Tributyrin in Inflammation: Does White Adipose Tissue Affect Colorectal Cancer?

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Procedures
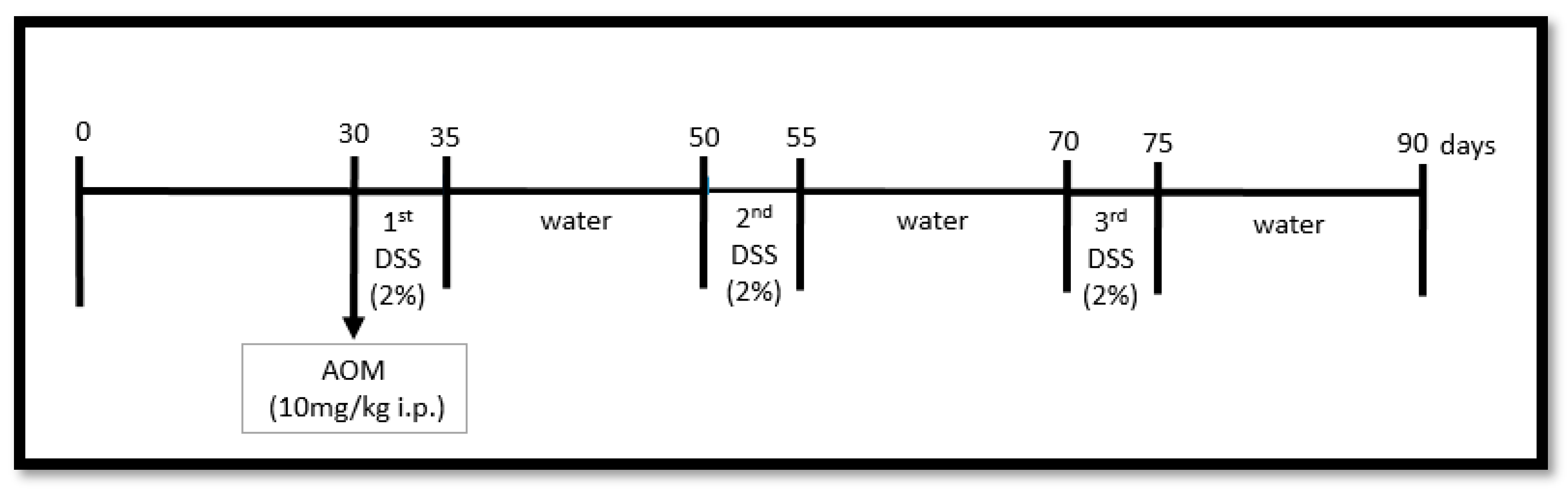
2.3. Colon Carcinogenesis Induction (AOM/DSS)
2.4. Lipid and Glucose Levels
2.5. Glucose Test Tolerance
2.6. Enzyme Linked Immunosorbent Assay (ELISA)
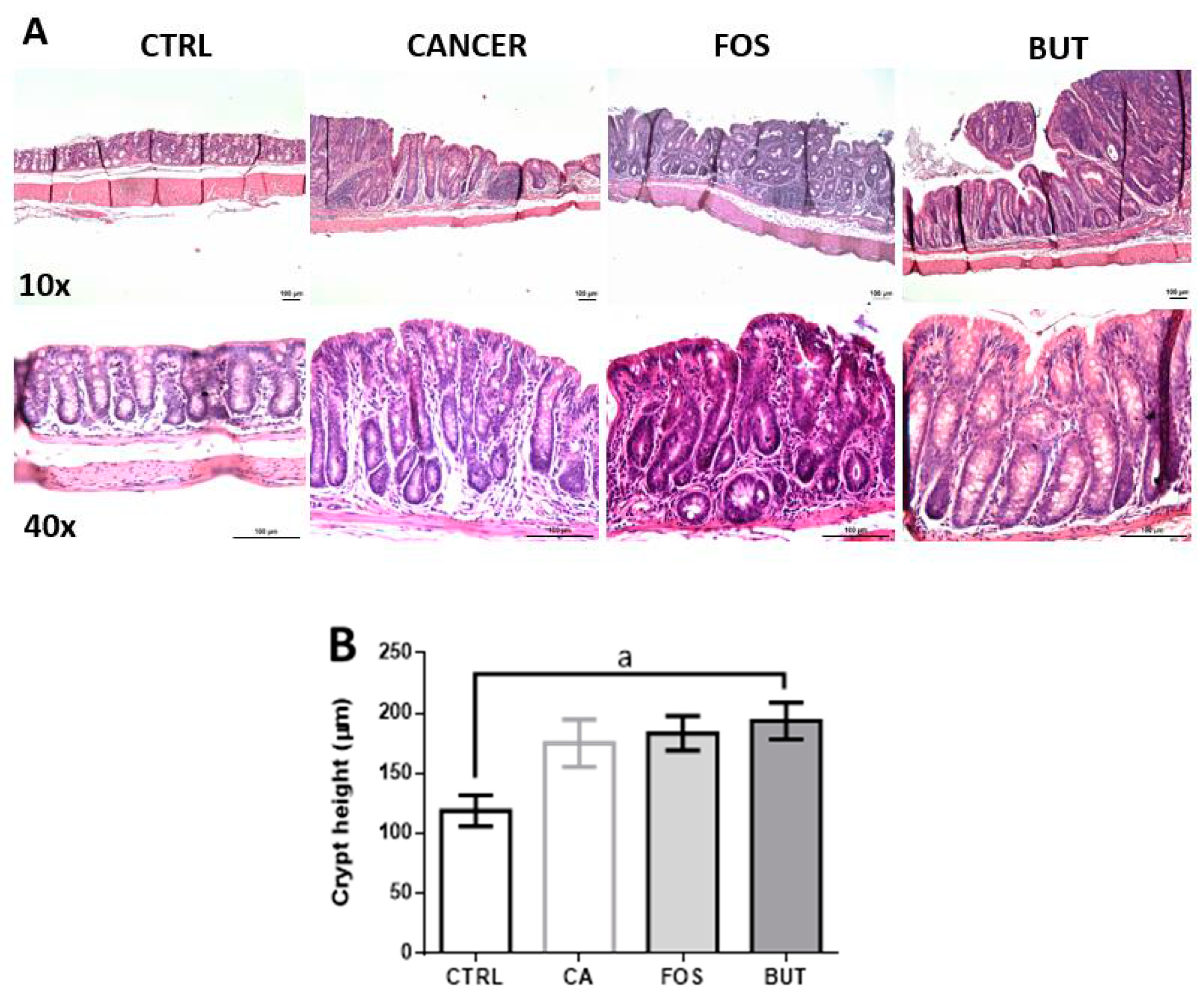
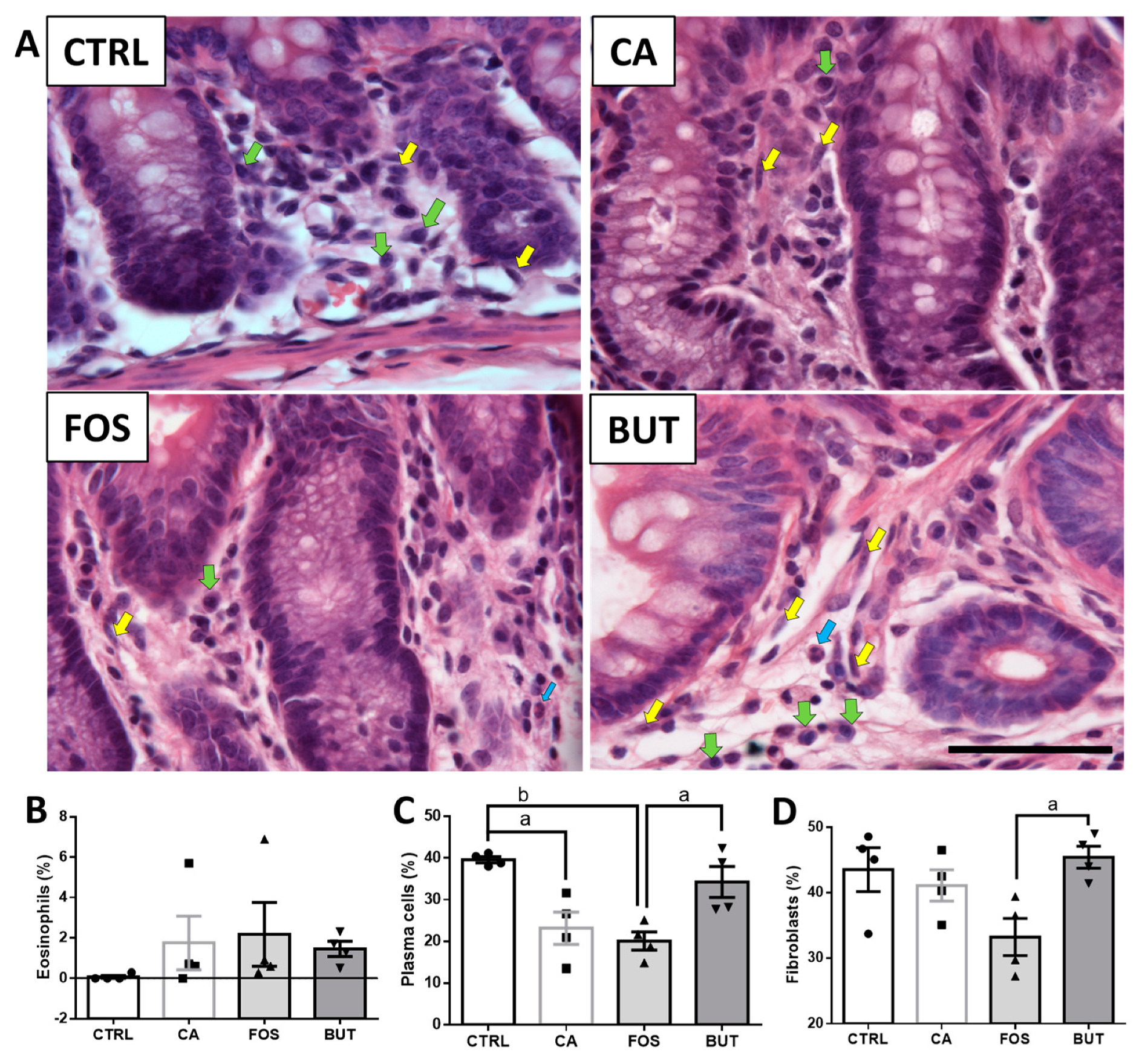
2.7. Colon Histology
2.8. Statistical Analysis
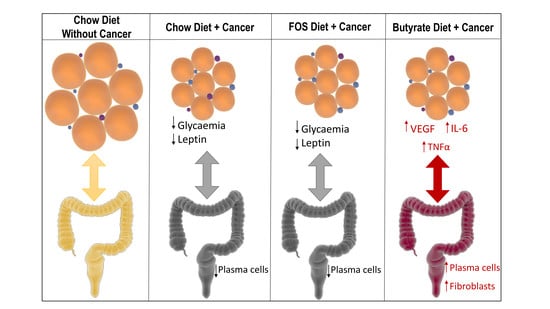
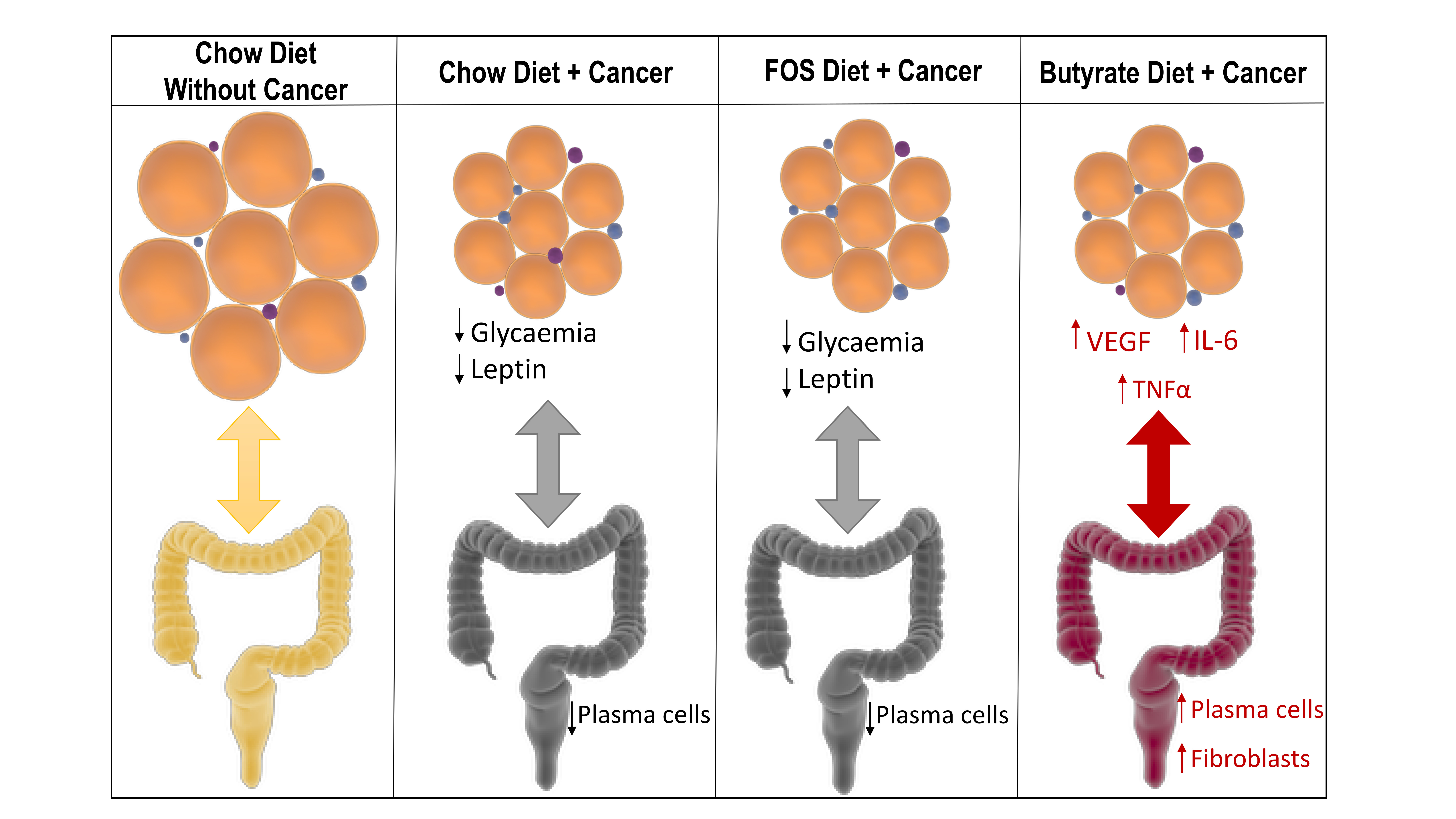
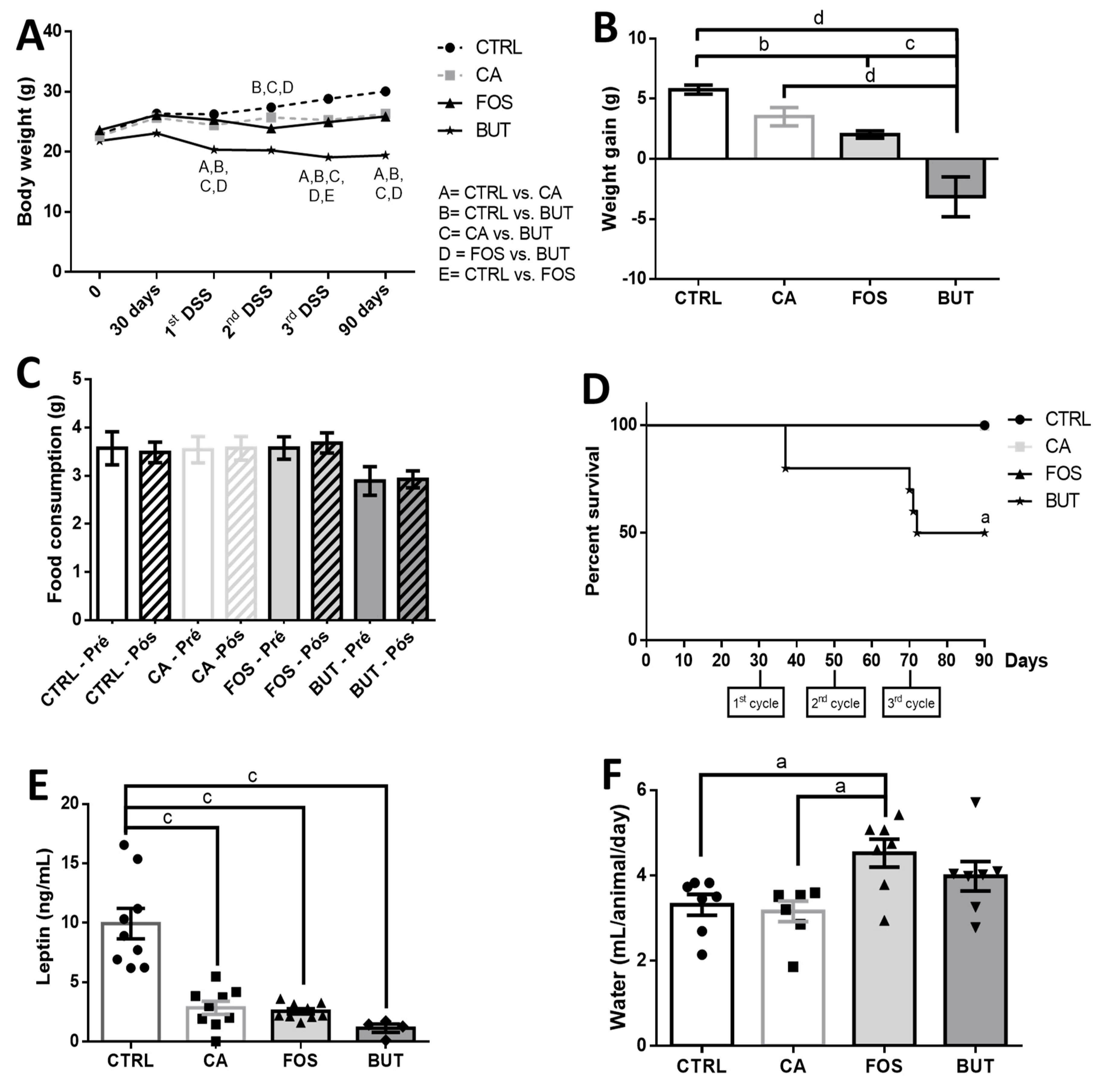
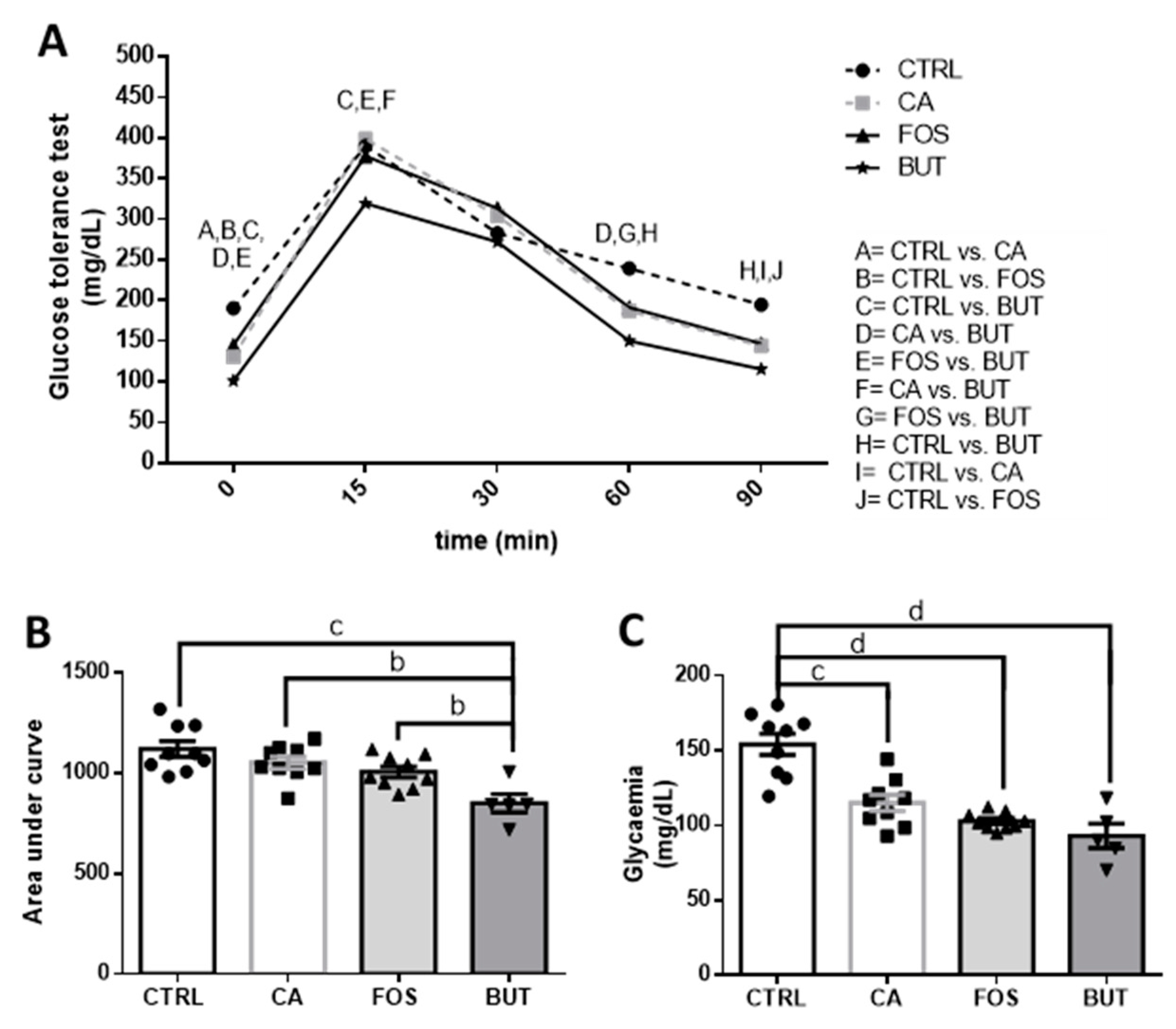
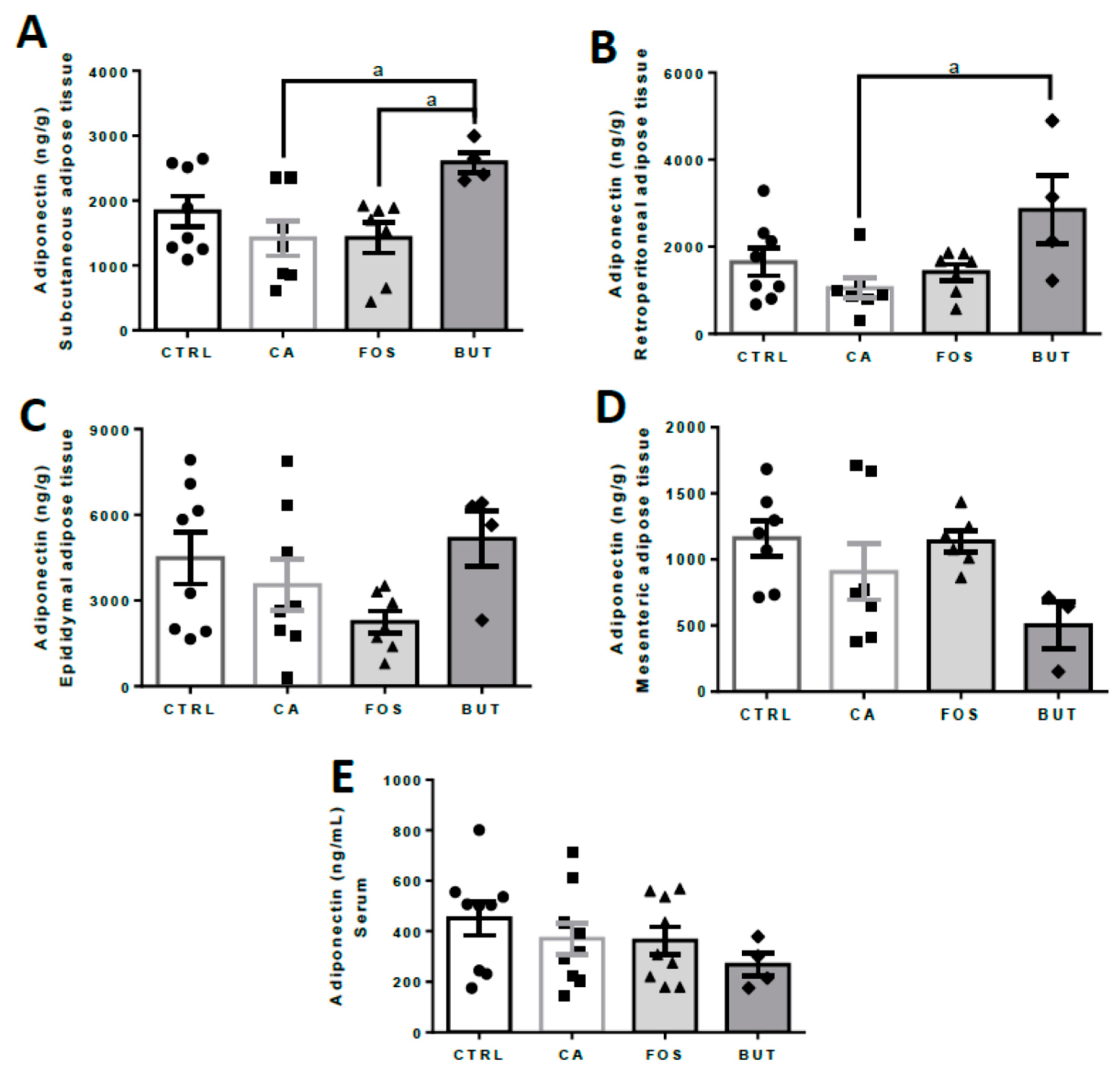
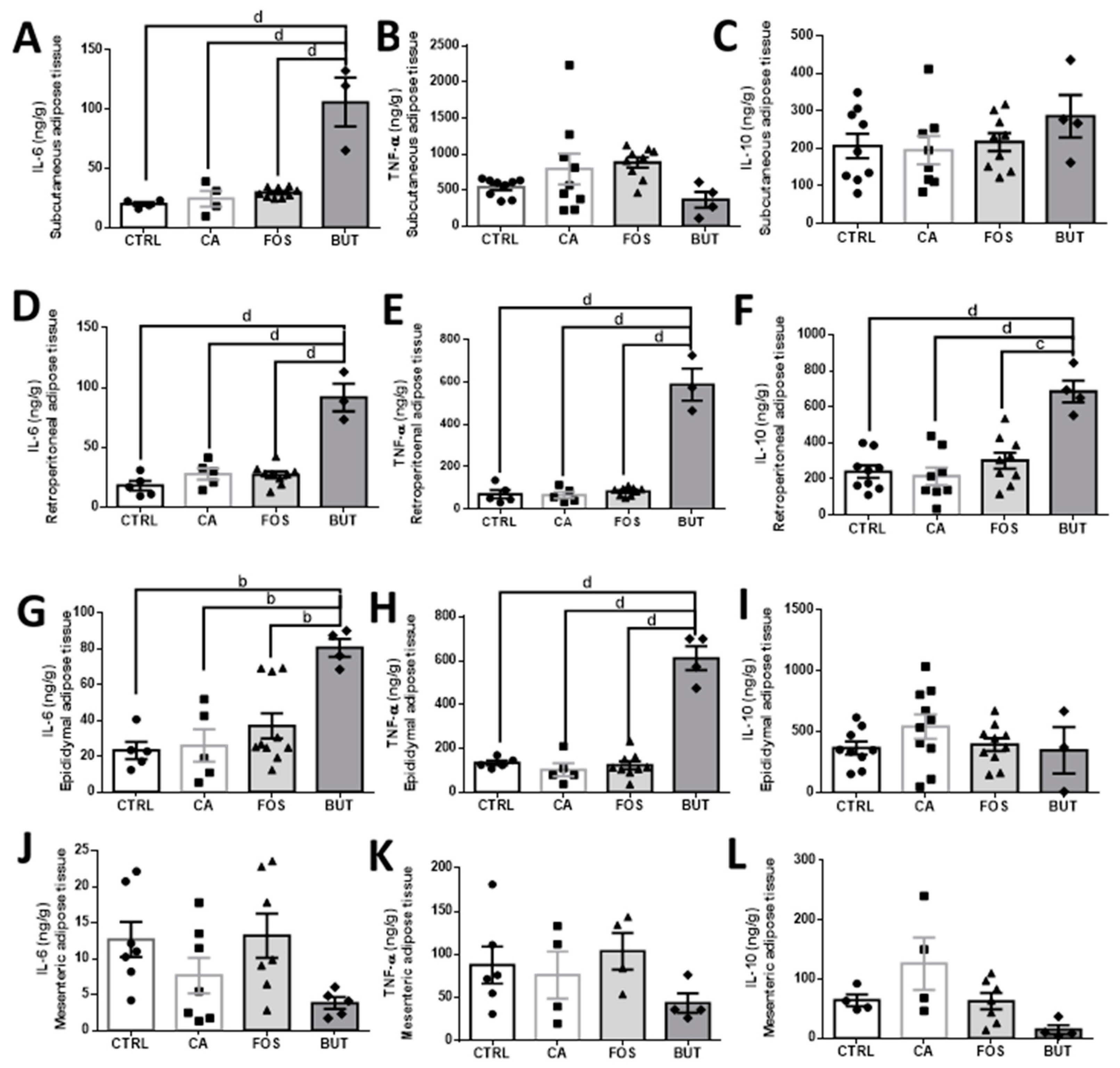
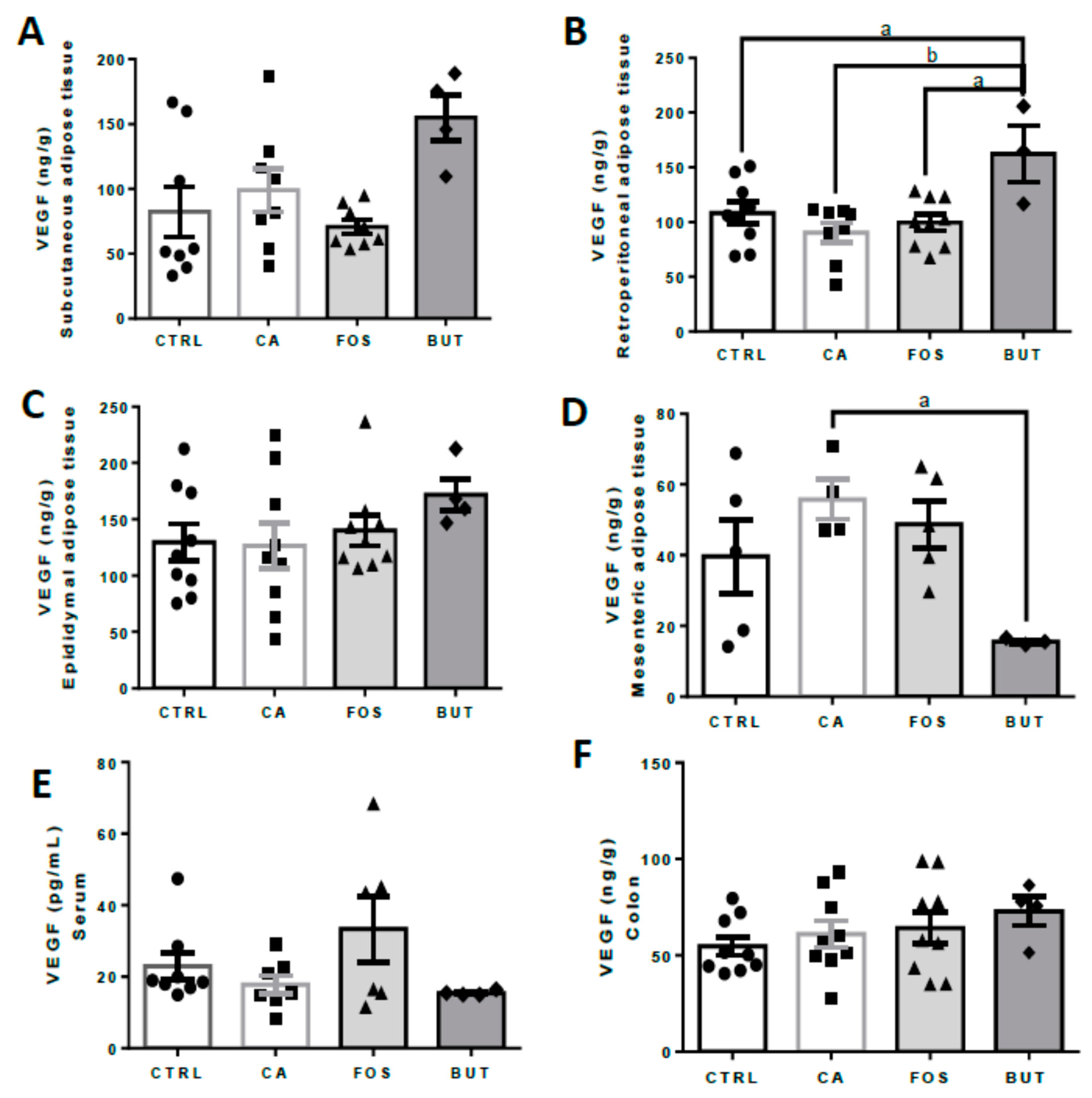
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ulrich, C.M.; Himbert, C.; Holowatyj, A.N.; Hursting, S.D. Energy balance and gastrointestinal cancer: risk, interventions, outcomes and mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 683–698. [Google Scholar] [CrossRef]
- Birt, D.F.; Phillips, G.J. Diet, genes, and microbes: Complexities of colon cancer prevention. Toxicol. Pathol. 2014, 42, 182–188. [Google Scholar] [CrossRef]
- Lofano, K.; Principi, M.; Scavo, M.P.; Pricci, M.; Ierardi, E.; Di, A.L. Dietary lifestyle and colorectal cancer onset, recurrence, and survival: Myth or reality? J. Gastrointest. Cancer 2013, 44, 1–11. [Google Scholar] [CrossRef]
- Murphy, N.; Norat, T.; Ferrari, P.; Jenab, M.; Bueno-de-Mesquita, B.; Skeie, G.; Dahm, C.C.; Overvad, K.; Olsen, A.; Tjønneland, A.; et al. Dietary fibre intake and risks of cancers of the colon and rectum in the European prospective investigation into cancer and nutrition (EPIC). PLoS ONE 2012, 7, e39361. [Google Scholar] [CrossRef]
- Aune, D.; Chan, D.S.M.; Lau, R.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Dietary fibre, whole grains, and risk of colorectal cancer: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2011, 343, d6617. [Google Scholar] [CrossRef]
- Vulcan, A.; Brändstedt, J.; Manjer, J.; Jirström, K.; Ohlsson, B.; Ericson, U. Fibre intake and incident colorectal cancer depending on fibre source, sex, tumour location and Tumour, Node, Metastasis stage. Br. J. Nutr. 2015, 114, 959–969. [Google Scholar] [CrossRef]
- Caetano, B.F.R.; de Moura, N.A.; Almeida, A.P.S.; Dias, M.C.; Sivieri, K.; Barbisan, L.F. Yacon (Smallanthus sonchifolius) as a Food Supplement: Health-Promoting Benefits of Fructooligosaccharides. Nutrients 2016, 8, 436. [Google Scholar] [CrossRef]
- Goto, H.; Takemura, N.; Ogasawara, T.; Sasajima, N.; Watanabe, J.; Ito, H.; Morita, T.; Sonoyama, K. Effects of fructo-oligosaccharide on DSS-induced colitis differ in mice fed nonpurified and purified diets. J. Nutr. 2010, 140, 2121–2127. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Ryu, S.H.; Koues, O.I.; Collins, P.L.; Solnica-Krezel, L.; Pearce, E.J.; Pearce, E.L.; Oltz, E.M.; Stappenbeck, T.S. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 2016, 165, 1708–1720. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Encarnação, J.C.; Abrantes, A.M.; Pires, A.S.; Botelho, M.F. Revisit dietary fiber on colorectal cancer: Butyrate and its role on prevention and treatment. Cancer Metastasis Rev. 2015, 34, 465–478. [Google Scholar] [CrossRef]
- Pierre, F.; Perrin, P.; Bassonga, E.; Bornet, F.; Meflah, K.; Menanteau, J. T cell status influences colon tumor occurrence in min mice fed short chain fructo-oligosaccharides as a diet supplement. Carcinogenesis 1999, 20, 1953–1956. [Google Scholar] [CrossRef]
- Conley, B.A.; Egorin, M.J.; Tait, N.; Rosen, D.M.; Sausville, E.A.; Dover, G.; Fram, R.J.; Van Echo, D.A. Phase I study of the orally administered butyrate prodrug, tributyrin, in patients with solid tumors. Clin. Cancer Res. 1998, 4, 629–634. [Google Scholar]
- Clarke, K.O.; Feinman, R.; Harrison, L.E. Tributyrin, an oral butyrate analogue, induces apoptosis through the activation of caspase-3. Cancer Lett. 2001, 171, 57–65. [Google Scholar] [CrossRef]
- Perrin, P.; Pierre, F.; Patry, Y.; Champ, M.; Berreur, M.; Pradal, G.; Bornet, F.; Meflah, K.; Menanteau, J. Only fibres promoting a stable butyrate producing colonic ecosystem decrease the rate of aberrant crypt foci in rats. Gut 2001, 48, 53–61. [Google Scholar] [CrossRef]
- Coburn, L.A.; Singh, K.; Asim, M.; Barry, D.P.; Allaman, M.M.; Al-Greene, N.T.; Hardbower, D.M.; Polosukhina, D.; Williams, C.S.; Delgado, A.G.; et al. Loss of solute carrier family 7 member 2 exacerbates inflammation-associated colon tumorigenesis. Oncogene 2018. [Google Scholar] [CrossRef]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef]
- Herrinton, L.J.; Liu, L.; Levin, T.R.; Allison, J.E.; Lewis, J.D.; Velayos, F. Incidence and mortality of colorectal adenocarcinoma in persons with inflammatory bowel disease from 1998 to 2010. Gastroenterology 2012, 143, 382–389. [Google Scholar] [CrossRef]
- Ebadi, M.; Mazurak, V.C. Evidence and mechanisms of fat depletion in cancer. Nutrients 2014, 6, 5280–5297. [Google Scholar] [CrossRef]
- Wolf, I.; Sadetzki, S.; Kanety, H.; Kundel, Y.; Pariente, C.; Epstein, N.; Oberman, B.; Catane, R.; Kaufman, B.; Shimon, I. Adiponectin, ghrelin, and leptin in cancer cachexia in breast and colon cancer patients. Cancer 2006, 106, 966–973. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef]
- Copeland, W.C.; Wachsman, J.T.; Johnson, F.M.; Penta, J.S. Mitochondrial DNA alterations in cancer. Cancer Investig. 2002, 20, 557–569. [Google Scholar] [CrossRef]
- Kannen, V.; Moreira, M.C.S.; Waaga-Gasser, A.M.; Modiano, P.; Elias Junior, J.; Fernandes, C.R.; Garcia, S.B. Partial lipectomy reduces dimethylhydrazine-induced carcinogenic initiation in the colon of rats. Toxicology 2014, 316, 9–13. [Google Scholar] [CrossRef]
- Enos, R.T.; Velázquez, K.T.; McClellan, J.L.; Cranford, T.L.; Nagarkatti, M.; Nagarkatti, P.S.; Davis, J.M.; Murphy, E.A. High-fat diets rich in saturated fat protect against azoxymethane/dextran sulfate sodium-induced colon cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G906–G919. [Google Scholar] [CrossRef]
- Boddicker, R.L.; Whitley, E.; Birt, D.F.; Spurlock, M.E. Early lesion formation in colorectal carcinogenesis is associated with adiponectin status whereas neoplastic lesions are associated with diet and sex in C57BL/6J mice. Nutr. Cancer 2011, 63, 1297–1306. [Google Scholar] [CrossRef]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar]
- Laviano, A.; Inui, A.; Marks, D.L.; Meguid, M.M.; Pichard, C.; Rossi Fanelli, F.; Seelaender, M. Neural control of the anorexia-cachexia syndrome. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1000–E1008. [Google Scholar] [CrossRef]
- Hu, Z.; Cha, S.H.; Chohnan, S.; Lane, M.D. Hypothalamic malonyl-CoA as a mediator of feeding behavior. Proc. Natl. Acad. Sci. USA 2003, 100, 12624–12629. [Google Scholar] [CrossRef]
- Endo, H.; Hosono, K.; Uchiyama, T.; Sakai, E.; Sugiyama, M.; Takahashi, H.; Nakajima, N.; Wada, K.; Takeda, K.; Nakagama, H.; et al. Leptin acts as a growth factor for colorectal tumours at stages subsequent to tumour initiation in murine colon carcinogenesis. Gut 2011, 60, 1363–1371. [Google Scholar] [CrossRef]
- Chang, L.; Villacorta, L.; Li, R.; Hamblin, M.; Xu, W.; Dou, C.; Zhang, J.; Wu, J.; Zeng, R.; Chen, Y.E. Loss of perivascular adipose tissue on peroxisome proliferator-activated receptor-γ deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation 2012, 126, 1067–1078. [Google Scholar] [CrossRef]
- Kliewer, K.L.; Ke, J.-Y.; Tian, M.; Cole, R.M.; Andridge, R.R.; Belury, M.A. Adipose tissue lipolysis and energy metabolism in early cancer cachexia in mice. Cancer Biol. Ther. 2015, 16, 886–897. [Google Scholar] [CrossRef]
- Bultman, S.J. Molecular pathways: Gene-environment interactions regulating dietary fiber induction of proliferation and apoptosis via butyrate for cancer prevention. Clin. Cancer Res. 2014, 20, 799–803. [Google Scholar] [CrossRef]
- Feng, W.; Cui, G.; Tang, C.-W.; Zhang, X.-L.; Dai, C.; Xu, Y.-Q.; Gong, H.; Xue, T.; Guo, H.-H.; Bao, Y. Role of glucose metabolism related gene GLUT1 in the occurrence and prognosis of colorectal cancer. Oncotarget 2017, 8, 56850–56857. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, E.C.; da Silva, J.F.; Navia-Pelaez, J.M.; Leonel, A.J.; Lopes, L.G.; Menezes-Garcia, Z.; Ferreira, A.V.M.; Capettini, L.D.S.A.; Teixeira, L.G.; Lemos, V.S.; et al. Sodium butyrate modulates adipocyte expansion, adipogenesis, and insulin receptor signaling by upregulation of PPAR-γ in obese Apo E knockout mice. Nutrition 2018, 47, 75–82. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef]
- Shehzad, A.; Iqbal, W.; Shehzad, O.; Lee, Y.S. Adiponectin: Regulation of its production and its role in human diseases. Hormones 2012, 11, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Ealey, K.N.; Archer, M.C. Elevated circulating adiponectin and elevated insulin sensitivity in adiponectin transgenic mice are not associated with reduced susceptibility to colon carcinogenesis. Int. J. Cancer 2009, 124, 2226–2230. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Baliga, M.S.; Ponemone, V.; Kaur, K.; Larsen, B.; Fletcher, E.; Greene, J.; Fayad, R. Mucus and adiponectin deficiency: Role in chronic inflammation-induced colon cancer. Int. J. Colorectal Dis. 2013, 28, 1267–1279. [Google Scholar] [CrossRef]
- Perše, M.; Cerar, A. The dimethylhydrazine induced colorectal tumours in rat- experimental colorectal carcinogenesis. Radiol. Oncol. 2005, 39, 61–70. [Google Scholar]
- Gálfi, P.; Bokori, J. Feeding trial in pigs with a diet containing sodium n-butyrate. Acta Vet. Hung. 1990, 38, 3–17. [Google Scholar]
- Kim, M.; Qie, Y.; Park, J.; Kim, C.H. Gut Microbial Metabolites Fuel Host Antibody Responses. Cell Host Microbe 2016, 20, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Muir, J.G.; Gibson, P.R. Does butyrate protect from colorectal cancer? J. Gastroenterol. Hepatol. 2006, 21, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, A.; Hayashi, M.; Andharia, N.; Matsuda, H. Involvement of butyrate in electrogenic K+ secretion in rat rectal colon. Pflug. Arch. 2018. [Google Scholar] [CrossRef] [PubMed]
- Graboń, W.; Otto-Ślusarczyk, D.; Chrzanowska, A.; Mielczarek-Puta, M.; Joniec-Maciejak, I.; Słabik, K.; Barańczyk-Kuźma, A. Lactate Formation in Primary and Metastatic Colon Cancer Cells at Hypoxia and Normoxia. Cell Biochem. Funct. 2016, 34, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Jahnel, J.; Fickert, P.; Hauer, A.C.; Högenauer, C.; Avian, A.; Trauner, M. Inflammatory bowel disease alters intestinal bile acid transporter expression. Drug Metab. Dispos. 2014, 42, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Gassler, N.; Rohr, C.; Schneider, A.; Kartenbeck, J.; Bach, A.; Obermüller, N.; Otto, H.F.; Autschbach, F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am. J. Physiol. 2001, 281, G216–G228. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Alli, R.; Vogel, P.; Geiger, T.L. IL-10 modulates DSS-induced colitis through a macrophage- ROS—NO axis. Mucosal Immunol. 2014, 7, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Wehling-Henricks, M.; Villalta, S.A.; Wang, Y.; Tidball, J.G. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol. 2012, 189, 3669–3680. [Google Scholar] [CrossRef]
- Rossowska, J.; Anger, N.; Szczygieł, A.; Mierzejewska, J.; Pajtasz-Piasecka, E. Reprogramming the murine colon cancer microenvironment using lentivectors encoding shRNA against IL-10 as a component of a potent DC-based chemoimmunotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 126. [Google Scholar] [CrossRef]
- Jo, H.; Eom, Y.W.; Kim, H.-S.; Park, H.J.; Kim, H.M.; Cho, M.-Y. Regulatory Dendritic Cells Induced by Mesenchymal Stem Cells Ameliorate Dextran Sodium Sulfate-Induced Chronic Colitis in Mice. Gut Liver 2018, 12, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; Busquets, S.; López-Soriano, F.J. Cytokines in the pathogenesis of cancer cachexia. Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Deschner, E.E.; Ruperto, J.F.; Lupton, J.R.; Newmark, H.L. Dietary butyrate (tributyrin) does not enhance AOM-induced colon tumorigenesis. Cancer Lett. 1990, 52, 79–82. [Google Scholar] [CrossRef]
- Moon, H.-S.; Liu, X.; Nagel, J.M.; Chamberland, J.P.; Diakopoulos, K.N.; Brinkoetter, M.T.; Hatziapostolou, M.; Wu, Y.; Robson, S.C.; Iliopoulos, D.; et al. Salutary effects of adiponectin on colon cancer: In vivo and in vitro studies in mice. Gut 2013, 62, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Cascio, S.; Ferla, R.; D’Andrea, A.; Gerbino, A.; Bazan, V.; Surmacz, E.; Russo, A. Expression of angiogenic regulators, VEGF and leptin, is regulated by the EGF/PI3K/STAT3 pathway in colorectal cancer cells. J. Cell. Physiol. 2009, 221, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Fenton, J.I.; Birmingham, J.M. Adipokine regulation of colon cancer: Adiponectin attenuates interleukin-6-induced colon carcinoma cell proliferation via STAT-3. Mol. Carcinog. 2010, 49, 700–709. [Google Scholar] [CrossRef]
- Tsoli, M.; Schweiger, M.; Vanniasinghe, A.S.; Painter, A.; Zechner, R.; Clarke, S.; Robertson, G. Depletion of White Adipose Tissue in Cancer Cachexia Syndrome Is Associated with Inflammatory Signaling and Disrupted Circadian Regulation. PLoS ONE 2014, 9, e92966. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, B.; Yehuda-Shnaidman, E. Putative Role of Adipose Tissue in Growth and Metabolism of Colon Cancer Cells. Front. Oncol. 2014, 4, 164. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Izaguirre, M.; Hernández-Lizoain, J.L.; Baixauli, J.; Martí, P.; Valentí, V.; Moncada, R.; et al. Increased Obesity-Associated Circulating Levels of the Extracellular Matrix Proteins Osteopontin, Chitinase-3 Like-1 and Tenascin C Are Associated with Colon Cancer. PLoS ONE 2016, 11, e0162189. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Ortega, V.A.; Hernández-Lizoain, J.L.; Baixauli, J.; Becerril, S.; Rotellar, F.; Valentí, V.; et al. IL-32α-induced inflammation constitutes a link between obesity and colon cancer. Oncoimmunology 2017, 6, e1328338. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Ghossein, R.A.; Morris, L.G.; Zhou, X.K.; Kochhar, A.; Morris, P.G.; Pfister, D.G.; Patel, S.G.; Boyle, J.O.; Hudis, C.A.; et al. White adipose tissue inflammation and cancer-specific survival in patients with squamous cell carcinoma of the oral tongue. Cancer 2016, 122, 3794–3802. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Products | Chow Diet (g/Kg) | Tributyrin Diet (g/Kg) | Fructooligosaccharides Diet (g/Kg) |
|---|---|---|---|
| Cornstarch | 465 | 465 | 465 |
| Dextrinized cornstarch | 155 | 155 | 155 |
| Casein | 140 | 140 | 140 |
| Sucrose | 100 | 100 | 100 |
| Soybean oil | 40 | 40 | 40 |
| Cellulose | 50 | 50 | 50 |
| Minerals mix | 35 | 35 | 35 |
| Vitamins mix | 10 | 10 | 10 |
| Choline bitartrate | 2.5 | 2.5 | 2.5 |
| L-cystine | 1.8 | 1.8 | 1.8 |
| Tributyrin | 0 | 100 | 0 |
| Fructooligosaccharides | 0 | 0 | 60 |
| Total energy (Kcal/Kg) | 3474 | 4074 | 3474 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biondo, L.A.; Teixeira, A.A.S.; Silveira, L.S.; Souza, C.O.; Costa, R.G.F.; Diniz, T.A.; Mosele, F.C.; Rosa Neto, J.C. Tributyrin in Inflammation: Does White Adipose Tissue Affect Colorectal Cancer? Nutrients 2019, 11, 110. https://doi.org/10.3390/nu11010110
Biondo LA, Teixeira AAS, Silveira LS, Souza CO, Costa RGF, Diniz TA, Mosele FC, Rosa Neto JC. Tributyrin in Inflammation: Does White Adipose Tissue Affect Colorectal Cancer? Nutrients. 2019; 11(1):110. https://doi.org/10.3390/nu11010110
Chicago/Turabian StyleBiondo, Luana Amorim, Alexandre Abilio S. Teixeira, Loreana S. Silveira, Camila O. Souza, Raquel G. F. Costa, Tiego A. Diniz, Francielle C. Mosele, and José Cesar Rosa Neto. 2019. "Tributyrin in Inflammation: Does White Adipose Tissue Affect Colorectal Cancer?" Nutrients 11, no. 1: 110. https://doi.org/10.3390/nu11010110
APA StyleBiondo, L. A., Teixeira, A. A. S., Silveira, L. S., Souza, C. O., Costa, R. G. F., Diniz, T. A., Mosele, F. C., & Rosa Neto, J. C. (2019). Tributyrin in Inflammation: Does White Adipose Tissue Affect Colorectal Cancer? Nutrients, 11(1), 110. https://doi.org/10.3390/nu11010110