, K.D.; Manalo, S. Chronic stress and obesity: A new view of “comfort food”. Proc. Natl. Acad. Sci. USA 2003, 100, 11696–11701. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, L.; Wolf, O.T. Stress prompts habit behavior in humans. J. Neurosci. 2009, 29, 7191–7198. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.; Ingram, M.C.; Anderson, N.H.; Morrison, C.; Davies, E.; Connell, J.M. Cortisol effects on body mass, blood pressure, and cholesterol in the general population. Hypertension 1999, 33, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Tataranni, P.A.; Larson, D.E.; Snitker, S.; Young, J.B.; Flatt, J.P.; Ravussin, E. Effects of glucocorticoids on nergy metabolism and food intake in humans. Am. J. Physiol. 1996, 271, E317–E325. [Google Scholar] [PubMed]
- Epel, E.S.; Lapidus, R.; McEwen, B.; Brownell, K. Stress may add bite to appetite in women: A laboratory study of stress-induced cortisol and eating behavior. Psychoneuroendocrinology 2001, 26, 37–49. [Google Scholar] [CrossRef]
- Sinha, R.; Jastreboff, A.M. Stress as a common risk factor for obesity and addiction. Biol. Psychiatry 2013, 73, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Björntorp, P. Do stress reactions cause abdominal obesity and comorbidities? Obes. Rev. 2001, 2, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Shibli-Rahhal, A.; Van Beek, M.; Schlechte, J.A. Cushing’s syndrome. Clin. Dermatol. 2006, 24, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Björntorp, P.; Rosmond, R. The metabolic syndrome: A neuroendocrine disorder? Br. J. Nutr. 2000, 83, S49–S57. [Google Scholar] [CrossRef]
- Coppack, S.W. Pro-inflammatory cytokines and adipose tissue. Proc. Nutr. Soc. 2001, 3, 349–356. [Google Scholar] [CrossRef]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose tissue in obesity-related inflammation and insulin resistance: Cells, cytokines, and chemokines. ISRN Inflamm. 2013, 2013, 139239. [Google Scholar] [CrossRef] [PubMed]
- Björntorp, P.; Bergman, H.; Varnauskas, E. Plasma free fatty acid turnover rate in obesity. Acta Med. Scand. 1969, 185, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Contreras, C.; González-García, I.; Martínez-Sánchez, N.; Seoane-Collazo, P.; Jacas, J.; Morgan, D.A.; Serra, D.; Gallego, R.; Gonzalez, F.; Casals, N.; et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep. 2014, 9, 366–377. [Google Scholar] [CrossRef]
- McFadden, J.W.; Aja, S.; Li, Q.; Bandaru, V.V.; Kim, E.K.; Haughey, N.J.; Kuhajda, F.P.; Ronnett, G.V. Increasing fatty acid oxidation remodels the hypothalamic neurometabolome to mitigate stress and inflammation. PLoS ONE 2014, 9, e115642. [Google Scholar] [CrossRef] [PubMed]
- Viader, A.; Sasaki, Y.; Kim, S.; Strickland, A.; Workman, C.S.; Yang, K.; Gross, R.W.; Milbrandt, J. Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron 2013, 77, 886–898. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Schneider, J.A.; Tangney, C.; Tremblay-Mercier, J.; Fortier, M.; Bennett, D.A.; Morris, M.C. Plasma and brain fatty acid profiles in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 2012, 29, 691–697. [Google Scholar] [CrossRef]
- Elias, M.F.; Elias, P.K.; Sullivan, L.M.; Wolf, P.A.; D’Agostino, R.B. Obesity, diabetes and cognitive deficit: The Framingham Heart Study. Neurobiol. Aging 2005, 26, S11–S16. [Google Scholar] [CrossRef]
- Cournot, M.; Marquié, J.C.; Ansiau, D.; Martinaud, C.; Fonds, H.; Ferrières, J.; Ruidavets, J.B. Relation between body mass index and cognitive function in healthy midd-le-aged men and women. Neurology 2006, 67, 1208–1214. [Google Scholar] [CrossRef]
- Sabia, S.; Kivimaki, M.; Shipley, M.J.; Marmot, M.G.; Singh-Manoux, A. Body mass index over the adult life course and cognition in late midlife: The Whitehall II Cohort Study. Am. J. Clin. Nutr. 2009, 89, 601–607. [Google Scholar] [CrossRef]
- Hassing, L.B.; Dahl, A.K.; Pedersen, N.L.; Johansson, B. Overweight in midlife is related to lower cognitive function 30 years later: A prospective study with longitudinal assessments. Dement. Geriatr. Cogn. Disord. 2010, 29, 543–552. [Google Scholar] [CrossRef]
- Dahl, A.K.; Hassing, L.B.; Fransson, E.I.; Gatz, M.; Reynolds, C.A.; Pedersen, N.L. Body mass index across midlife and cogni-tive change in late life. Int. J. Obes. 2013, 37, 296–302. [Google Scholar] [CrossRef]
- Anstey, K.J.; Cherbuin, N.; Budge, M.; Young, J. Body mass index in midlife and late-life as a risk factor for dementia: A meta-analysis of prospective studies. Obes. Rev. 2011, 12, e426–e437. [Google Scholar] [CrossRef]
- Pedditizi, E.; Peters, R.; Beckett, N. The risk of overweight/obesity in mid-life and late life for the development of de-mentia: A systematic review and meta-analysis of longitudinal studies. Age Ageing 2016, 45, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Cheke, L.G.; Simons, J.S.; Clayton, N.S. Higher body mass index is associated with episodic memory deficits in young adults. Q. J. Exp. Psychol. 2016, 69, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Navas, J.F.; Vilar-López, R.; Perales, J.C.; Steward, T.; Fernández-Aranda, F.; Verdejo-García, A. Altered Decision-Making under Risk in Obesity. PLoS ONE 2016, 11, e0155600. [Google Scholar] [CrossRef] [PubMed]
- Cortese, S.; Vincenzi, B. Obesity and ADHD: Clinical and neurobiological implications. Curr. Top. Behav. Neurosci. 2012, 9, 199–218. [Google Scholar] [PubMed]
- Holtkamp, K.; Konrad, K.; Müller, B.; Heussen, N.; Herpertz, S.; Herpertz-Dahlmann, B.; Hebebrand, J. Overweight and obesity in children with attention-deficit/hyperactivity disorder. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Kummer, A.; Barbosa, I.G.; Rodrigues, D.H.; Rocha, N.P.; Rafael Mda, S.; Pfeilsticker, L.; Silva, A.C.; Teixeira, A.L. Frequency of overweight and obesity in children and adolescents with autism and attention deficit/hyperactivity disorder. Rev. Paul. Pediatr. 2016, 34, 71–77. [Google Scholar] [CrossRef]
- Fitzpatrick, S.; Gilbert, S.; Serpell, L. Systematic review: Are overweight and obese individuals impaired on behavioural tasks of executive functioning? Neuropsychol. Rev. 2013, 23, 138–156. [Google Scholar] [CrossRef]
- Yau, P.L.; Kang, E.H.; Javier, D.C.; Convit, A. Preliminary evidence of cognitive and brain abnormalities in uncomplicated adolescent obesity. Obesity 2014, 22, 1865–1871. [Google Scholar] [CrossRef]
- Boeka, A.G.; Lokken, K.L. Neuropsychological performance of a clinical sample of extremely obese individuals. Arch. Clin. Neuropsychol. 2008, 23, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W.; Harvey, D.; Mungas, D.; Haan, M. Central obesity and the aging brain. Arch. Neurol. 2005, 62, 1545–1548. [Google Scholar] [CrossRef] [PubMed]
- Raji, C.A.; Ho, A.J.; Parikshak, N.N.; Becker, J.T.; Lopez, O.L.; Kuller, L.H.; Hua, X.; Leow, A.D.; Toga, A.W.; Thompson, P.M. Brain structure and obesity. Hum. Brain Mapp. 2010, 31, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Molteni, R.; Barnard, R.J.; Ying, Z.; Roberts, C.K.; Gomez-Pinilla, F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience 2002, 112, 803–814. [Google Scholar] [CrossRef]
- Jurdak, N.; Lichtenstein, A.H.; Kanarek, R.B. Diet-induced obesity and spatial cognition in young male rats. Nutr. Neurosci. 2008, 11, 48–54. [Google Scholar] [CrossRef]
- Farr, S.A.; Yamada, K.A.; Butterfield, D.A.; Abdul, H.M.; Xu, L.; Miller, N.E.; Banks, W.A.; Morley, J.E. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology 2008, 149, 2628–2636. [Google Scholar] [CrossRef]
- Hargrave, S.L.; Davidson, T.L.; Zheng, W.; Kinzig, K.P. Western diets induce blood-brain barrier leakage and alter spatial strategies in rats. Behav. Neurosci. 2016, 130, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Buckman, L.B.; Hasty, A.H.; Flaherty, D.K.; Buckman, C.T.; Thompson, M.M.; Matlock, B.K.; Weller, K.; Ellacott, K.L. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav. Immun. 2014, 35, 33–42. [Google Scholar] [CrossRef]
- Sims-Robinson, C.; Bakeman, A.; Glasser, R.; Boggs, J.; Pacut, C.; Feldman, E.L. The role of endoplasmic reticulum stress in hippocampal insulin resistance. Exp. Neurol. 2016, 277, 261–267. [Google Scholar] [CrossRef]
- Cai, M.; Wang, H.; Li, J.J.; Zhang, Y.L.; Xin, L.; Li, F.; Lou, S.J. The signaling mechanisms of hippocampal endoplasmic reticulum stress affecting neu-ronal plasticity-related protein levels in high fat diet-induced obese rats and the regulation of aerobic exercise. Brain Behav. Immun. 2016, 57, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Wu, J.; Nesil, T.; Li, M.D.; Aylor, K.W.; Liu, Z. Long-term high-fat diet induces hippo-campal microvascular insulin resistance and cognitive dysfunction. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E89–E97. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Moraes, J.C.; Coope, A.; Morari, J.; Cintra, D.E.; Roman, E.A.; Pauli, J.R.; Romanatto, T.; Carvalheira, J.B.; Oliveira, A.L.; Saad, M.J.; et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS ONE 2009, 4, e5045. [Google Scholar] [CrossRef] [PubMed]
- Hryhorczuk, C.; Sharma, S.; Fulton, S.E. Metabolic disturbances connecting obesity and depression. Front. Neurosci. 2013, 7, 177. [Google Scholar] [CrossRef] [PubMed]
- Carey, M.; Small, H.; Yoong, S.L.; Boyes, A.; Bisquera, A.; Sanson-Fisher, R. Prevalence of comorbid depression and obesity in general practice: A cross-sectional survey. Br. J. Gen. Pract. 2014, 64, e122–e127. [Google Scholar] [CrossRef] [PubMed]
- de Wit, L.; Luppino, F.; van Straten, A.; Penninx, B.; Zitman, F.; Cuijpers, P. Depression and obesity: A meta-analysis of community-based studies. Psychiatry Res. 2010, 178, 230–235. [Google Scholar] [CrossRef]
- Lambert, E.; Sar, C.I.; Dawood, T.; Nguyen, J.; McGrane, M.; Eikelis, N.; Chopra, R.; Wong, C.; Chatzivlastou, K.; Head, G.; et al. Sympathetic nervous system activity is associated with obesity-induced subcli-nical organ damage in young adults. Hypertension 2010, 56, 351–358. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Hinder, L.M.; Callaghan, B.C.; Feldman, E.L. Neurological consequences of obesity. Lancet Neurol. 2017, 16, 465–477. [Google Scholar] [CrossRef]
- Ylitalo, K.R.; Sowers, M.; Heeringa, S. Peripheral vascular disease and peripheral neuropathy in individuals with cardio-metabolic clustering and obesity: National Health and Nutrition Examination Survey 2001–2004. Diabetes Care 2011, 34, 1642–1647. [Google Scholar] [CrossRef]
- Tesfaye, S.; Chaturvedi, N.; Eaton, S.E.; Ward, J.D.; Manes, C.; Ionescu-Tirgoviste, C.; Witte, D.R.; Fuller, J.H.; EURODIAB Prospective Complications Study Group. Vascular risk factors and diabetic neuropathy. N. Engl. J. Med. 2005, 352, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.; Rathmann, W.; Dickhaus, T.; Meisinger, C.; Mielck, A.; KORA Study Group. Prevalence of polyneuropathy in pre-diabetes and diabetes is associated with abdominal obesity and macroangiopathy: The MONICA/KORA Augsburg Surveys S2 and S3. Diabetes Care 2008, 31, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Singleton, J.; Volckmann, E.; Graham, T.; Smith, A. Neuropathy Associated with Nondiabetic Obesity. Neurology 2014, 82, S10. [Google Scholar]
- Jayaraman, A.; Lent-Schochet, D.; Pike, C.J. Diet-induced obesity and low testosterone increase neuroinflammation and impair neural function. J. Neuroinflamm. 2014, 11, 162. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; von Hehn, C.A.; Woolf, C.J. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat. Neurosci. 2012, 15, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Russell, J.; Feldman, E.L.; Goldstein, J.; Peltier, A.; Smith, S.; Hamwi, J.; Pollari, D.; Bixby, B.; Howard, J.; et al. Lifestyle intervention for pre-diabetic neuropathy. Diabetes Care 2006, 29, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Zuloaga, K.L.; Kugelman, T.L.; Mader, K.S.; Morré, J.T.; Zuloaga, D.G.; Weber, S.; Marzulla, T.; Mulford, A.; Button, D.; et al. Amelioration of metabolic syndrome-associated cognitive impair-ments in mice via a reduction in dietary fat content or infusion of non-diabetic plasma. EBioMedicine 2016, 3, 26–42. [Google Scholar] [CrossRef]
- Kim, H.; Kang, H.; Heo, R.W.; Jeon, B.T.; Yi, C.O.; Shin, H.J.; Kim, J.; Jeong, S.Y.; Kwak, W.; Kim, W.H.; et al. Caloric restriction improves diabetes-induced cognitive deficits by attenuating neurogranin-associated calcium signaling in high-fat diet-fed mice. J. Cereb. Blood Flow Metab. 2016, 36, 1098–1110. [Google Scholar] [CrossRef]
- Sims-Robinson, C.; Bakeman, A.; Bruno, E.; Jackson, S.; Glasser, R.; Murphy, G.G.; Feldman, E.L. Dietary reversal ameliorates short- and long-term memory deficits in-duced by high-fat diet early in life. PLoS ONE 2016, 11, e0163883. [Google Scholar] [CrossRef]
- Miller, L.A.; Crosby, R.D.; Galioto, R.; Strain, G.; Devlin, M.J.; Wing, R.; Cohen, R.A.; Paul, R.H.; Mitchell, J.E.; Gunstad, J. Bariatric surgery patients exhibit improved memory function 12 months po-stoperatively. Obes. Surg. 2013, 23, 1527–1535. [Google Scholar] [CrossRef]
- Archie, E.A.; Theis, K.R. Animal behavior meets microbial ecology. Anim. Behav. 2011, 82, 425–436. [Google Scholar] [CrossRef]
- Natividad, J.M.M.; Verdu, E.F. Modulation of intestinal barrier by intestinal microbiota: Pathological and therapeutic implications. Pharmacol. Res. 2013, 69, 42–51. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Bäumler, A.J.; Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 2016, 535, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Repáraz, J.; Kasper, L.H. Gutmicrobiome and the risk factors in central nervous system autoimmunity. FEBS Lett. 2014, 588, 4214–4222. [Google Scholar] [CrossRef] [PubMed]
- Albenberg, L.G.; Wu, G.D. Diet and the intestinal microbiome: Associations, functions, and implications for health and disease. Gastroenterology 2014, 146, 1564–1572. [Google Scholar] [CrossRef]
- Russo, E.; Bacci, G.; Chiellini, C.; Fagorzi, C.; Niccolai, E.; Taddei, A.; Ricci, F.; Ringressi, M.N.; Borrelli, R.; Melli, F.; et al. Preliminary Comparison of Oral and Intestinal Human Microbiota in Patients with Colorectal Cancer: A Pilot Study. Front. Microbiol. 2018, 8, 2699. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut mi-crobiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar]
- Mayer, E.A. Gut feelings: The emerging biology of gut-brain communication. Nat. Rev. Neurosci. 2011, 12, 453–466. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Hyland, N.P.; Dinan, T.G.; Cryan, J.F. Maternal separation as a model of brain-gut axis dysfunction. Psychopharmacology 2011, 214, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Desbonnet, L.; Garrett, L.; Clarke, G.; Bienenstock, J.; Dinan, T.G. The probiotic Bifidobacteria infantis: Anassessmentofpotential antidepressantpropertiesinthe rat. J. Psychiatr. Res. 2009, 43, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, P.; Bienenstock, J. Immunomodulationby commensalandprobioticbacteria. Immunol. Investig. 2010, 39, 429–448. [Google Scholar] [CrossRef] [PubMed]
- Duerkop, B.A.; Vaishnava, S.; Hooper, L.V. Immune responsestothemicrobiotaatthe intestinalmucosalsur-face. Immunity 2009, 31, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinalflora and gastrointestinalsta-tusinchil-drenwithautism–comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.H.; Meeking, M.M.; Mepham, J.R.; Tichenoff, L.; Possmayer, F.; Liu, S.; MacFabe, D.F. The entericbacterialme-tabolitepropi-onic acidaltersbrainandplasma phospholipid molecularspecies:fur-ther developmentofarodentmodel of autismspectrumdisorders. J. Neuroinflamm. 2012, 9, 153. [Google Scholar]
- Grimaldi, R.; Gibson, G.R.; Vulevic, J.; Giallourou, N.; Castro-Mejía, J.L.; Hansen, L.H.; Gibson, E.L.; Nielsen, D.S.; Costabile, A. A prebiotic intervention study in children with autism spectrum disorders (ASDs). BMC Microb. 2018, 6, 133. [Google Scholar] [CrossRef]
- Foster, J.A.; Mc Vey Neufeld, K.A. Gut–brain axis: How the microbiome influences anxiety and depression. Trend Neurosci. 2013, 36, 305–312. [Google Scholar] [CrossRef]
- Manco, M. Gut microbiota and developmental programming of the brain: From evidence in behavioral endophenotypes to novel perspective in obesity. Front. Cell. Infect. Microbiol. 2012, 2, 109. [Google Scholar] [CrossRef]
- Davey, K.J.; O’Mahony, S.M.; Schellekens, H.; O’Sullivan, O.; Bienenstock, J.; Cotter, P.D.; Dinan, T.G.; Cryan, J.F. Gender-dependent consequences of chronic olanzapine in the rat: Effects on body weight, inflammatory, metabolic and microbiota parameters. Psychopharmacology 2012, 221, 155–169. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Gordon, J.I. The core gut microbiome, energy balance and obesity. J. Physiol. 2009, 587, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Thompson, G.R.; Trexler, P.C. Gastrointestinal structure and function in germ-free or gnotobiotic animals. Gut 1971, 12, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Wostmann, B.S. Germ Free and Gnotobiotic Animal Models: Background and Applications; CRC Press: Boca Raton, FL, USA, 1996; pp. 1–188. [Google Scholar]
- McCracken, V.; Lorenz, R. The gastrointestinal ecosystem: A precarious alliance among epithelium, immunity and microbiota. Cell. Microbiol. 2001, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Duca, F.A.; Swartz, T.D.; Sakar, Y.; Covasa, M. Increased oral detection, but decreased intestinal signaling for fats in mice lacking gut microbiota. PLoS ONE 2012, 7, e39748. [Google Scholar] [CrossRef] [PubMed]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Karlsson, F.; Werling, M.; Ståhlman, M.; Kovatcheva-Datchary, P.; Olbers, T.; Fändriks, L.; le Roux, C.W.; Nielsen, J.; Bäckhed, F. Roux-en-Y gastric bypass and vertical banded gastroplasty induce long-term changes on the human gut microbiome contributing to fat mass regulation. Cell. Metab. 2015, 22, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6–14. [Google Scholar] [CrossRef]
- Brinkworth, G.D.; Noakes, M.; Clifton, P.M.; Bird, A.R. Comparative effects of very low-carbohydrate, high-fat and high-carbohydrate, low-fat weight-loss diets on bowel habit and faecal short-chain fatty acids and bacterial populations. Br. J. Nutr. 2009, 101, 1493–1502. [Google Scholar] [CrossRef]
- de La Serre, C.B.; Ellis, C.L.; Lee, J.; Hartman, A.L.; Rutledge, J.C.; Raybould, H.E. Propensity to high-fat diet-induced obesity in rats is associ-ated with changes in the gut microbiota and gut inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Angelakis, E.; Armougom, F.; Million, M.; Raoult, D. The relationship between gut microbiota and weight gain in humans. Future Microbiol. 2012, 7, 91–109. [Google Scholar] [CrossRef] [PubMed]
- Ravussin, Y.; Koren, O.; Spor, A.; LeDuc, C.; Gutman, R.; Stombaugh, J.; Knight, R.; Ley, R.E.; Leibel, R.L. Responses of gut microbiota to diet composition and weight loss in lean and obese mice. Obesity 2012, 20, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, K.R.; Hauck, L.; Jeffrey, B.M.; Elias, V.; Humphrey, A.; Nath, R.; Perrone, A.; Bermudez, L.E. Relationships between diet-related changes in the gut microbiome and cognitive flexibility. Neuroscience 2015, 300, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.S.; Chambers, E.S.; Morrison, D.J.; Frost, G. The role of short chain fatty acids in appetite regulation and energy homeostasis. Int. J. Obes. 2015, 39, 1331–1338. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide utilization by gut bacteria: Potential for new insights from ge-nomic analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypo-thalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schafer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Louis, P.; Young, P.; Holtrop, G.; Flint, H.J. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA: Acetate CoA-transferase gene. Environ. Microbiol. 2010, 12, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome-brain-β cell axis promoting metabolic syndrome. Nature 2016, 534, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Boulange, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Müller, M.; Groen, A.K.; Hooiveld, G.J.; Bakker, B.M.; et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G900–G910. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, E.; Grootaert, C.; Verstraete, W.; Van de Wiele, T. Propionate as a health-promoting microbial metabolite in the human gut. Nutr. Rev. 2011, 69, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Berggren, A.M.; Nyman, E.M.; Lundquist, I.; Björck, I.M. Influence of orally and rectally administered propionate on cholesterol and glucose metabolism in obese rats. Br. J. Nutr. 1996, 76, 287–294. [Google Scholar] [CrossRef]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef]
- Sahuri-Arisoylu, M.; Brody, L.P.; Parkinson, J.R.; Parkes, H.; Navaratnam, N.; Miller, A.D.; Thomas, E.L.; Frost, G.; Bell, J.D. Reprogramming of hepatic fat accumulation and ‘browning’ of adipose tissue by the short-chain fatty acid acetate. Int. J. Obes. 2016, 40, 955–963. [Google Scholar] [CrossRef]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef]
- Everard, A.; Lazarevic, V.; Gaïa, N.; Johansson, M.; Ståhlman, M.; Backhed, F.; Delzenne, N.M.; Schrenzel, J.; François, P.; Cani, P.D. Microbiome of prebiotic-treated mice reveals novel targets involved in host response during obesity. ISME J. 2014, 8, 2116–2130. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [PubMed]
- DiBaise, J.K.; Zhang, H.; Crowell, M.D.; Krajmalnik-Brown, R.; Decker, G.A.; Rittmann, B.E. Gut microbiota and its possible relationship with obesity. Mayo Clin. Proc. 2008, 83, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Bjursell, M.K.; Himrod, J.; Deng, S.; Carmichael, L.K.; Chiang, H.C.; Hooper, L.V.; Gordon, J.I. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 2003, 299, 2074–2076. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Torres-Fuentes, C.; Schellekens, H.; Dinan, T.G.; Cryan, J.F. The microbiota-gut-brain axis in obesity. Lancet Gastroenterol. Hepatol. 2017, 2, 747–756. [Google Scholar] [CrossRef]
- Delzenne, N.M.; Neyrinck, A.M.; Backhed, F.; Cani, P.D. Targeting gut microbiota in obesity: Effects of prebiotics and probiotics. Nat. Rev. Endocrinol. 2011, 7, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Obanda, D.; Page, R.; Guice, J.; Raggio, A.M.; Husseneder, C.; Marx, B.; Stout, R.W.; Welsh, D.A.; Taylor, C.M.; Luo, M. CD Obesity-Prone Rats, but not Obesity-Resistant Rats, Robustly Ferment Resistant Starch Without Increased Weight or Fat Accretion. Obesity 2018, 2, 570–577. [Google Scholar] [CrossRef]
- Zhou, J.; Martin, R.J.; Raggio, A.M.; Shen, L.; McCutcheon, K.; Keenan, M.J. The importance of GLP-1 and PYY in resistant starch’s effect on body fat in mice. Mol. Nutr. Food Res. 2015, 59, 1000–1003. [Google Scholar] [CrossRef]
- Patterson, E.; Ryan, P.M.; Cryan, J.F.; Dinan, T.G.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C. Gut microbiota, obesity and diabetes. Postgrad. Med. J. 2016, 92, 286–300. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. Mood by microbe: Towards clinical translation. Genome Med. 2016, 8, 36. [Google Scholar] [CrossRef]
- Sandhu, K.V.; Sherwin, E.; Schellekens, H.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Feeding the microbiota-gut-brain axis: Diet, microbiome, and neuropsychiatry. Transl. Res. 2017, 179, 223–244. [Google Scholar] [CrossRef]
- Camilleri, M.; Toouli, J.; Herrera, M.F.; Kulseng, B.; Kow, L.; Pantoja, J.P.; Marvik, R.; Johnsen, G.; Billington, C.J.; Moody, F.G.; et al. Intra-abdominal vagal blocking (VBLOC therapy): Clinical results with a new implantable medical device. Surgery 2008, 143, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Sawchenko, P.E.; Gold, R.M.; Leibowitz, S.F. Evidence for vagal involvement in the eating elicited by adrenergic stimulation of the paraventricular nucleus. Brain Res. 1981, 225, 249–269. [Google Scholar] [CrossRef]
- Kollai, M.; Bonyhay, I.; Jokkel, G.; Szonyi, L. Cardiac vagal hyperactivity in adolescent anorexia nervosa. Eur. Heart J. 1994, 15, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and en-teric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar] [PubMed]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef]
- Shah, M.; Vella, A. Effects of GLP-1 on appetite and weight. Rev. Endocr. Metab. Disord. 2014, 15, 181–187. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef]
- Xiong, Y.; Miyamoto, N.; Shibata, K.; Valasek, M.A.; Motoike, T.; Kedzierski, R.M.; Yanagisawa, M. Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proc. Natl. Acad. Sci. USA 2004, 101, 1045–1050. [Google Scholar] [CrossRef]
- Nohr, M.K.; Pedersen, M.H.; Gille, A.; Egerod, K.L.; Engelstoft, M.S.; Husted, A.S.; Sichlau, R.M.; Grunddal, K.V.; Poulsen, S.S.; Han, S.; et al. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology 2013, 154, 3552–3564. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, C.B.; Gabe, M.B.N.; Svendsen, B.; Dragsted, L.O.; Rosenkilde, M.M.; Holst, J.J. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G53–G65. [Google Scholar] [CrossRef] [PubMed]
- Silberbauer, C.J.; Surina-Baumgartner, D.M.; Arnold, M.; Langhans, W. Prandial lactate infusion inhibits spontaneous feeding in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R646–R653. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Stilling, R.M.; Kennedy, P.J.; Stanton, C.; Cryan, J.F.; Dinan, T.G. Minireview: Gut microbiota: The neglected endocrine organ. Mol. Endocrinol. 2014, 28, 1221–1238. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Aneman, A.; Friberg, P.; Hooper, D.; Fåndriks, L.; Lonroth, H.; Hunyady, B.; Mezey, E. Substantial production of dopamine in the human gastrointestinal tract. J. Clin. Endocrinol. Metab. 1997, 82, 3864–3871. [Google Scholar] [CrossRef] [PubMed]
- Lyte, M. Probiotics function mechanistically as delivery vehicles for neuroactive compounds: Microbial endocrinology in the design and use of probiotics. Bioessays 2011, 33, 574–581. [Google Scholar] [CrossRef]
- Tsavkelova, E.A.; Klimova, Slu.; Cherdyntseva, T.A.; Netrusov, A.I. Hormones and hormone-like substances of microorganisms: A review. Prikl. Biokhim. Mikrobiol. 2006, 42, 261–268. [Google Scholar] [CrossRef]
- Desbonnet, L.; Garrett, L.; Clarke, G.; Kiely, B.; Cryan, J.F.; Dinan, T.G. Effects of the probiotic Bifidobacterium infantis in the maternal separation model of depression. Neuroscience 2010, 170, 1179–1188. [Google Scholar] [CrossRef]
- Heisler, L.K.; Jobst, E.E.; Sutton, G.M.; Zhou, L.; Borok, E.; Thornton-Jones, Z.; Liu, H.Y.; Zigman, J.M.; Balthasar, N.; Kishi, T.; et al. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron 2006, 51, 239–249. [Google Scholar] [CrossRef]
- Xu, Y.; Jones, J.E.; Kohno, D.; Williams, K.W.; Lee, C.E.; Choi, M.J.; Anderson, J.G.; Heisler, L.K.; Zigman, J.M.; Lowell, B.B.; et al. 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate energy homeostasis. Neuron 2008, 60, 582–589. [Google Scholar] [CrossRef]
- Thomas, C.M.; Hong, T.; van Pijkeren, J.P.; Hemarajata, P.; Trinh, D.V.; Hu, W.; Britton, R.A.; Kalkum, M.; Versalovic, J. Histamine derived from probiotic Lactobacillus reuteri suppresses TNF via modulation of PKA and ERK signaling. PLoS ONE 2012, 7, e31951. [Google Scholar] [CrossRef] [PubMed]
- Lyte, M.; Vulchanova, L.; Brown, D.R. Stress at the intestinal surface: Catecholamines and mucosa-bacteria interactions. Cell Tissue Res. 2011, 343, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Delgado, T.C. Glutamate and GABA in appetite regulation. Front. Endocrinol. 2013, 4, 103. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Cenit, M.C.; Sanz, Y.; Codoñer-Franch, P. Influence of gut microbiota on neuropsychiatric disorders. World J. Gastroenterol. 2017, 23, 5486–5498. [Google Scholar] [CrossRef]
- Kelly, J.R.; Borre, Y.; O’Brien, C.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G.; et al. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Schachter, J.; Martel, J.; Lin, C.S.; Chang, C.J.; Wu, T.R.; Lu, C.C.; Ko, Y.F.; Lai, H.C.; Ojcius, D.M.; Young, J.D. Effects of obesity on depression: A role for inflammation and the gut microbiota. Brain Behav. Immun. 2018, 69, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ohland, C.L.; Kish, L.; Bell, H.; Thiesen, A.; Hotte, N.; Pankiv, E.; Madsen, K.L. Effects of Lactobacillus helveticus on murine behavior are dependent on diet and genotype and correlate with alterations in the gut microbiome. Psychoneuroendocrinology 2013, 38, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, A.; Elfving, B.; Hokland, M.; Wegener, G.; Lund, S. Probiotic treatment reduces depressive-like behaviour in rats inde-pendently of diet. Psychoneuroendocrinology 2017, 79, 40–48. [Google Scholar] [CrossRef]
- Alcock, J.; Maley, C.C.; Aktipis, C.A. Is eating behavior manipulated by the gastrointestinal microbiota? Evolutionary pressures and potential mechanisms. Bioessays 2014, 36, 940–949. [Google Scholar] [CrossRef]
- Njoroge, J.; Sperandio, V. Enterohemorrhagic Escherichia coli virulence regulation by two bacterial adrenergic kinases, QseC and QseE. Infect. Immun. 2012, 80, 688–703. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Heesters, B.A.; Ghasemlou, N.; Von Hehn, C.A.; Zhao, F.; Tran, J.; Wainger, B.; Strominger, A.; Muralidharan, S.; Horswill, A.R. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013, 501, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Gazzaniga, F.S.; Kasper, D.L. Veggies and Intact Grains a Day Keep the Pathogens Away. Cell 2016, 167, 1161–1162. [Google Scholar] [CrossRef]
- Amaral, F.A.; Sachs, D.; Costa, V.V.; Fagundes, C.T.; Cisalpino, D.; Cunha, T.M.; Ferreira, S.H.; Cunha, F.Q.; Silva, T.A.; Nicoli, J.R. Commensal microbiota is fundamental for the development of inflammatory pain. Proc. Natl. Acad. Sci. USA 2008, 105, 2193–2197. [Google Scholar] [CrossRef]
- Khasar, S.G.; Reichling, D.B.; Green, P.G.; Isenberg, W.M.; Levine, J.D. Fasting is a physiological stimulus of vagus-mediated enhance-ment of nociception in the female rat. Neuroscience 2003, 119, 215–221. [Google Scholar] [CrossRef]
- Cani, P.D.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids—At the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef]
- Jager, G.; Witkamp, R.F. The endocannabinoid system and appetite: Relevance for food reward. Nutr. Res. Rev. 2014, 27, 172–185. [Google Scholar] [CrossRef]
- Schellekens, H.; Finger, B.C.; Dinan, T.G.; Cryan, J.F. Ghrelin signalling and obesity: At the interface of stress, mood and food reward. Pharmacol. Ther. 2012, 135, 316–326. [Google Scholar] [CrossRef]
- Byrne, C.S.; Chambers, E.S.; Alhabeeb, H.; Chhina, N.; Morrison, D.J.; Preston, T.; Tedford, C.; Fitzpatrick, J.; Irani, C.; Busza, A.; et al. Increased colonic propionate reduces anticipatory reward responses in the human striatum to high-energy foods. Am. J. Clin. Nutr. 2016, 104, 5–14. [Google Scholar] [CrossRef]
- Buffington, S.A.; Di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762–1775. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.S.; Egan, J.M. The endocrinology of taste receptors. Nat. Rev. Endocrinol. 2015, 11, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Swartz, T.D.; Duca, F.A.; de Wouters, T.; Sakar, Y.; Covasa, M.B. Up-regulation of intestinal type 1 taste receptor 3 and sodium glucose luminal transporter-1 expression and increased sucrose intake in mice lacking gut microbiota. J. Nutr. 2012, 107, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, C.; Thuru, X.; Gelot, A.; Barnich, N.; Neut, C.; Dubuquoy, L.; Dubuquoy, C.; Merour, E.; Geboes, K.; Chamaillard, M.; et al. Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nat. Med. 2007, 13, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Rezzi, S.; Ramadan, Z.; Martin, F.P.; Fay, L.B.; van Bladeren, P.; Lindon, J.C.; Nicholson, J.K.; Kochhar, S. Human metabolic pheno-types link directly to specific dietary preferences in healthy individuals. J. Proteome Res. 2007, 6, 4469–4477. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, L.; Sellaro, R.; van Hemert, S.; Bosch, J.A.; Colzato, L.S. A randomized controlled trial to test the effect of multispecies pro-biotics on cognitive reactivity to sad mood. Brain Behav. Immun. 2015, 48, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.C.; Rehman, A.; Yu, S.; Andino, N.M. Brain fogginess, gas and bloating: A link between SIBO, probiotics and metabolic acidosis. Clin. Transl. Gastroenterol. 2018, 9, 162. [Google Scholar] [CrossRef]
- Goodhand, J.R.; Wahed, M.; Mawdsley, J.E.; Farmer, A.D.; Aziz, Q.; Rampton, D.S. Mood disorders in inflammatory bowel disease: Relation to diagnosis, disease activity, perceived stress, and other factors. Inflamm. Bowel Dis. 2012, 18, 2301–2309. [Google Scholar] [CrossRef]
- Addolorato, G.; Capristo, E.; Stefanini, G.F.; Gasbarrini, G. Inflammatory bowel disease: A study of the association between anxiety and depression, physical morbidity, and nutritional status. Scand. J. Gastroenterol. 1997, 32, 1013–1021. [Google Scholar] [CrossRef]
- Kovacs, Z.; Kovacs, F. Depressive and anxiety symptoms, dysfunctional attitudes and social aspects in irritable bowel syndrome and inflammatory bowel disease. Int. J. Psychiatry Med. 2007, 37, 245–255. [Google Scholar] [CrossRef]
- Mardini, H.E.; Kip, K.E.; Wilson, J.W. Crohn’s disease: A two-year prospective study of the association between psy-chological distress and disease activity. Dig. Dis. Sci. 2004, 49, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Niccolai, E.; Ricci, F.; Russo, E.; Nannini, G.; Emmi, G.; Taddei, A.; Ringressi, M.N.; Melli, F.; Miloeva, M.; Cianchi, F.; et al. The Different Functional Distribution of “Not Effector” T Cells (Treg/Tnull) in Colorectal Cancer. Front. Immunol. 2017, 8, 1900. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Slyepchenko, A.; Maes, M.; Jacka, F.N.; Köhler, C.A.; Barichello, T.; McIntyre, R.S.; Berk, M.; Grande, I.; Foster, J.A.; Vieta, E.; et al. Gut microbiota, bacterial translocation, and interactions with diet: Pathophysiological links between major depressive disorder and non-communicable medical comorbidities. Psychother. Psychosom. 2017, 86, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Hamada, K.; Sumitomo, N.; Okamoto, H.; Sakakibara, B. Serum amyloid A, cytokines, and corticosterone responses in germfree and conventional mice after lipopolysaccharide injection. Biosci. Biotechnol. Biochem. 1999, 63, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Souza, D.G.; Vieira, A.T.; Soares, A.C.; Pinho, V.; Nicoli, J.R.; Vieira, L.Q.; Teixeira, M.M. The essential role of the inte-stinal microbiota in facilitating acute inflammatory responses. J. Immunol. 2004, 173, 4137–4146. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Seo, S.U.; Kamada, N.; Muñoz-Planillo, R.; Kim, Y.G.; Kim, D.; Koizumi, Y.; Hasegawa, M.; Himpsl, S.D.; Browne, H.P.; Lawley, T.D.; et al. Distinct commensals induce interleukin-1β via NLRP3 inflammasome in inflammatory mo-nocytes to promote intestinal inflammation in response to injury. Immunity 2015, 42, 744–755. [Google Scholar] [CrossRef]
- Sen, T.; Cawthon, C.R.; Ihde, B.T.; Hajnal, A.; DiLorenzo, P.M.; de La Serre, C.B.; Czaja, K. Diet-driven microbiota dysbiosis is associated with vagal remodeling and obesity. Physiol. Behav. 2017, 173, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Sanz, Y.; Moya-Perez, A. Microbiota, inflammation and obesity. Adv. Exp. Med. Biol. 2014, 817, 291–317. [Google Scholar] [PubMed]
- Maes, M.; Kubera, M.; Mihaylova, I.; Geffard, M.; Galecki, P.; Leunis, J.C.; Berk, M. Increased autoimmune responses against auto-epitopes modified by oxidative and nitrosative damage in depression: Implications for the pathways to chronic depression and neuroprogression. J. Affect. Disord. 2013, 149, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Riches, D.W. Ifn-gamma + Lps induction of inos is modulated by Erk, Jnk/Sapk, and P38 Mapk in a mouse ma-crophage cell line. Am. J. Physiol. Cell Physiol. 2001, 280, C441–C450. [Google Scholar] [CrossRef] [PubMed]
- Wischmeyer, P.E. Glutamine: Role in gut protection in critical illness. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 607–612. [Google Scholar] [CrossRef]
- Lucas, K.; Maes, M. Role of the toll like receptor (Tlr) radical cycle in chronic inflammation: Possible treat-ments targeting the Tlr4 pathway. Mol. Neurobiol. 2013, 48, 190–204. [Google Scholar] [CrossRef]
- Maes, M.; Kubera, M.; Leunis, J.C. The gut-brain barrier in major depression: Intestinal mucosal dysfunction with an increased translocation of lps from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro Endocrinol. Lett. 2008, 29, 117–124. [Google Scholar]
- Maes, M.; Mihaylova, I.; Leunis, J.C. Increased serum iga and igm against lps of enterobacteria in chronic fatigue syndrome (Cfs): Indication for the involvement of gram-negative enterobacteria in the etiology of cfs and for the presence of an increased gut-intestinal permeability. J. Affect. Disord. 2007, 99, 237–240. [Google Scholar] [CrossRef]
- Hamilton, M.K.; Boudry, G.; Lemay, D.G.; Raybould, H.E. Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G840–G851. [Google Scholar] [CrossRef]
- deMelo, L.G.P.; Nunes, S.O.V.; Anderson, G.; Vargas, H.O.; Barbosa, D.S.; Galecki, P.; Carvalho, A.F.; Maes, M. Shared metabolic and immune-inflammatory, oxidative and nitrosative stress pathways in the metabolic syndrome and mood disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 78, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Keller, A.J.; Salbaum, J.M.; Luo, M.; Blanchard, E.; Taylor, C.M.; Welsh, D.A.; Berthoud, H.R. Obese-type gut microbiota induce neurobehavioral changes in the absence of obesity. Biol. Psychiatry 2015, 77, 607–615. [Google Scholar] [CrossRef]
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, saboteur, or something else? Science 2013, 339, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Kingwell, K. Neurodegenerative disease: Microglia in early disease stages. Nat. Rev. Neurol. 2012, 8, 475. [Google Scholar] [CrossRef]
- Vaughn, A.C.; Cooper, E.M.; DiLorenzo, P.M.; O’Loughlin, L.J.; Konkel, M.E.; Peters, J.H.; Hajnal, A.; Sen, T.; Lee, S.H.; de La Serre, C.B.; et al. Energy-dense diet triggers changes in gut microbiota, reorganization of gut-brain vagal communication and increases body fat accumulation. Acta Neurobiol. Exp. 2017, 77, 18–30. [Google Scholar] [CrossRef]
- Amedei, A.; Boem, F. I’ve Gut A Feeling: Microbiota Impacting the Conceptual and Experimental Perspectives of Personalized Medicine. Int. J. Mol. Sci. 2018, 19, 3756. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study | Sample Size | Patients’ Features | Findings | Ref. |
|---|---|---|---|---|
| Central nervous system disorders | ||||
| Holtkamp et al., 2004 | 97 men | Children with attention-deficit/hyperactivity disorder (ADHD) | Obesity development independent of ADHD diagnosis | [69] |
| Elias et al., 2005 | 551 men, 872 women | Individuals with healthy body weight, overweight, obese | In men, obesity association with adverse cognitive effects | [59] |
| Cournot et al., 2006 | 1660 men, 1576 women | Healthy workers (32–62 years old) | Higher BMI association with lower cognitive scoresand higher cognitive decline | [60] |
| Boeka et al., 2008 | 20 men, 48 women | Caucasian and African American extremely obese patients | Evidence of specific cognitive dysfunction in extremely obese individuals | [73] |
| Sabia et al., 2009 | 3788 men, 1343 women | White individuals | Multiple effects of obesity on cognition | [61] |
| Hassing et al., 2010 | 140 men, 277 women | Swedish twin registry | Midlife overweight association to lower overall cognitive function in old age | [62] |
| Anstey et al., 2011 | 71529 individuals | Participants evaluated for any type of dementia | Overweight and obesity in midlife increase dementia risk | [64] |
| Dahl et al., 2013 | 280 men, 377 women | Swedish adoption and twin study of ageing | Midlife overweight or obesity responsible of lower cognitive function and cognitive decline in late life | [63] |
| Yau et al., 2014 | 30 obese, 30 lean adolescents | Obese without insulin resistance or metabolic syndrome | Uncomplicated obesity may result in subtle brain alterations | [72] |
| Cheke et al., 2016 | 14 men, 36 women | 8 Obese individuals, 16 overweight, 26 lean | Higher BMI association with lower performance on the what-where-when memory task | [66] |
| Navas et al., 2016 | 35 men, 44 women | 38 Normal weight, 21 overweight, 20 obese | Obesity is linked to a propensity to make risky decisions | [67] |
| Kummer et al., 2016 | 92 patients and 19 controls | Children and adolescents: autism spectrum disorder (ASD) andADHD | Higher risk of overweight and obesity in ASD and ADHD | [70] |
| Peripheral nervous system diseases | ||||
| Ylitalo et al., 2011 | 2514 adults aged ≥ 40 years | Individuals with peripheral neuropathy,peripheral vascular disease (PVD), alower-extremity diseases (LEDs). | Obesity and cardiometabolic clustering markedly increased the likelihood of LEDs | [92] |
| Tesfaye et al., 2005 | 1172 patients | Patients with type 1 diabetes mellitus. | Higher BMI independently associated with the incidence of neuropathy. | [93] |
| Ziegler et al., 2008 | 195 patients and 198 controls | Population-based MONICA/KORA Augsburg Surveys aged 25–74 years. | Waist circumference association with peripheral arterial disease (PAD) | [94] |
| Singleton et al., 2014 | 21 obese, 51 lean controls | Non-diabetic obese patients referred for Roux en Y bariatric surgery compared with lean controls. | Asymptomatic neuropathy is common in very obese patients independent of glucose control | [95] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niccolai, E.; Boem, F.; Russo, E.; Amedei, A. The Gut–Brain Axis in the Neuropsychological Disease Model of Obesity: A Classical Movie Revised by the Emerging Director “Microbiome”. Nutrients 2019, 11, 156. https://doi.org/10.3390/nu11010156
Niccolai E, Boem F, Russo E, Amedei A. The Gut–Brain Axis in the Neuropsychological Disease Model of Obesity: A Classical Movie Revised by the Emerging Director “Microbiome”. Nutrients. 2019; 11(1):156. https://doi.org/10.3390/nu11010156
Chicago/Turabian StyleNiccolai, Elena, Federico Boem, Edda Russo, and Amedeo Amedei. 2019. "The Gut–Brain Axis in the Neuropsychological Disease Model of Obesity: A Classical Movie Revised by the Emerging Director “Microbiome”" Nutrients 11, no. 1: 156. https://doi.org/10.3390/nu11010156
APA StyleNiccolai, E., Boem, F., Russo, E., & Amedei, A. (2019). The Gut–Brain Axis in the Neuropsychological Disease Model of Obesity: A Classical Movie Revised by the Emerging Director “Microbiome”. Nutrients, 11(1), 156. https://doi.org/10.3390/nu11010156