Omega-3 Fatty Acids Prevent Early Pancreatic Carcinogenesis via Repression of the AKT Pathway


 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. MTT Assay
2.3. Western Blotting
2.4. Diet Studies
2.5. Histology
2.6. Immunohistochemistry
2.7. Apoptosis Assay by Annexin V-FITC and Propidium Iodide (PI) Staining
2.8. Statistical Analysis
3. Results
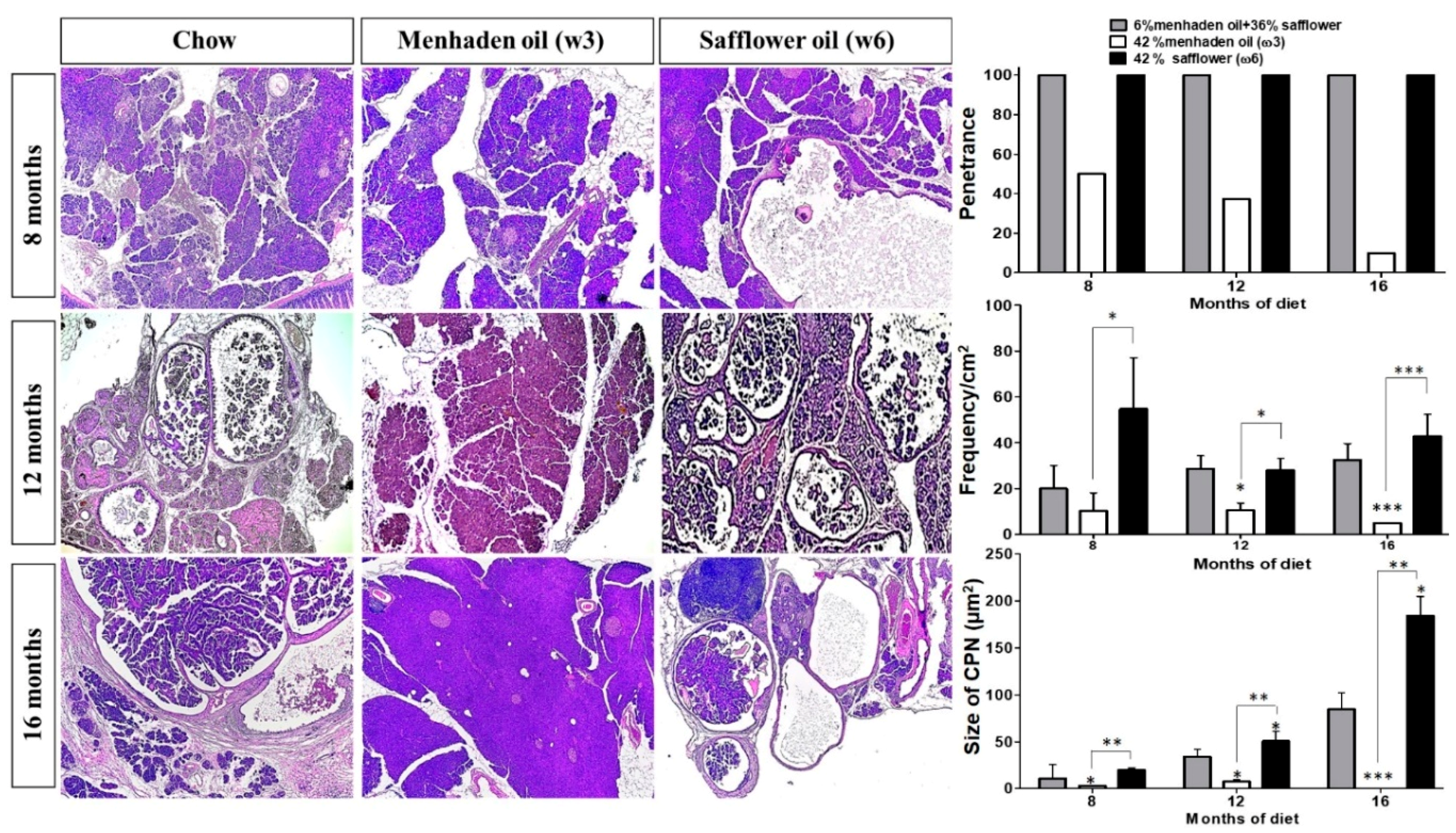
3.1. Omega-3 Enriched Diets Protect Against, Whereas ω6-Enriched Diets Accelerate Pancreatic Tumorigenesis In Vivo
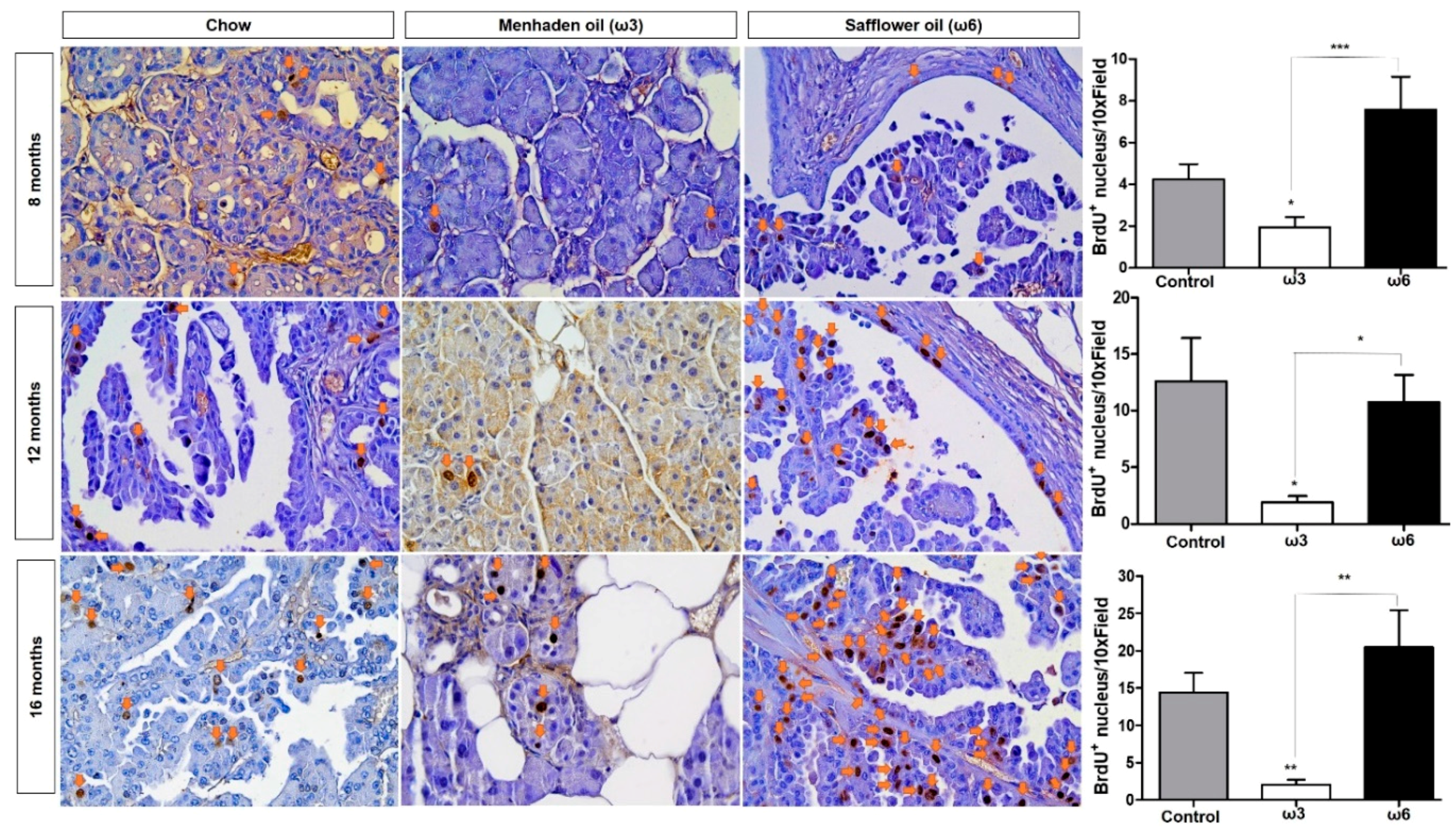
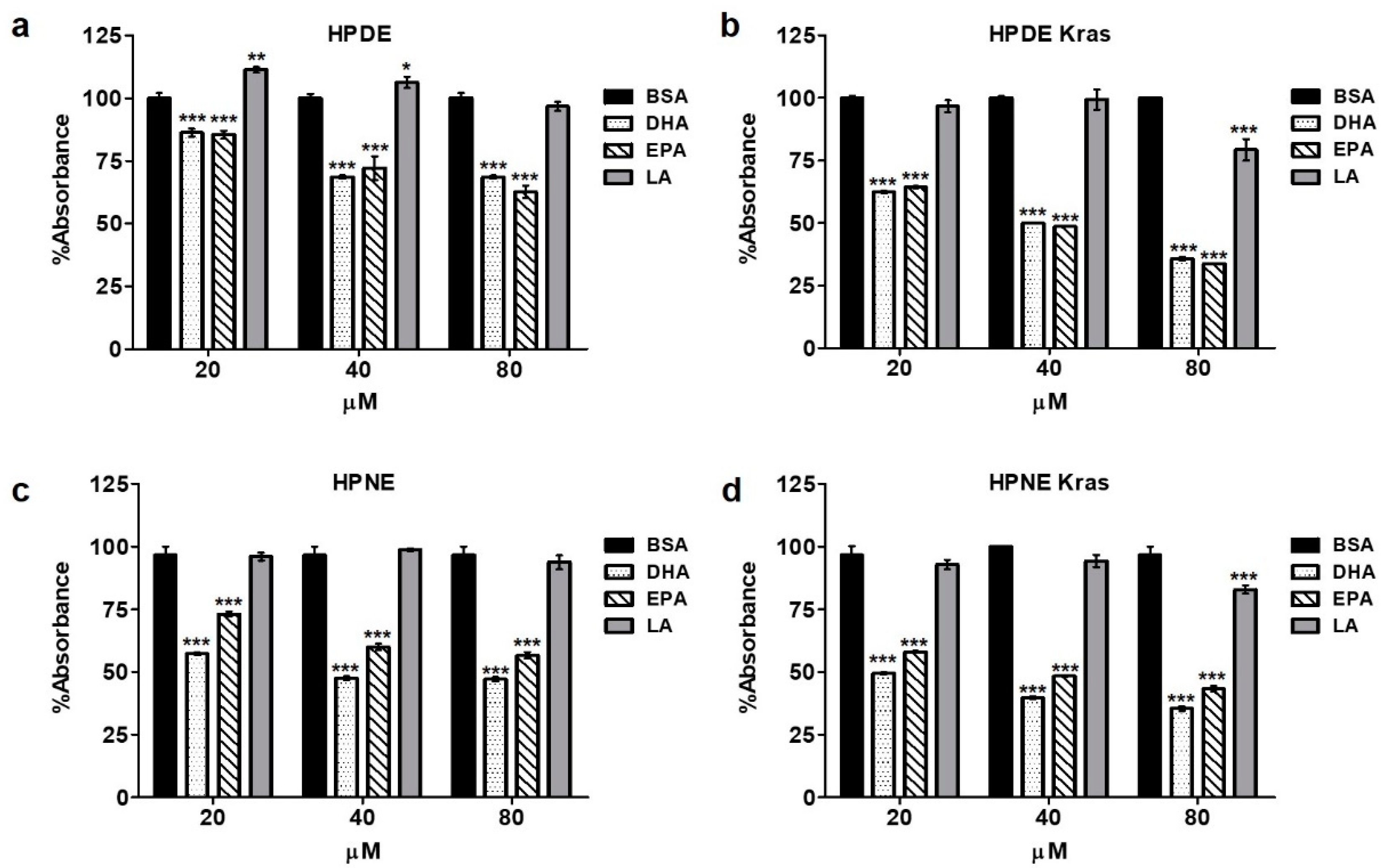
3.2. Omega-3 Fatty Acids Reduce Epithelial Cell Proliferation/Viability
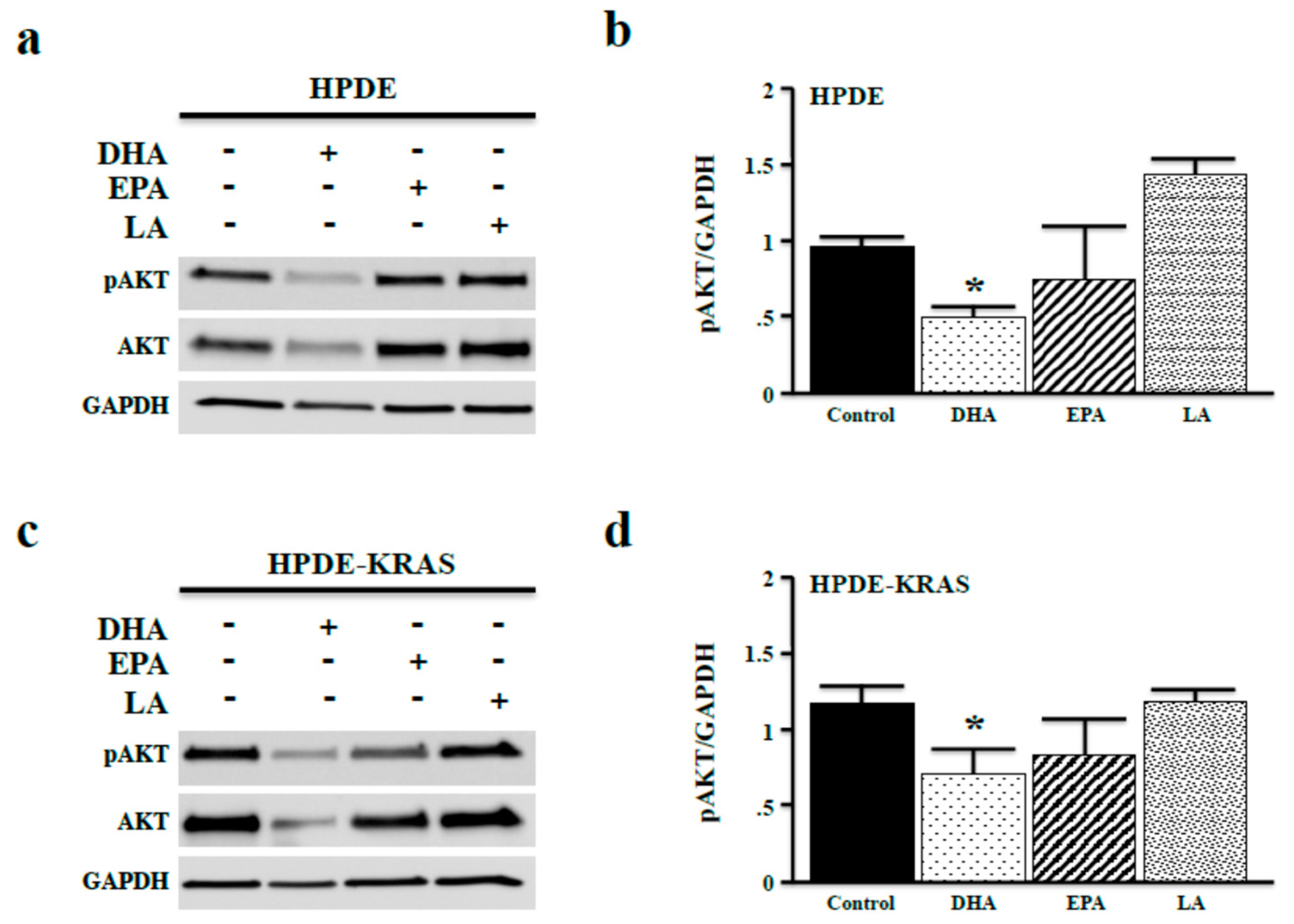
3.3. Omega-3 Fatty Acids Inhibit AKT Phosphorylation
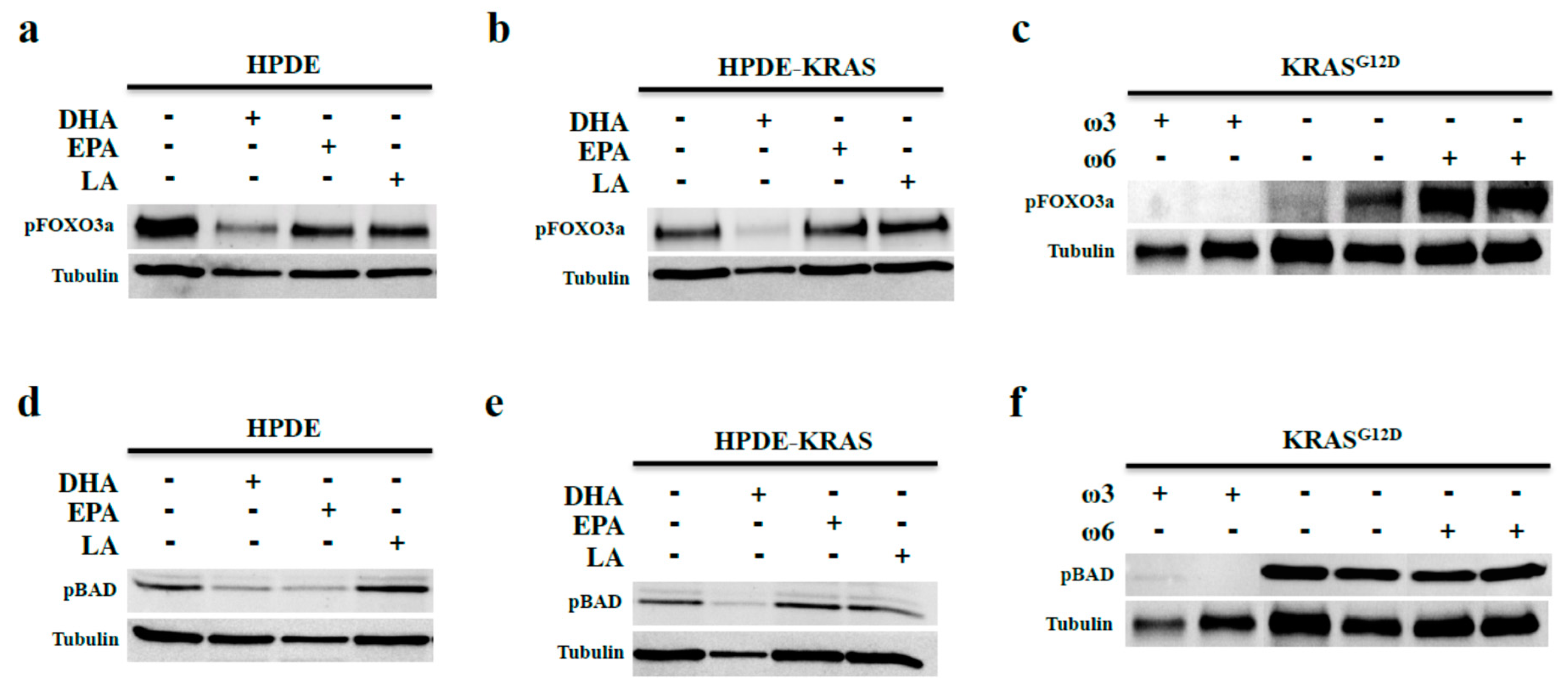
3.4. Omega-3 Fatty Acids Prevent FOX3a and BAD Phosphorylation In Vitro and In Vivo
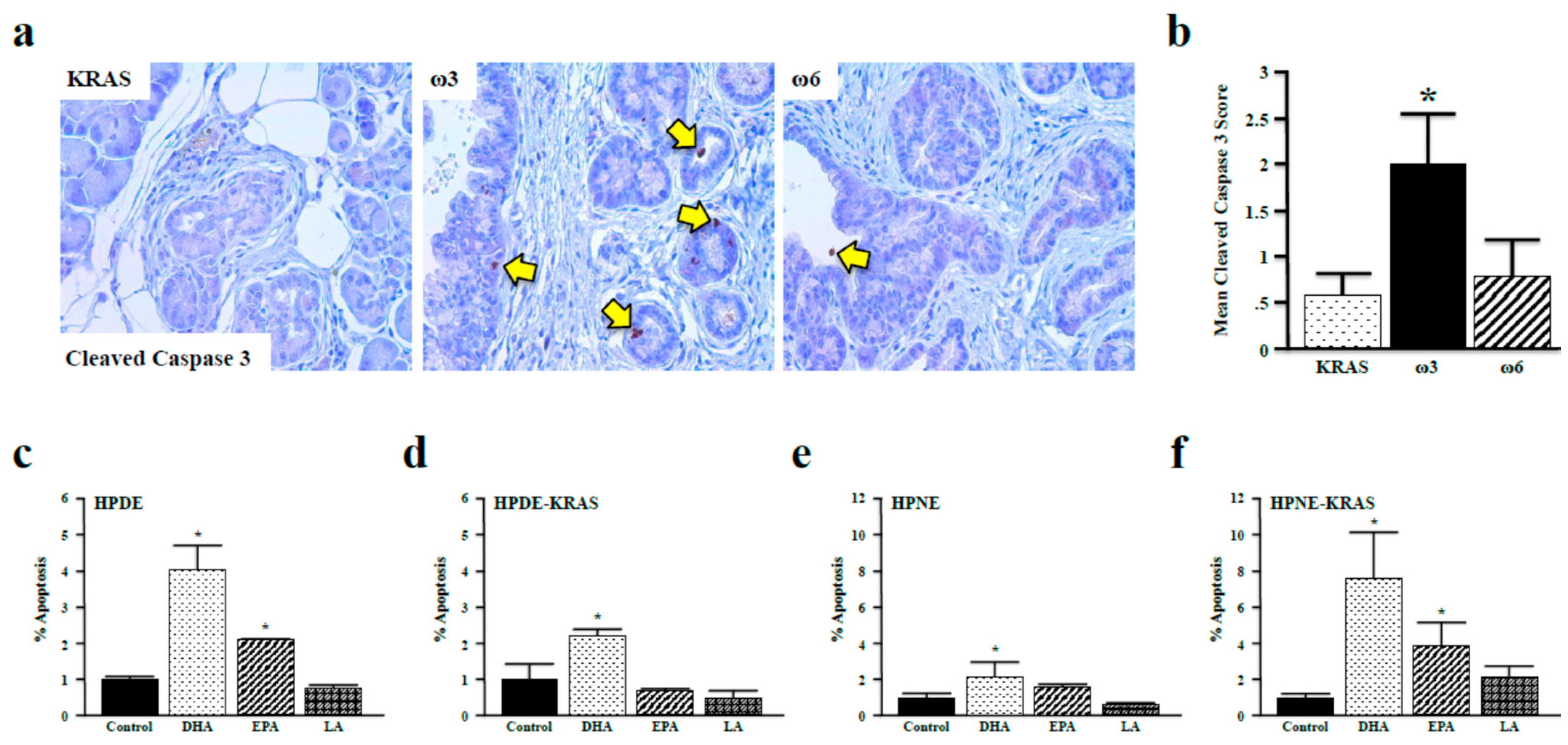
3.5. Omega-3 Fatty Acids Promote Apoptosis in Vitro and in Vivo
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Warshaw, A.L.; Castillo, C.F. Pancreatic Carcinoma. N. Engl. J. Med. 1992, 326, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Magee, C.J.; Ghaneh, P.; Neoptolemos, J.P. Surgical and medical therapy for pancreatic carcinoma. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 435–455. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, B.; Correa, A.M.; Ho, L. Survival in pancreatic carcinoma based on tumor size. Pancreas 2008, 36, e15–e20. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Boyd, L.; Walker, L.C. The fraction of cancer attributable to lifestyle and environmental factors in the UK in 2010. Br. J. Cancer 2011, 105, S77–S81. [Google Scholar] [CrossRef] [PubMed]
- World Cancer Research Fund; American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective; American Institute for Cancer Research: Washington, DC, USA, 2007; ISBN 9780972252225. [Google Scholar]
- Chan, J.M.; Gong, Z.; Holly, E.A.; Bracci, P.M. Dietary patterns and risk of pancreatic cancer in a large population-based case-control study in the San Francisco Bay Area. Nutr. Cancer 2013, 65, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Lipids and Cell Membranes. In Biochemistry; W H Freeman: New York, NY, USA, 2002; pp. 568–591. ISBN 978-0716746843. [Google Scholar]
- Torres, C.; Diaz, A.M.; Principe, D.R.; Grippo, P.J. The complexity of omega-3 fatty acid modulation of signaling pathways related to pancreatic cancer. Curr. Med. Chem. 2018, 25, 2608–2623. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Essential fatty acids: Biochemistry, physiology and pathology. Biotechnol. J. 2006, 1, 420–439. [Google Scholar] [CrossRef] [PubMed]
- Arshad, A.; Al-Leswas, D.; Stephenson, J.; Metcalfe, M.; Dennison, A. Potential applications of fish oils rich in n-3 fatty acids in the palliative treatment of advanced pancreatic cancer. Br. J. Nutr. 2011, 106, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Fay, M.P.; Freedman, L.S.; Clifford, C.K.; Midthune, D.N. Effect of different types and amounts of fat on the development of mammary tumors in rodents: A review. Cancer Res. 1997, 57, 3979–3988. [Google Scholar] [PubMed]
- Akinsete, J.A.; Ion, G.; Witte, T.R.; Hardman, W.E. Consumption of high ω-3 fatty acid diet suppressed prostate tumorigenesis in C3(1) Tag mice. Carcinogenesis 2012, 33, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Colomer, R.; Moreno-Nogueira, J.M.; García-Luna, P.P.; García-Peris, P.; García-de-Lorenzo, A.; Zarazaga, A.; Quecedo, L.; del Llano, J.; Usán, L.; Casimiro, C. N-3 fatty acids, cancer and cachexia: A systematic review of the literature. Br. J. Nutr. 2007, 97, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Dekoj, T.; Lee, S.; Desai, S.; Trevino, J.; Babcock, T.A.; Helton, W.S.; Espat, N.J. G2/M cell-cycle arrest and apoptosis by n-3 fatty acids in a pancreatic cancer model. J. Surg. Res. 2007, 139, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Merendino, N.; Molinari, R.; Loppi, B.; Pessina, G.; D’Aquino, M.; Tomassi, G.; Velotti, F. Induction of Apoptosis in Human Pancreatic Cancer Cells by Docosahexaenoic Acid. Ann. N. Y. Acad. Sci. 2003, 1010, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Shirota, T.; Haji, S.; Yamasaki, M.; Iwasaki, T.; Hidaka, T.; Takeyama, Y.; Shiozaki, H.; Ohyanagi, H. Apoptosis in human pancreatic cancer cells induced by eicosapentaenoic acid. Nutrition 2005, 21, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Hering, J.; Garrean, S.; Dekoj, T.; Razzak, A.; Saied, A.; Trevino, J.; Babcock, T.; Espat, N. Inhibition of Proliferation by Omega-3 Fatty Acids in Chemoresistant Pancreatic Cancer Cells. Ann. Surg. Oncol. 2007, 14, 3620–3628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Long, Y.; Zhang, J.; Wang, C. Modulatory effects of EPA and DHA on proliferation and apoptosis of pancreatic cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2007, 27, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Boutros, C.; Somasundar, P.; Razzak, A.; Helton, S.; Espat, N.J. Omega-3 fatty acids: Investigations from cytokine regulation to pancreatic cancer gene suppression. Arch. Surg. 2010, 145, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Lim, J.W.; Kim, H. Inhibitory mechanism of omega-3 fatty acids in pancreatic inflammation and apoptosis. Ann. N. Y. Acad. Sci. 2009, 1171, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Merendino, N.; Loppi, B.; D’Aquino, M.; Molinari, R.; Pessina, G.; Romano, C.; Velotti, F. Docosahexaenoic acid induces apoptosis in the human PaCa-44 pancreatic cancer cell line by active reduced glutathione extrusion and lipid peroxidation. Nutr. Cancer 2005, 52, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Maingay, J.P.; Fearon, K.C.H.; Sangster, K.; Powell, J.J. Eicosapentaenoic acid perturbs signalling via the NFkappaB transcriptional pathway in pancreatic tumour cells. Int. J. Oncol. 2003, 23, 1733–1738. [Google Scholar] [PubMed]
- Funahashi, H.; Satake, M.; Hasan, S.; Sawai, H.; Newman, R.A.; Reber, H.A.; Hines, O.J.; Eibl, G. Opposing effects of n-6 and n-3 polyunsaturated fatty acids on pancreatic cancer growth. Pancreas 2008, 36, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Cheon, E.C.; Strouch, M.J.; Barron, M.R.; Ding, Y.; Melstrom, L.G.; Krantz, S.B.; Mullapudi, B.; Adrian, K.; Rao, S.; Adrian, T.E.; et al. Alteration of strain background and a high omega-6 fat diet induces earlier onset of pancreatic neoplasia in EL-Kras transgenic mice. Int. J. Cancer 2011, 128, 2783–2792. [Google Scholar] [CrossRef] [PubMed]
- Grippo, P.J.; Nowlin, P.S.; Demeure, M.J.; Longnecker, D.S.; Sandgren, E.P. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003, 63, 2016–2019. [Google Scholar] [PubMed]
- Boreddy, S.R.; Pramanik, K.C.; Srivastava, S.K. Pancreatic tumor suppression by benzyl isothiocyanate is associated with inhibition of PI3K/AKT/FOXO pathway. Clin. Cancer Res. 2011, 17, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; DeCant, B.; Mascariñas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.; et al. TGFβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef] [PubMed]
- McCleary-Wheeler, A.L.; Lomberk, G.A.; Weiss, F.U.; Schneider, G.; Fabbri, M.; Poshusta, T.L.; Dusetti, N.J.; Baumgart, S.; Iovanna, J.L.; Ellenrieder, V.; et al. Insights into the epigenetic mechanisms controlling pancreatic carcinogenesis. Cancer Lett. 2013, 328, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.A.; Slingerland, J.M. Cytokines, Obesity, and Cancer: New Insights on Mechanisms Linking Obesity to Cancer Risk and Progression. Annu. Rev. Med. 2013, 64, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Hammamieh, R.; Chakraborty, N.; Miller, S.-A.; Waddy, E.; Barmada, M.; Das, R.; Peel, S.A.; Day, A.A.; Jett, M. Differential Effects of Omega-3 and Omega-6 fatty Acids on Gene Expression in Breast Cancer Cells. Breast Cancer Res. Treat. 2007, 101, 7–16. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, M.; Ma, D.W.L. Role of dietary fatty acids in mammary gland development and breast cancer. Breast Cancer Res. 2010, 12, 211. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Calder, P.C. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006, 83, 1505S–1519S. [Google Scholar] [CrossRef] [PubMed]
- Wigmore, S.J.; Fearon, K.C.; Maingay, J.P.; Ross, J.A. Down-regulation of the acute-phase response in patients with pancreatic cancer cachexia receiving oral eicosapentaenoic acid is mediated via suppression of interleukin-6. Clin. Sci. (Lond.) 1997, 92, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.D.; Ross, J.A.; Voss, A.C.; Tisdale, M.J.; Fearon, K.C. The effect of an oral nutritional supplement enriched with fish oil on weight-loss in patients with pancreatic cancer. Br. J. Cancer 1999, 81, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Zuijdgeest-Van Leeuwen, S.D.; Dagnelie, P.C.; Wattimena, J.L.; Van den Berg, J.W.; Van der Gaast, A.; Swart, G.R.; Wilson, J.H. Eicosapentaenoic acid ethyl ester supplementation in cachectic cancer patients and healthy subjects: Effects on lipolysis and lipid oxidation. Clin. Nutr. 2000, 19, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Wigmore, S.J.; Barber, M.D.; Ross, J.A.; Tisdale, M.J.; Fearon, K.C. Effect of oral eicosapentaenoic acid on weight loss in patients with pancreatic cancer. Nutr. Cancer 2000, 36, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.D.; Fearon, K.C.; Tisdale, M.J.; McMillan, D.C.; Ross, J.A. Effect of a fish oil-enriched nutritional supplement on metabolic mediators in patients with pancreatic cancer cachexia. Nutr. Cancer 2001, 40, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Persson, C.; Glimelius, B.; Rönnelid, J.; Nygren, P. Impact of fish oil and melatonin on cachexia in patients with advanced gastrointestinal cancer: A randomized pilot study. Nutrition 2005, 21, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Capra, S.; Battistutta, D.; Davidson, W.; Ash, S. Compliance with nutrition prescription improves outcomes in patients with unresectable pancreatic cancer. Clin. Nutr. 2005, 24, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Sellem, L.; Srour, B.; Guéraud, F.; Pierre, F.; Kesse-Guyot, E.; Fiolet, T.; Lavalette, C.; Egnell, M.; Latino-Martel, P.; Fassier, P.; et al. Saturated, mono- and polyunsaturated fatty acid intake and cancer risk: Results from the French prospective cohort NutriNet-Santé. Eur. J. Nutr. 2018. [Google Scholar] [CrossRef] [PubMed]
- Van Blarigan, E.L.; Fuchs, C.S.; Niedzwiecki, D.; Ye, X.; Zhang, S.; Song, M.; Saltz, L.B.; Mayer, R.J.; Mowat, R.B.; Whittom, R.; et al. Marine ω-3 Polyunsaturated Fatty Acid and Fish Intake after Colon Cancer Diagnosis and Survival: CALGB 89803 (Alliance). Cancer Epidemiol. Biomark. Prev. 2018, 27, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Fadelu, T.; Zhang, S.; Niedzwiecki, D.; Ye, X.; Saltz, L.B.; Mayer, R.J.; Mowat, R.B.; Whittom, R.; Hantel, A.; Benson, A.B.; et al. Nut consumption and survival in patients with stage III colon cancer: Results from CALGB 89803 (Alliance). J. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Lipoxins, resolvins, protectins, maresins and nitrolipids, and their clinical implications with specific reference to cancer: Part I. Clin. Lipidol. 2013, 8, 437–463. [Google Scholar] [CrossRef]
- Schley, P.D.; Jijon, H.B.; Robinson, L.E.; Field, C.J. Mechanisms of omega-3 fatty acid-induced growth inhibition in MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2005, 92, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhury, T.; Mandal, C.C.; Woodruff, K.; St Clair, P.; Fernandes, G.; Choudhury, G.G.; Ghosh-Choudhury, N. Fish oil targets PTEN to regulate NFκB for downregulation of anti-apoptotic genes in breast tumor growth. Breast Cancer Res. Treat. 2009, 118, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Covey, T.M.; Edes, K.; Fitzpatrick, F.A. Akt activation by arachidonic acid metabolism occurs via oxidation and inactivation of PTEN tumor suppressor. Oncogene 2007, 26, 5784–5792. [Google Scholar] [CrossRef] [PubMed]
- Miglio, U.; Oldani, A.; Mezzapelle, R.; Veggiani, C.; Paganotti, A.; Garavoglia, M.; Boldorini, R. KRAS mutational analysis in ductal adenocarcinoma of the pancreas and its clinical significance. Pathol. Res. Pract. 2014. [Google Scholar] [CrossRef] [PubMed]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, W.; Ruparel, S.B.; Marciniak, R.A.; Degraffenried, L. Omega-3 fatty acid inhibition of prostate cancer progression to hormone independence is associated with suppression of mTOR signaling and androgen receptor expression. Nutr. Cancer 2011, 63, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Brewer, M.; Duff, A.; Lightfoot, S.; Brush, R.S.; Anderson, R.E.; Rao, C.V. Endogenous n-3 polyunsaturated fatty acids delay progression of pancreatic ductal adenocarcinoma in Fat-1-p48(Cre/+)-LSL-Kras(G12D/+) mice. Neoplasia 2012, 14, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Seti, H.; Leikin-Frenkel, A.; Werner, H. Effects of omega-3 and omega-6 fatty acids on IGF-I receptor signalling in colorectal cancer cells. Arch. Physiol. Biochem. 2009, 115, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.A.; Mourtzakis, M.; Chu, Q.S.C.; Baracos, V.E.; Reiman, T.; Mazurak, V.C. Supplementation with fish oil increases first-line chemotherapy efficacy in patients with advanced nonsmall cell lung cancer. Cancer 2011, 117, 3774–3780. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Lara, K.; Turcott, J.G.; Juárez-Hernández, E.; Nuñez-Valencia, C.; Villanueva, G.; Guevara, P.; De la Torre-Vallejo, M.; Mohar, A.; Arrieta, O. Effects of an oral nutritional supplement containing eicosapentaenoic acid on nutritional and clinical outcomes in patients with advanced non-small cell lung cancer: RANDOMISED trial. Clin. Nutr. 2014, 33, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Liu, Z.; Wu, L.; Yuan, Y.; Hu, Y.; Zhang, X.; Wei, L.; Zu, Y. Tumor targeting with docosahexaenoic acid-conjugated docetaxel for inhibiting lung cancer metastasis to bone. Oncol. Lett. 2018, 16, 2911–2920. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | ω-6 | high fat con | ω-3 |
|---|---|---|---|
| Menhaden Oil | - | 25.5 | 189.5 |
| Safflower Oil | 189.5 | 164.0 | - |
| Soybean Oil | 13.0 | 13.0 | 13.0 |
| Casein | 200.0 | 200.0 | 200.0 |
| Cellulose | 50.0 | 50.0 | 50.0 |
| Corn Starch | 72.8 | 72.8 | 72.8 |
| Maltodextrin | 100.0 | 100.0 | 100.0 |
| Sucrose | 172.8 | 172.8 | 172.8 |
| Mineral Mix | 10.0 | 10.0 | 10.0 |
| Dicalcium Phos | 13.0 | 13.0 | 13.0 |
| Calcium Carb | 5.5 | 5.5 | 5.5 |
| Potassium Citrate | 16.5 | 16.5 | 16.5 |
| Vitamin Mix | 10.0 | 10.0 | 10.0 |
| L-Cystine | 3.0 | 3.0 | 3.0 |
| Choline Bitartrate | 2.0 | 2.0 | 2.0 |
| Total Amount | 858.95 g | 858.84 g | 858.15 g |
| Caloric Intake | 4057 Kcal | 4057 Kcal | 4057 Kcal |
| Protein | 23.7% | 23.7% | 23.7% |
| Fat | 23.6% | 23.6% | 23.6% |
| Carbohydrates | 41.4% | 41.4% | 41.4% |
| ω-3: ω-6 fa ratio | 1:125 | 1:15 | 2.5:1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Y.; Mullapudi, B.; Torres, C.; Mascariñas, E.; Mancinelli, G.; Diaz, A.M.; McKinney, R.; Barron, M.; Schultz, M.; Heiferman, M.; et al. Omega-3 Fatty Acids Prevent Early Pancreatic Carcinogenesis via Repression of the AKT Pathway. Nutrients 2018, 10, 1289. https://doi.org/10.3390/nu10091289
Ding Y, Mullapudi B, Torres C, Mascariñas E, Mancinelli G, Diaz AM, McKinney R, Barron M, Schultz M, Heiferman M, et al. Omega-3 Fatty Acids Prevent Early Pancreatic Carcinogenesis via Repression of the AKT Pathway. Nutrients. 2018; 10(9):1289. https://doi.org/10.3390/nu10091289
Chicago/Turabian StyleDing, Yongzeng, Bhargava Mullapudi, Carolina Torres, Emman Mascariñas, Georgina Mancinelli, Andrew M. Diaz, Ronald McKinney, Morgan Barron, Michelle Schultz, Michael Heiferman, and et al. 2018. "Omega-3 Fatty Acids Prevent Early Pancreatic Carcinogenesis via Repression of the AKT Pathway" Nutrients 10, no. 9: 1289. https://doi.org/10.3390/nu10091289
APA StyleDing, Y., Mullapudi, B., Torres, C., Mascariñas, E., Mancinelli, G., Diaz, A. M., McKinney, R., Barron, M., Schultz, M., Heiferman, M., Wojtanek, M., Adrian, K., DeCant, B., Rao, S., Ouellette, M., Tsao, M.-S., Bentrem, D. J., & Grippo, P. J. (2018). Omega-3 Fatty Acids Prevent Early Pancreatic Carcinogenesis via Repression of the AKT Pathway. Nutrients, 10(9), 1289. https://doi.org/10.3390/nu10091289