Serum Parathyroid Hormone, 25-Hydroxyvitamin D, and Risk of Alzheimer’s Disease: A Mendelian Randomization Study




Abstract
1. Introduction
2. Methods
2.1. Study Design and Data Sources
2.2. Mendelian Randomization Analyses
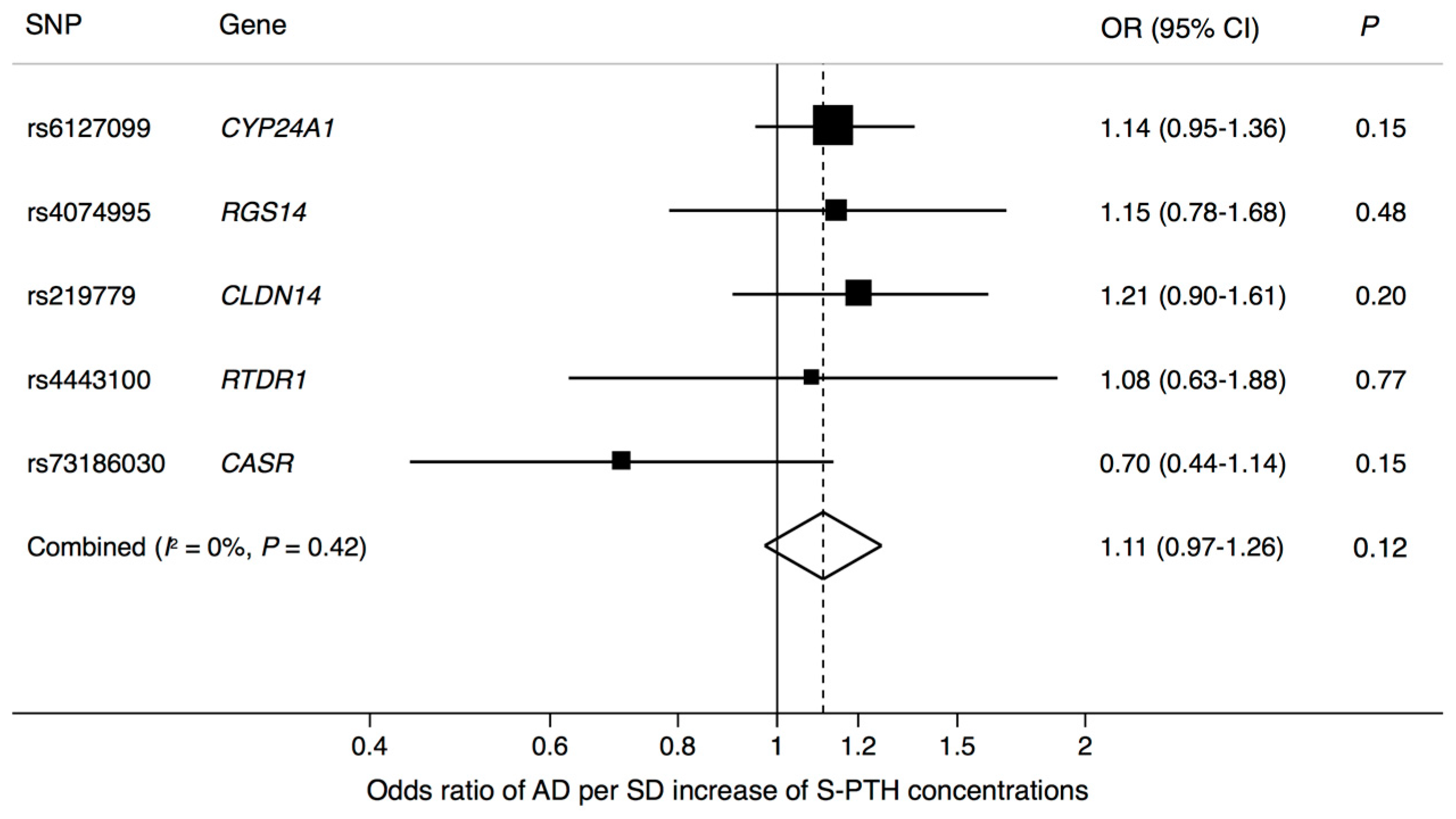
3. Results
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Holick, M.F. Vitamin D and brain health: The need for vitamin D supplementation and sensible sun exposure. J. Intern. Med. 2015, 277, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Dursun, E.; Feron, F.; Gezen-Ak, D.; Kalueff, A.V.; Littlejohns, T.; Llewellyn, D.J.; Millet, P.; Scott, T.; Tucker, K.L.; et al. ‘Vitamin D and cognition in older adults’: Updated international recommendations. J. Intern. Med. 2015, 277, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Morello, M.; Landel, V.; Lacassagne, E.; Baranger, K.; Annweiler, C.; Feron, F.; Millet, P. Vitamin D improves neurogenesis and cognition in a mouse model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 3463–6479. [Google Scholar] [CrossRef] [PubMed]
- Durk, M.R.; Han, K.; Chow, E.C.; Ahrens, R.; Henderson, J.T.; Fraser, P.E.; Pang, K.S. 1alpha, 25-Dihydroxyvitamin D3 reduces cerebral amyloid-beta accumulation and improves cognition in mouse models of Alzheimer’s disease. J. Neurosci. 2014, 34, 7091–7101. [Google Scholar] [CrossRef] [PubMed]
- Keeney, J.T.; Butterfield, D.A. Vitamin D deficiency and Alzheimer disease: Common links. Neurobiol. Dis. 2015, 84, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Afzal, S.; Bojesen, S.E.; Nordestgaard, B.G. Reduced 25-hydroxyvitamin D and risk of Alzheimer’s disease and vascular dementia. Alzheimers Dement. 2014, 10, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Littlejohns, T.J.; Henley, W.E.; Lang, I.A.; Annweiler, C.; Beauchet, O.; Chaves, P.H.; Fried, L.; Kestenbaum, B.R.; Kuller, L.H.; Langa, K.M.; et al. Vitamin D and the risk of dementia and Alzheimer disease. Neurology 2014, 83, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Feart, C.; Helmer, C.; Merle, B.; Herrmann, F.R.; Annweiler, C.; Dartigues, J.F.; Delcourt, C.; Samieri, C. Associations of lower vitamin D concentrations with cognitive decline and long-term risk of dementia and Alzheimer’s disease in older adults. Alzheimers Dement. 2017, 13, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Licher, S.; de Bruijn, R.; Wolters, F.J.; Zillikens, M.C.; Ikram, M.A.; Ikram, M.K. Vitamin D and the risk of dementia: The rotterdam study. J. Alzheimers Dis. 2017, 60, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Balion, C.; Griffith, L.E.; Strifler, L.; Henderson, M.; Patterson, C.; Heckman, G.; Llewellyn, D.J.; Raina, P. Vitamin D, cognition, and dementia: A systematic review and meta-analysis. Neurology 2012, 79, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Mokry, L.E.; Ross, S.; Morris, J.A.; Manousaki, D.; Forgetta, V.; Richards, J.B. Genetically decreased vitamin D and risk of Alzheimer disease. Neurology 2016, 87, 2567–2574. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Traylor, M.; Malik, R.; Dichgans, M.; Burgess, S.; Markus, H.S. Modifiable pathways in Alzheimer’s disease: Mendelian randomization analysis. BMJ 2017, 359, j5375. [Google Scholar] [CrossRef] [PubMed]
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J. Chem. Neuroanat. 2005, 29, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Usdin, T.B.; Gruber, C.; Bonner, T.I. Identification and functional expression of a receptor selectively recognizing parathyroid hormone, the PTH2 receptor. J. Biol. Chem. 1995, 270, 15455–15458. [Google Scholar] [CrossRef] [PubMed]
- Lourida, I.; Thompson-Coon, J.; Dickens, C.M.; Soni, M.; Kuzma, E.; Kos, K.; Llewellyn, D.J. Parathyroid hormone, cognitive function and dementia: A systematic review. PLoS ONE 2015, 10, e0127574. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, E.; Kilander, L.; Nylander, R.; Larsson, E.M.; Michaelsson, K.; Melhus, H.; Ahlstrom, H.; Johansson, L.; Lind, L.; Arnlov, J. Plasma parathyroid hormone is associated with vascular dementia and cerebral hyperintensities in two community-based cohorts. J. Clin. Endocrinol. Metab. 2014, 99, 4181–4189. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Cohen, C.; Lutsey, P.L.; Kleber, M.E.; Nielson, C.M.; Mitchell, B.D.; Bis, J.C.; Eny, K.M.; Portas, L.; Eriksson, J.; Lorentzon, M.; et al. Genetic variants associated with circulating parathyroid hormone. J. Am. Soc. Nephrol. 2017, 28, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; O’Reilly, P.F.; Aschard, H.; Hsu, Y.H.; Richards, J.B.; Dupuis, J.; Ingelsson, E.; Karasik, D.; Pilz, S.; Berry, D.; et al. Genome-wide association study in 79,366 European-ancestry individuals informs the genetic architecture of 25-hydroxyvitamin D levels. Nat. Commun. 2018, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Manousaki, D.; Dudding, T.; Haworth, S.; Hsu, Y.H.; Liu, C.T.; Medina-Gomez, C.; Voortman, T.; van der Velde, N.; Melhus, H.; Robinson-Cohen, C.; et al. Low-Frequency synonymous coding variation in CYP2R1 has large effects on vitamin D levels and risk of multiple sclerosis. Am. J. Hum. Genet. 2017, 101, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Bowden, J.; Fall, T.; Ingelsson, E.; Thompson, S.G. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology 2017, 28, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Michaëlsson, K.; Wolk, A.; Byberg, L.; Mitchell, A.; Mallmin, H.; Melhus, H. The seasonal importance of serum 25-hydroxyvitamin D for bone mineral density in older women. J. Intern. Med. 2017, 281, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Olsson, E.; Byberg, L.; Karlstrom, B.; Cederholm, T.; Melhus, H.; Sjogren, P.; Kilander, L. Vitamin D is not associated with incident dementia or cognitive impairment: An 18-y follow-up study in community-living old men. Am. J. Clin. Nutr. 2017, 105, 936–953. [Google Scholar] [CrossRef] [PubMed]
- Karakis, I.; Pase, M.P.; Beiser, A.; Booth, S.L.; Jacques, P.F.; Rogers, G.; DeCarli, C.; Vasan, R.S.; Wang, T.J.; Himali, J.J.; et al. Association of serum vitamin D with the risk of incident dementia and subclinical indices of brain aging: The framingham heart study. J. Alzheimers Dis. 2016, 51, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Maddock, J.; Zhou, A.; Cavadino, A.; Kuzma, E.; Bao, Y.; Smart, M.C.; Saum, K.U.; Schottker, B.; Engmann, J.; Kjaergaard, M.; et al. Vitamin D and cognitive function: A Mendelian randomization study. Sci. Rep. 2017, 7, 13230. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Herrmann, F.R.; Fantino, B.; Brugg, B.; Beauchet, O. Effectiveness of the combination of memantine plus vitamin D on cognition in patients with Alzheimer disease: A pre-post pilot study. Cogn. Behav. Neurol. 2012, 25, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.S.; Scherer, S.C.; Ladd, K.S.; Harrison, L.C. A randomized controlled trial of high-dose vitamin D2 followed by intranasal insulin in Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Przybelski, R.; Agrawal, S.; Krueger, D.; Engelke, J.A.; Walbrun, F.; Binkley, N. Rapid correction of low vitamin D status in nursing home residents. Osteoporos. Int. 2008, 19, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Rossom, R.C.; Espeland, M.A.; Manson, J.E.; Dysken, M.W.; Johnson, K.C.; Lane, D.S.; LeBlanc, E.S.; Lederle, F.A.; Masaki, K.H.; Margolis, K.L. Calcium and vitamin D supplementation and cognitive impairment in the women’s health initiative. J. Am. Geriatr. Soc. 2012, 60, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, I.; Sabir, M.S.; Dussik, C.M.; Whitfield, G.K.; Karrys, A.; Hsieh, J.C.; Haussler, M.R.; Meyer, M.B.; Pike, J.W.; Jurutka, P.W. 1,25-Dihydroxyvitamin D regulates expression of the tryptophan hydroxylase 2 and leptin genes: Implication for behavioral influences of vitamin D. FASEB J. 2015, 29, 4023–4035. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Markus, H.S. Branched-chain amino acids and Alzheimer’s disease: A Mendelian randomization analysis. Sci. Rep. 2017, 7, 13604. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, M.P.; Sorva, A.J.; Tilvis, R.S. Does elevated parathyroid hormone concentration predict cognitive decline in older people? Aging Clin. Exp. Res. 2010, 22, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Kipen, E.; Helme, R.D.; Wark, J.D.; Flicker, L. Bone density, vitamin D nutrition, and parathyroid hormone levels in women with dementia. J. Am. Geriatr. Soc. 1995, 43, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, T.; Miya, K.; Morimoto, S. Possible participation of calcium-regulating factors in senile dementia in elderly female subjects. Gerontology 1990, 36, 25–30. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Exposure | SNP | Chr | Nearby Gene | EA * | EAF | Associations with Exposure | Associations with AD | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| β † | SE | p | β ‡ | SE | p | ||||||
| S-PTH | rs6127099 | 20 | CYP24A1 | T | 0.34 | 0.07 | 0.003 | 2.4 × 10−72 | 0.028 | 0.019 | 0.155 |
| S-PTH | rs4074995 | 5 | RGS14 | G | 0.71 | 0.03 | 0.003 | 3.3 × 10−23 | 0.012 | 0.018 | 0.482 |
| S-PTH | rs219779 | 21 | CLDN14 | G | 0.75 | 0.04 | 0.003 | 8.9 × 10−22 | 0.023 | 0.018 | 0.202 |
| S-PTH | rs4443100 | 22 | RTDR1 | G | 0.32 | 0.02 | 0.003 | 4.1 × 10−11 | 0.005 | 0.017 | 0.773 |
| S-PTH | rs73186030 | 3 | CASR | T | 0.14 | 0.03 | 0.004 | 1.2 × 10−9 | −0.032 | 0.022 | 0.151 |
| S-25OHD | rs3755967 | 4 | GC | C | 0.72 | 0.089 | 0.002 | 4.74 × 10−343 | −0.036 | 0.017 | 0.034 |
| S-25OHD | rs117913124 | 11 | CYP2R1 | G | 0.975 | 0.430 # | 0.020 | 1.50 × 10−88 | −0.057 | 0.065 | 0.378 |
| S-25OHD | rs12785878 | 11 | DHCR7 | T | 0.75 | 0.036 | 0.002 | 3.80 × 10−62 | −0.022 | 0.018 | 0.208 |
| S-25OHD | rs10741657 | 11 | CYP2R1 | A | 0.40 | 0.031 | 0.002 | 2.05 × 10−46 | −0.011 | 0.016 | 0.492 |
| S-25OHD | rs17216707 | 20 | CYP24A1 | T | 0.79 | 0.026 | 0.003 | 8.14 × 10−23 | −0.045 | 0.022 | 0.038 |
| S-25OHD | rs10745742 § | 12 | AMDHD1 | T | 0.40 | 0.017 | 0.002 | 1.88 × 10−14 | −0.006 | 0.016 | 0.691 |
| S-25OHD | rs8018720 | 14 | SEC23A | G | 0.18 | 0.017 | 0.003 | 4.72 × 10−9 | −0.012 | 0.021 | 0.584 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larsson, S.C.; Traylor, M.; Markus, H.S.; Michaëlsson, K. Serum Parathyroid Hormone, 25-Hydroxyvitamin D, and Risk of Alzheimer’s Disease: A Mendelian Randomization Study. Nutrients 2018, 10, 1243. https://doi.org/10.3390/nu10091243
Larsson SC, Traylor M, Markus HS, Michaëlsson K. Serum Parathyroid Hormone, 25-Hydroxyvitamin D, and Risk of Alzheimer’s Disease: A Mendelian Randomization Study. Nutrients. 2018; 10(9):1243. https://doi.org/10.3390/nu10091243
Chicago/Turabian StyleLarsson, Susanna C., Matthew Traylor, Hugh S. Markus, and Karl Michaëlsson. 2018. "Serum Parathyroid Hormone, 25-Hydroxyvitamin D, and Risk of Alzheimer’s Disease: A Mendelian Randomization Study" Nutrients 10, no. 9: 1243. https://doi.org/10.3390/nu10091243
APA StyleLarsson, S. C., Traylor, M., Markus, H. S., & Michaëlsson, K. (2018). Serum Parathyroid Hormone, 25-Hydroxyvitamin D, and Risk of Alzheimer’s Disease: A Mendelian Randomization Study. Nutrients, 10(9), 1243. https://doi.org/10.3390/nu10091243