Induction of p53 Phosphorylation at Serine 20 by Resveratrol Is Required to Activate p53 Target Genes, Restoring Apoptosis in MCF-7 Cells Resistant to Cisplatin

 ,
,  ,
,  and
and 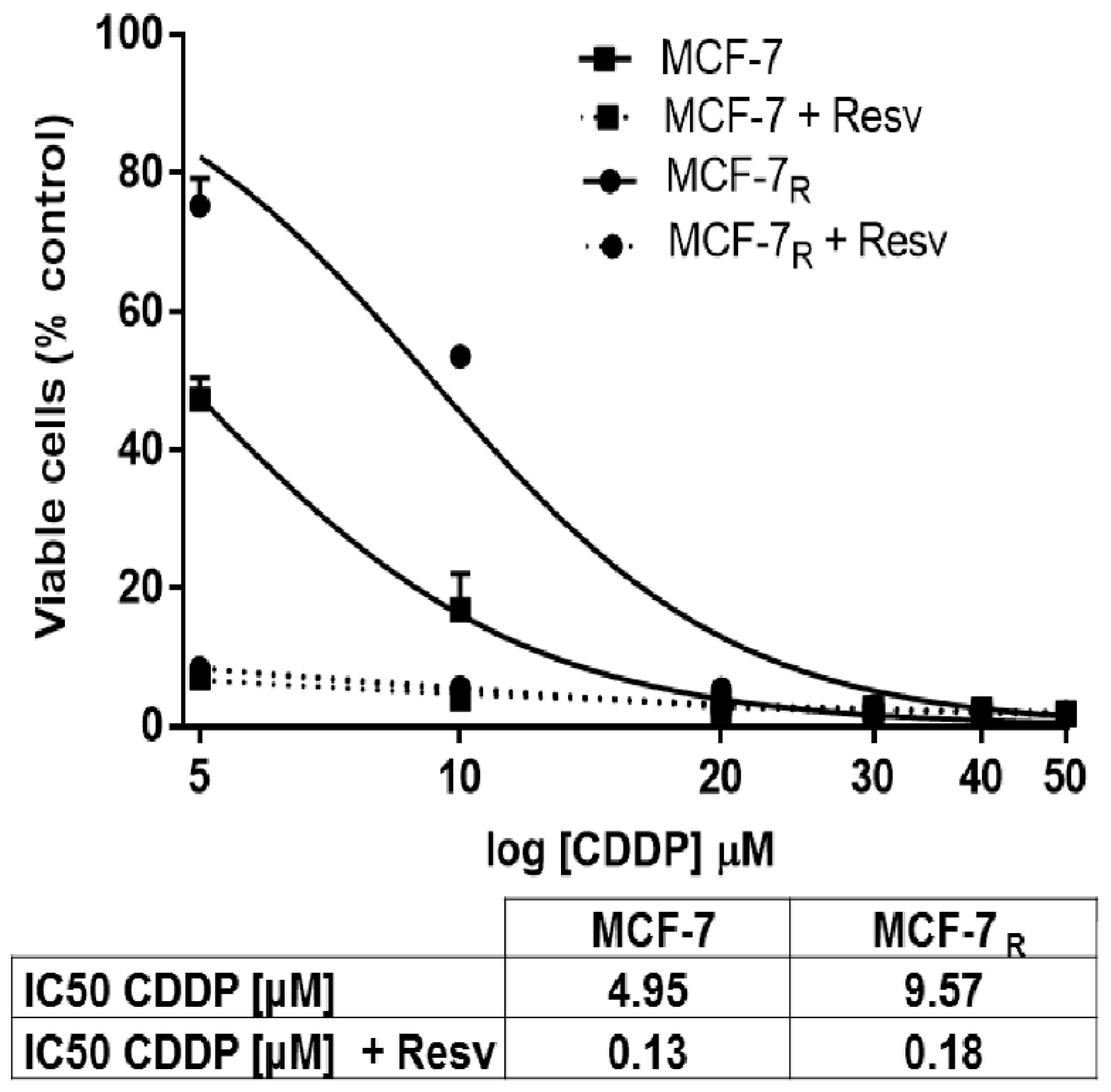{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Lines and Cell Culture
2.3. Generation of the CDDP-Resistant MCF-7 Cell Line Variant (MCF-7R)
2.4. Silencing of p53 Expression in MCF-7 and MCF-7R Cells by shRNA
2.5. Cell Viability Assay
2.6. Western Blot
2.7. Real-Time RT-PCR
2.8. Apoptosis Analysis
2.9. Statistical Analysis
3. Results
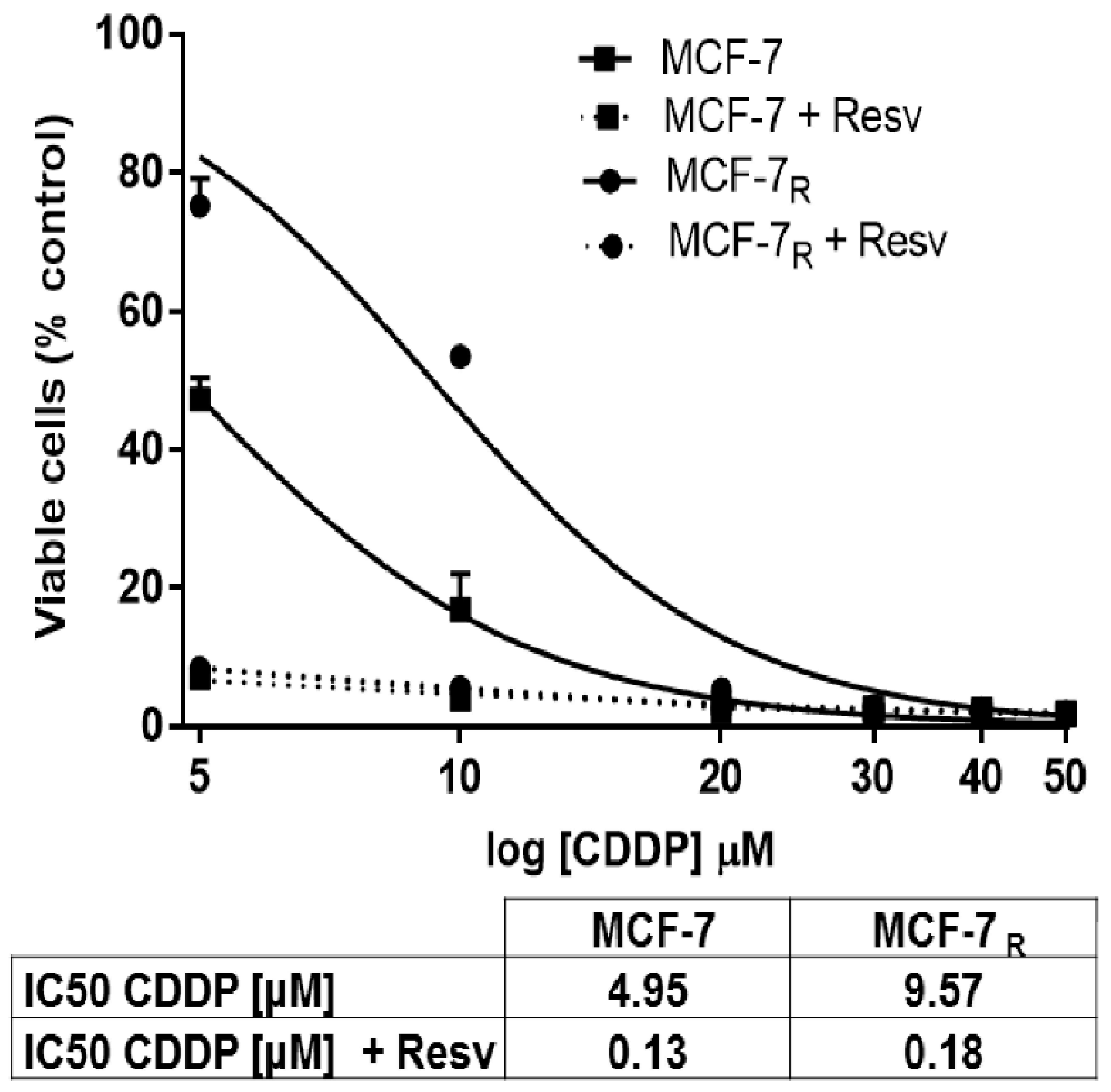
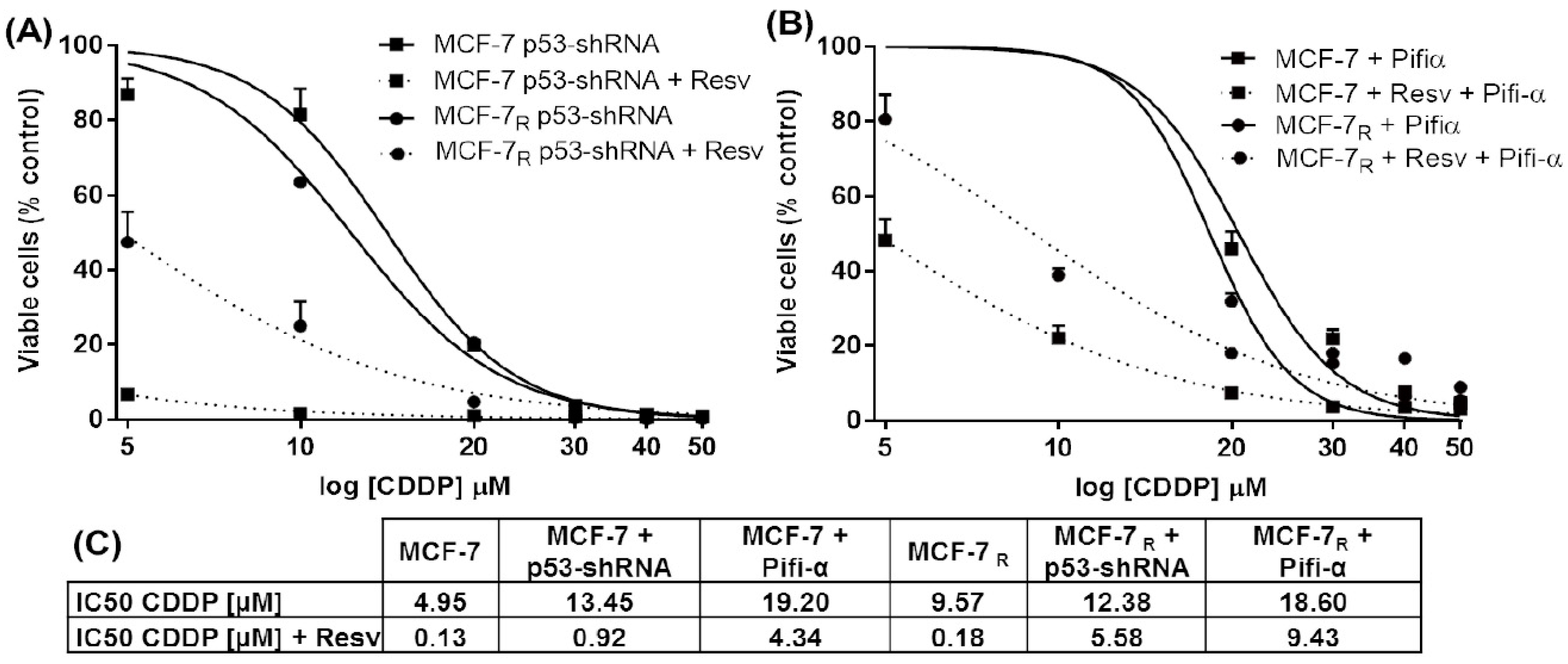
3.1. Resv Induces Sensitivity to CDDP in MCF-7R Cells
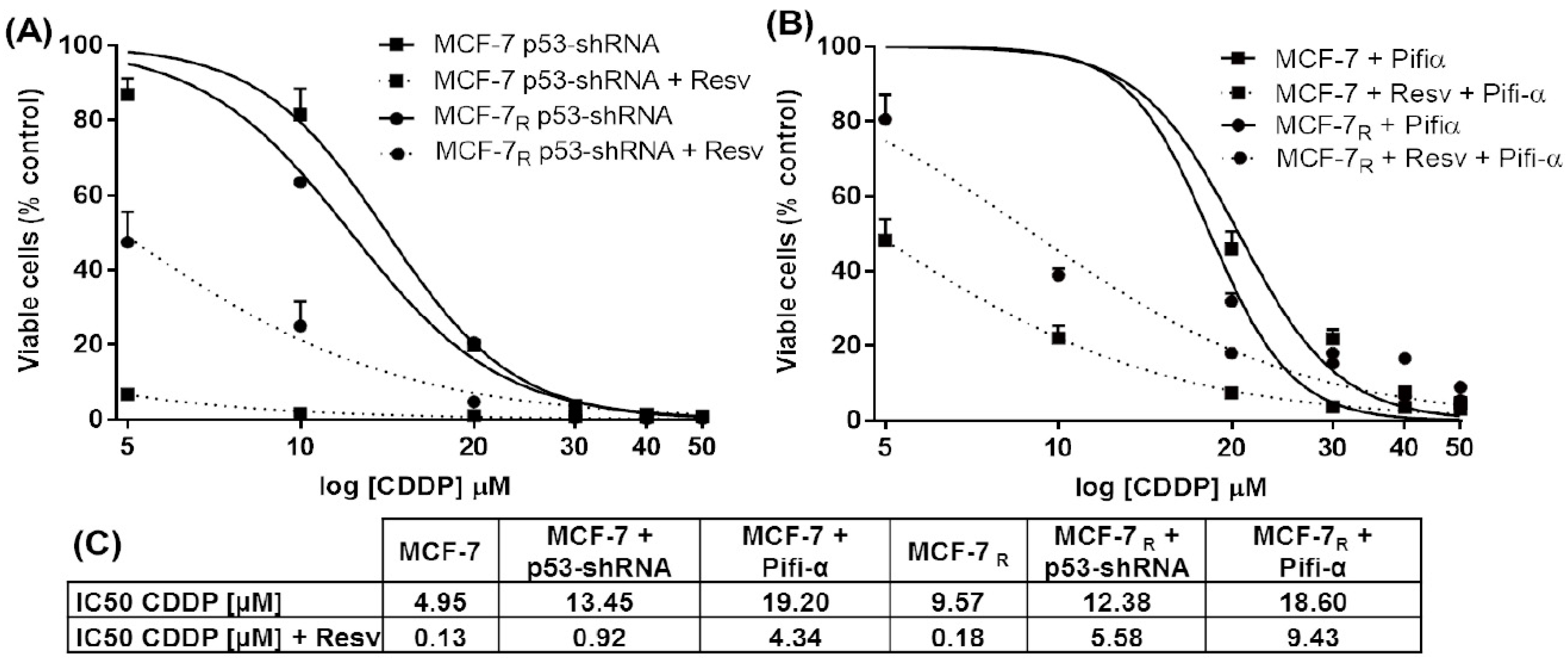
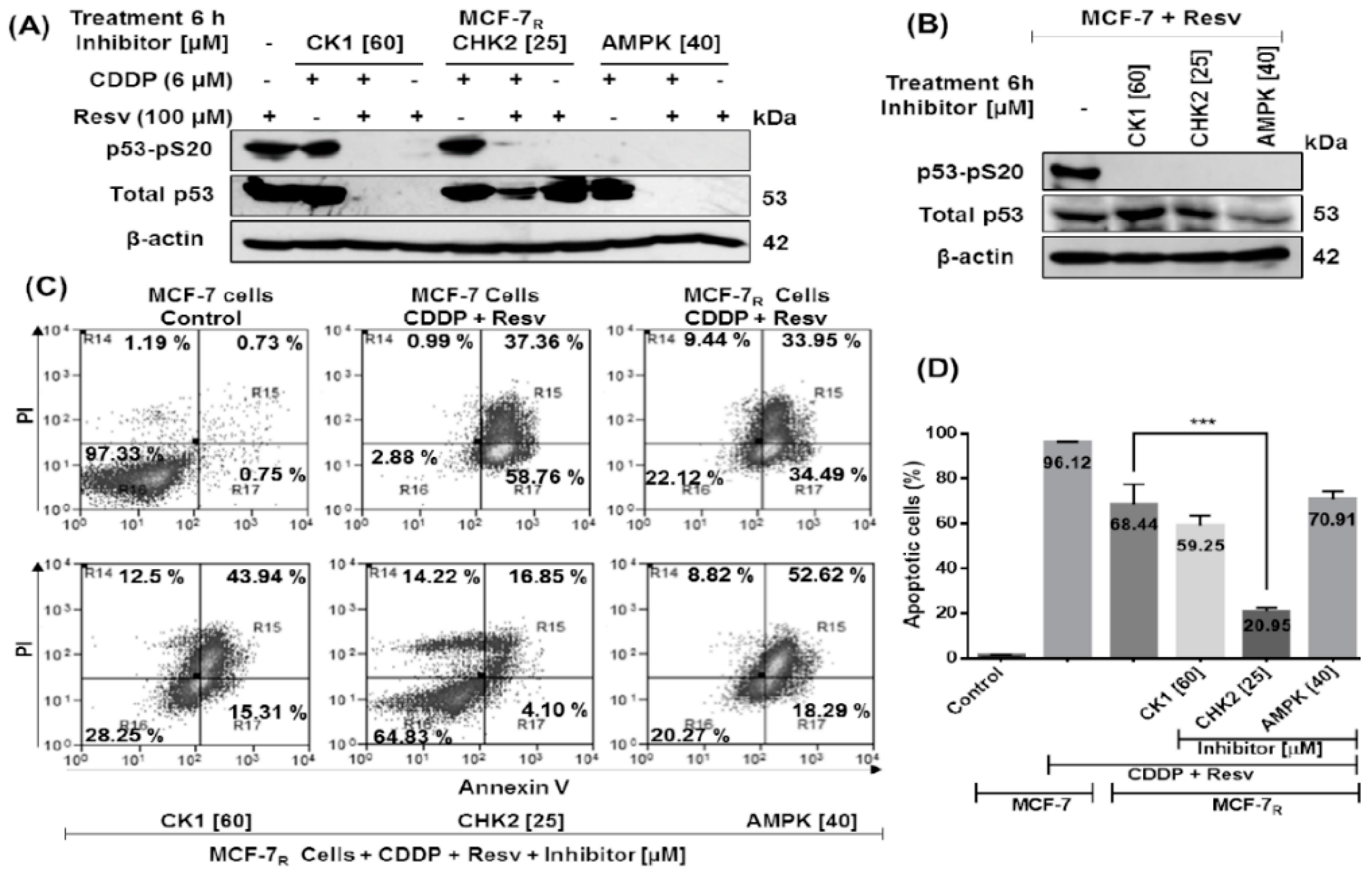
3.2. Down-Regulation of p53 Expression and Inhibition of the p53 Protein Activity Enhances Resistance to CDDP and Resv in MCF-7 and MCF-7R Cells
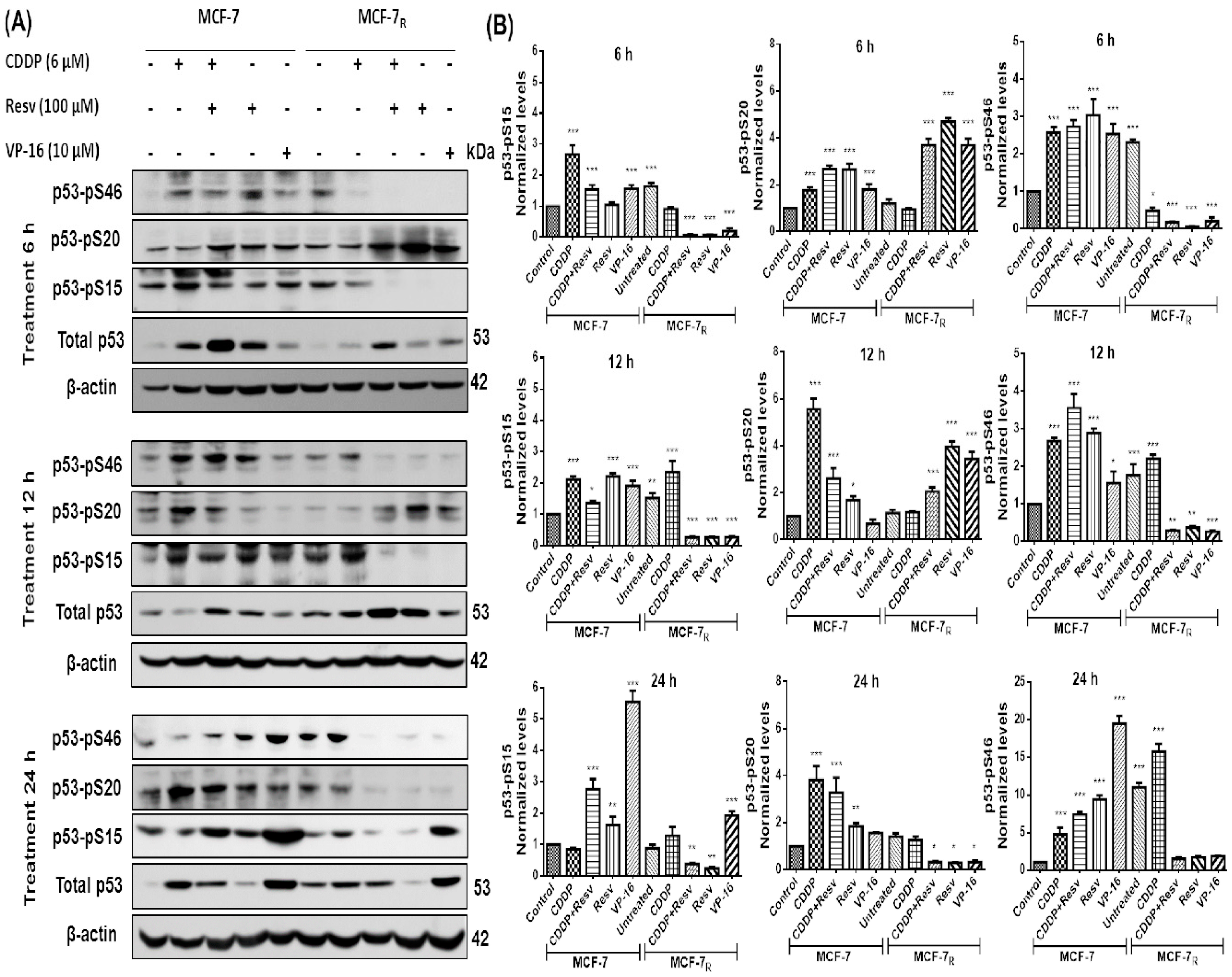
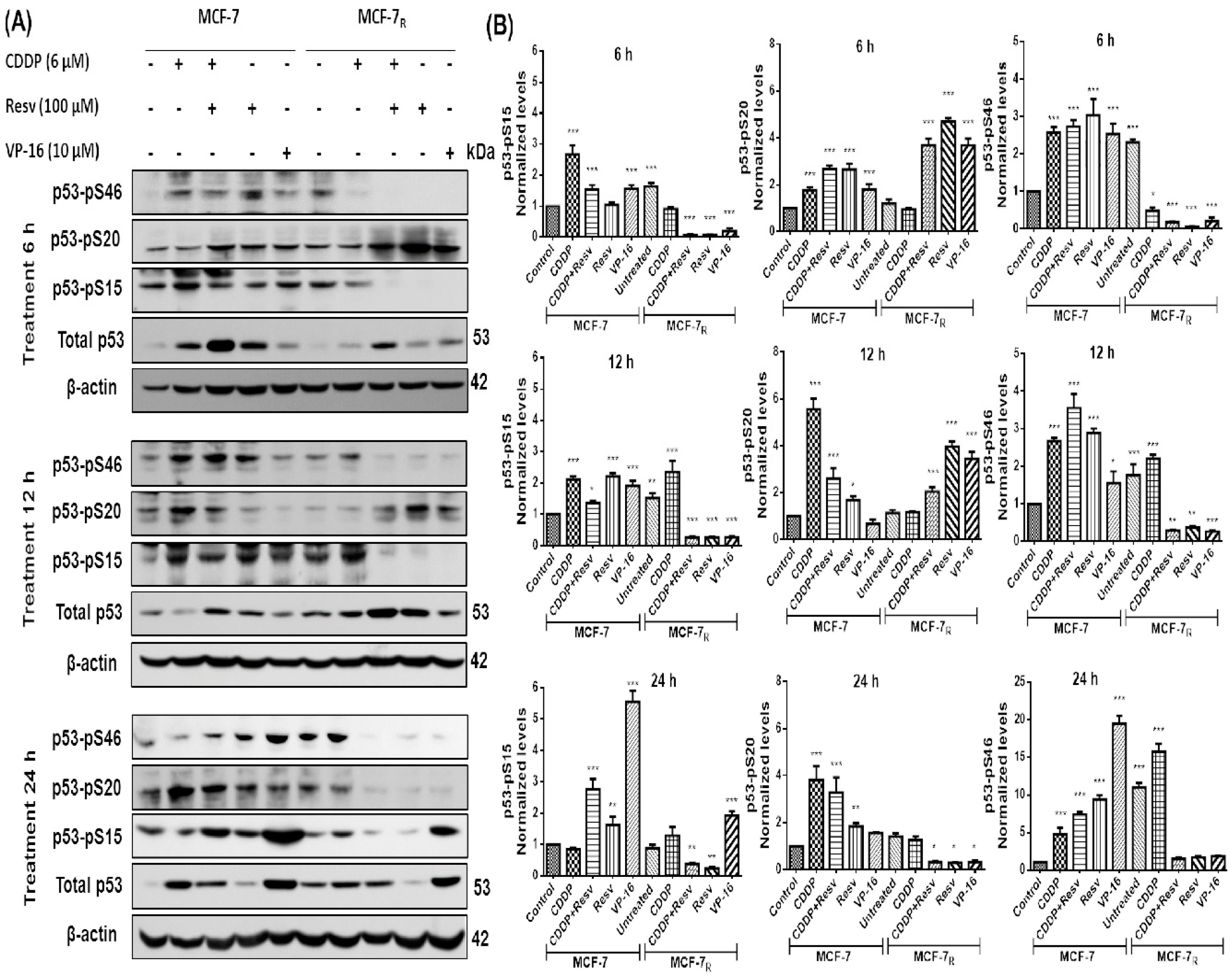
3.3. Resv Induces S20 Phosphorylation and Attenuates Phosphorylation of p53 in S15 and S46 in CDDP-Treated MCF-7R Cells
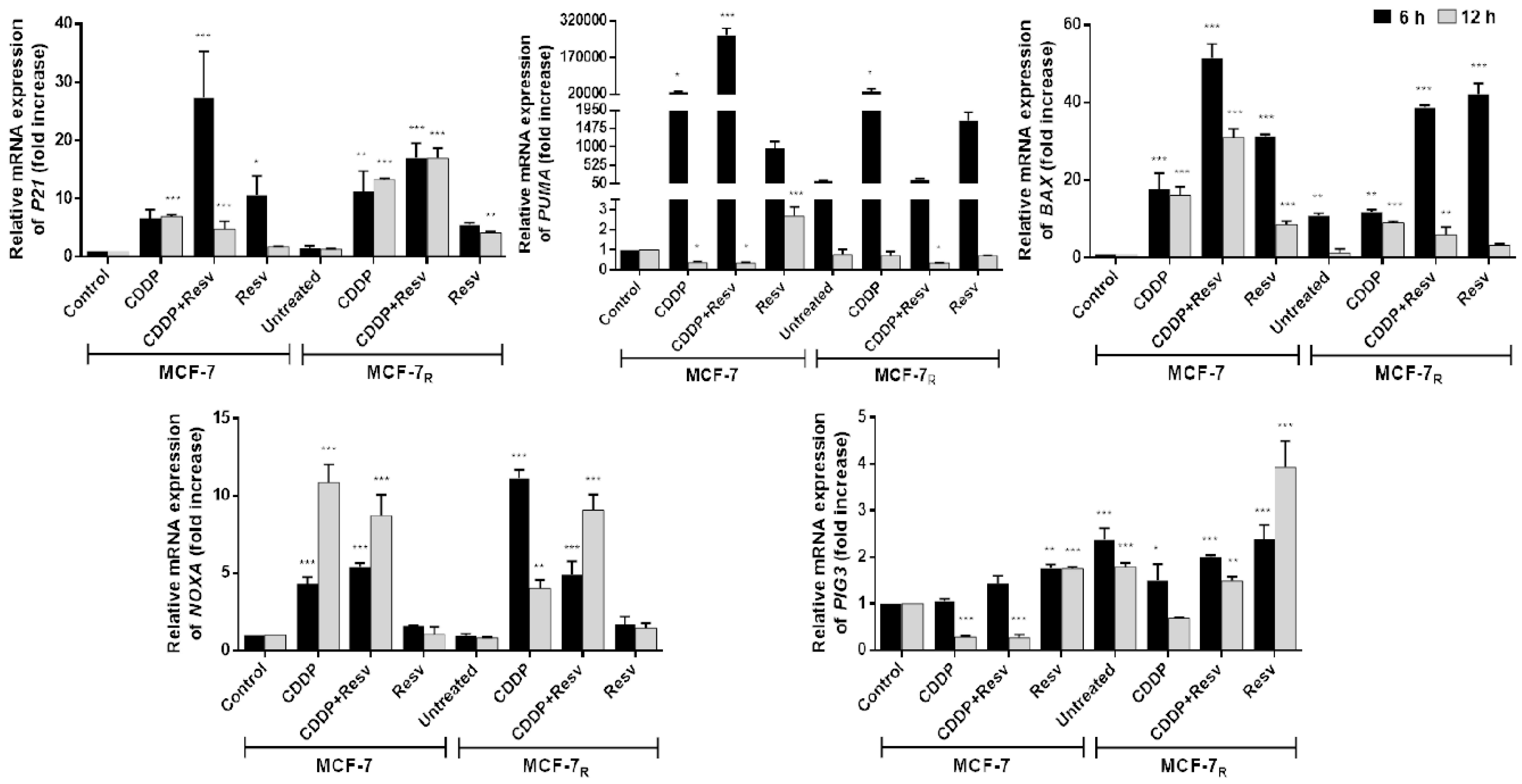
3.4. Early Phosphorylation of p53 in S20 Induced by Resv Is Sufficient to Activate p53-Dependent Gene Transcription in MCF-7R Cells
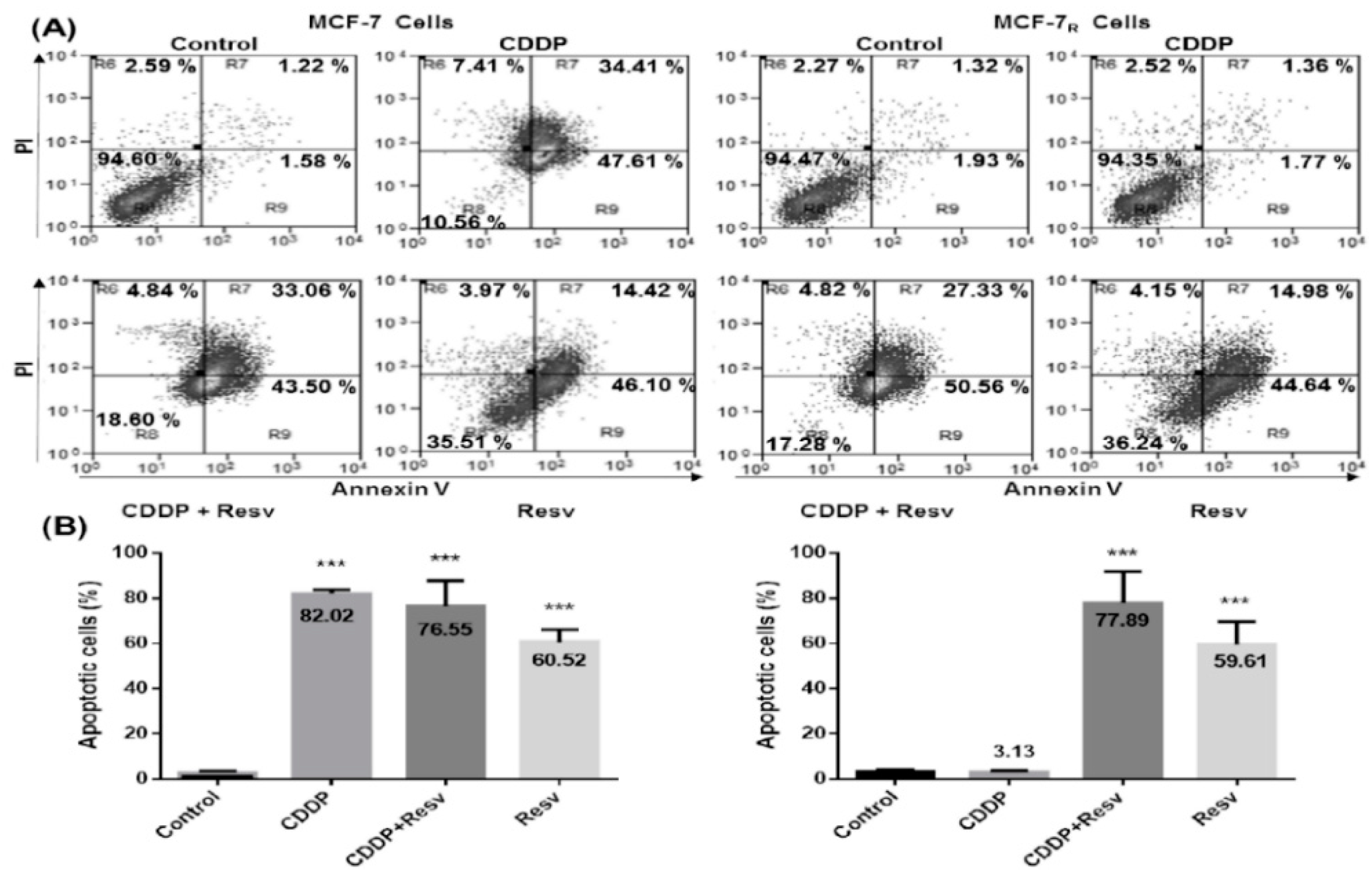
3.5. Resv Overcome CDDP-Resistance and Induces Apoptosis in MCF-7R Cells
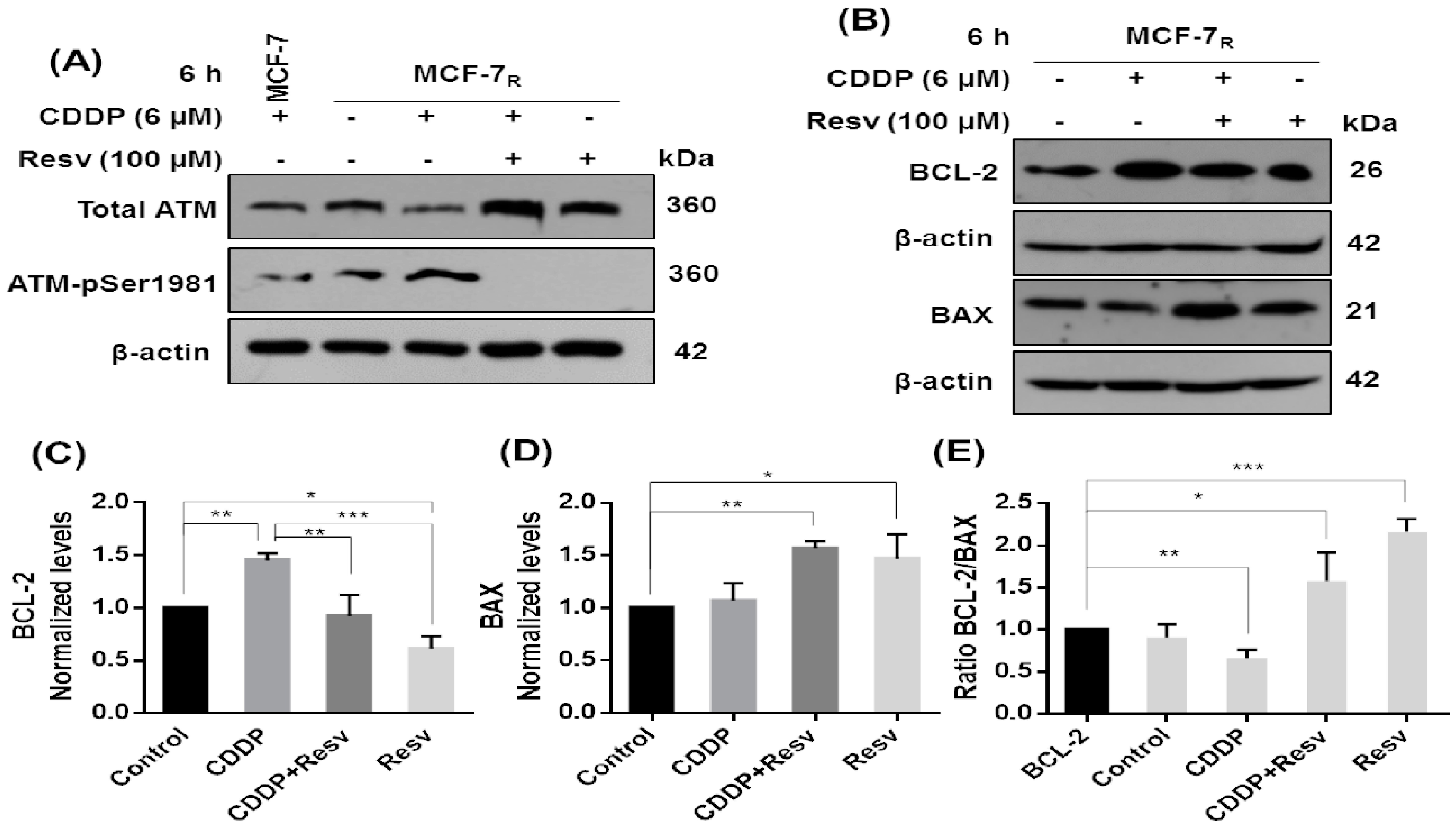
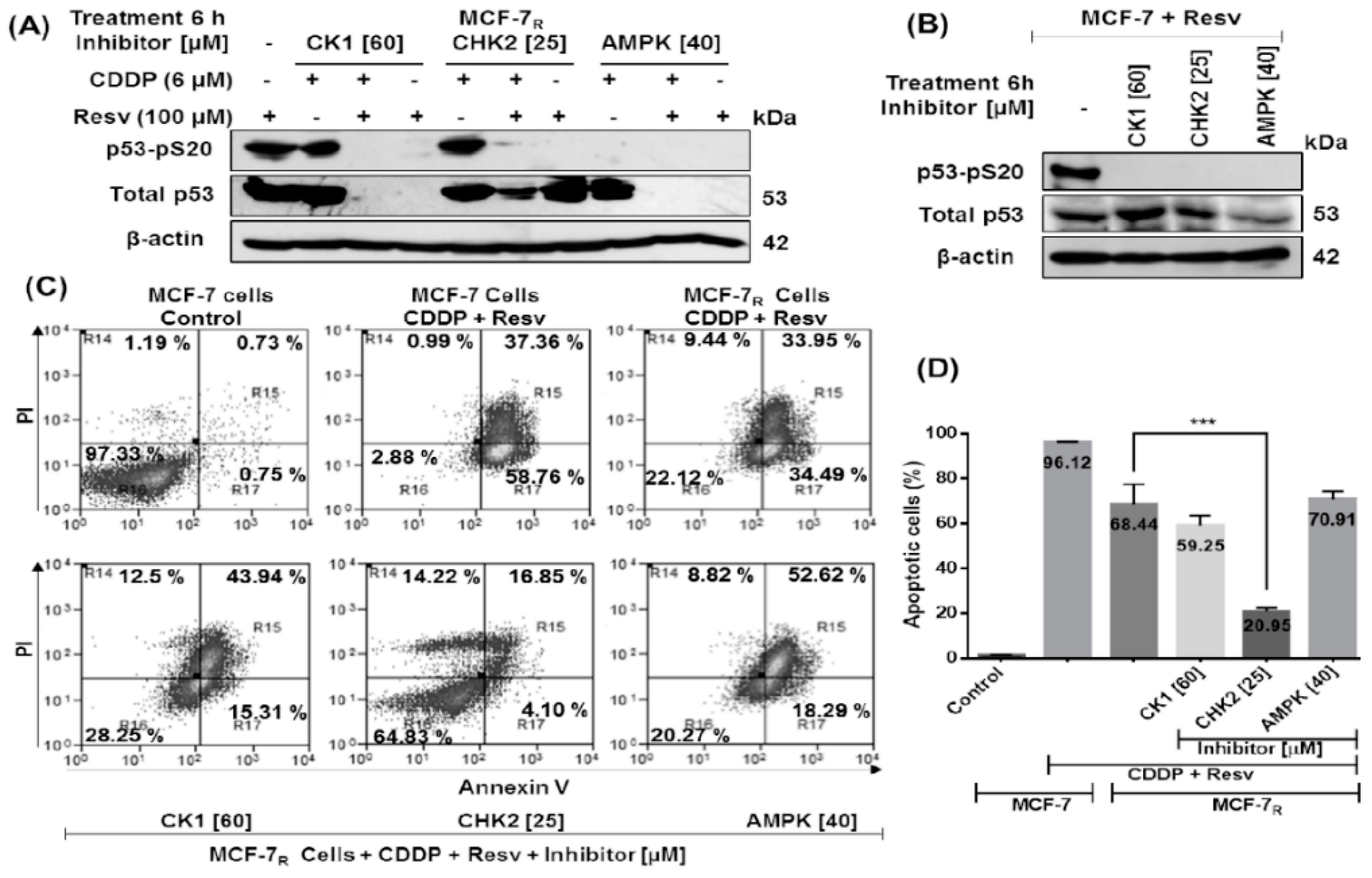
3.6. Early Phosphorylation of p53 in S20 Induced by Resv Is Necessary for p53-Stability in MCF-7R Cells
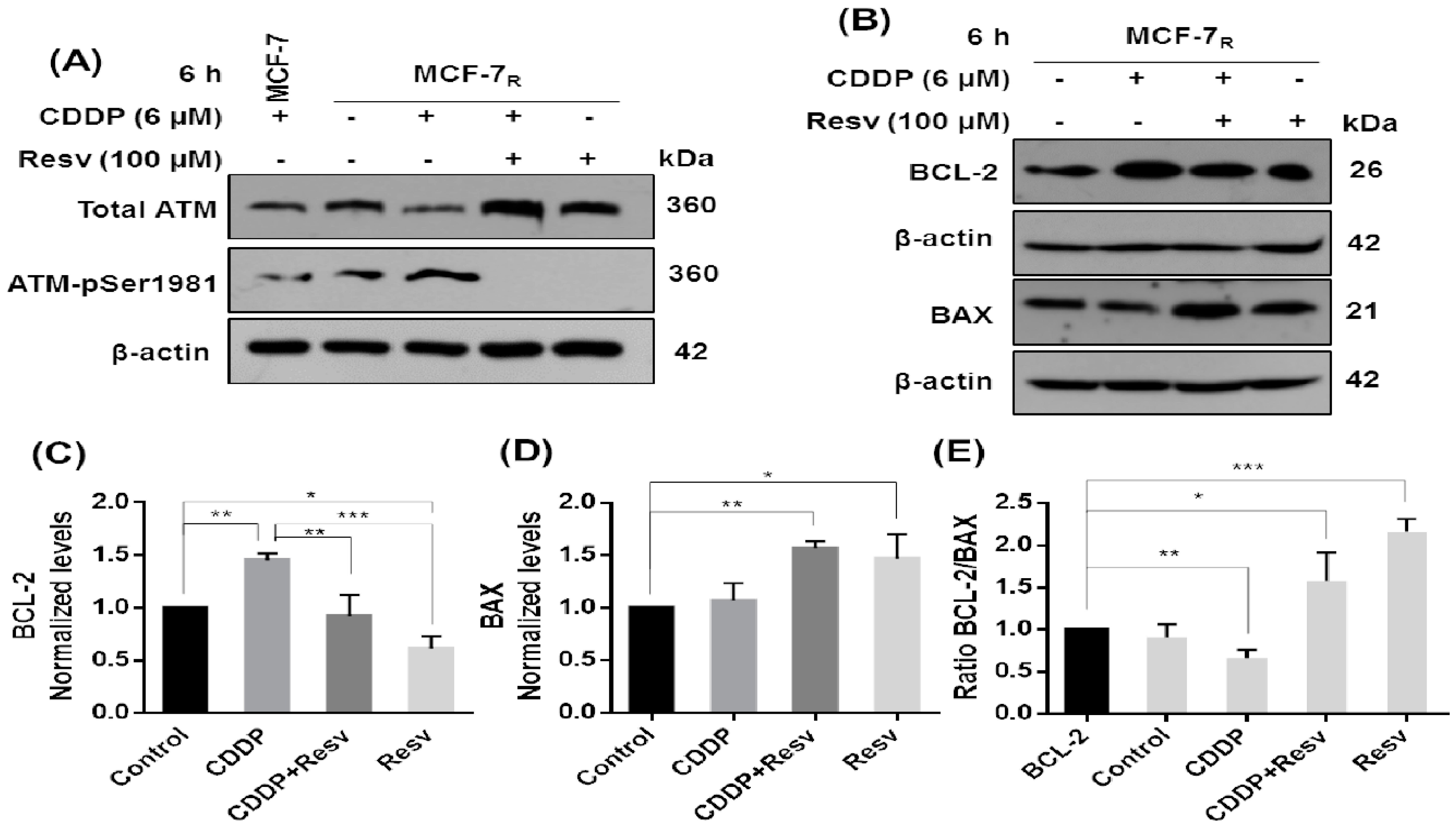
3.7. Resv Promotes Early Dephosphorylation of ATM, Inhibition of BCL-2, and Upregulation of BAX
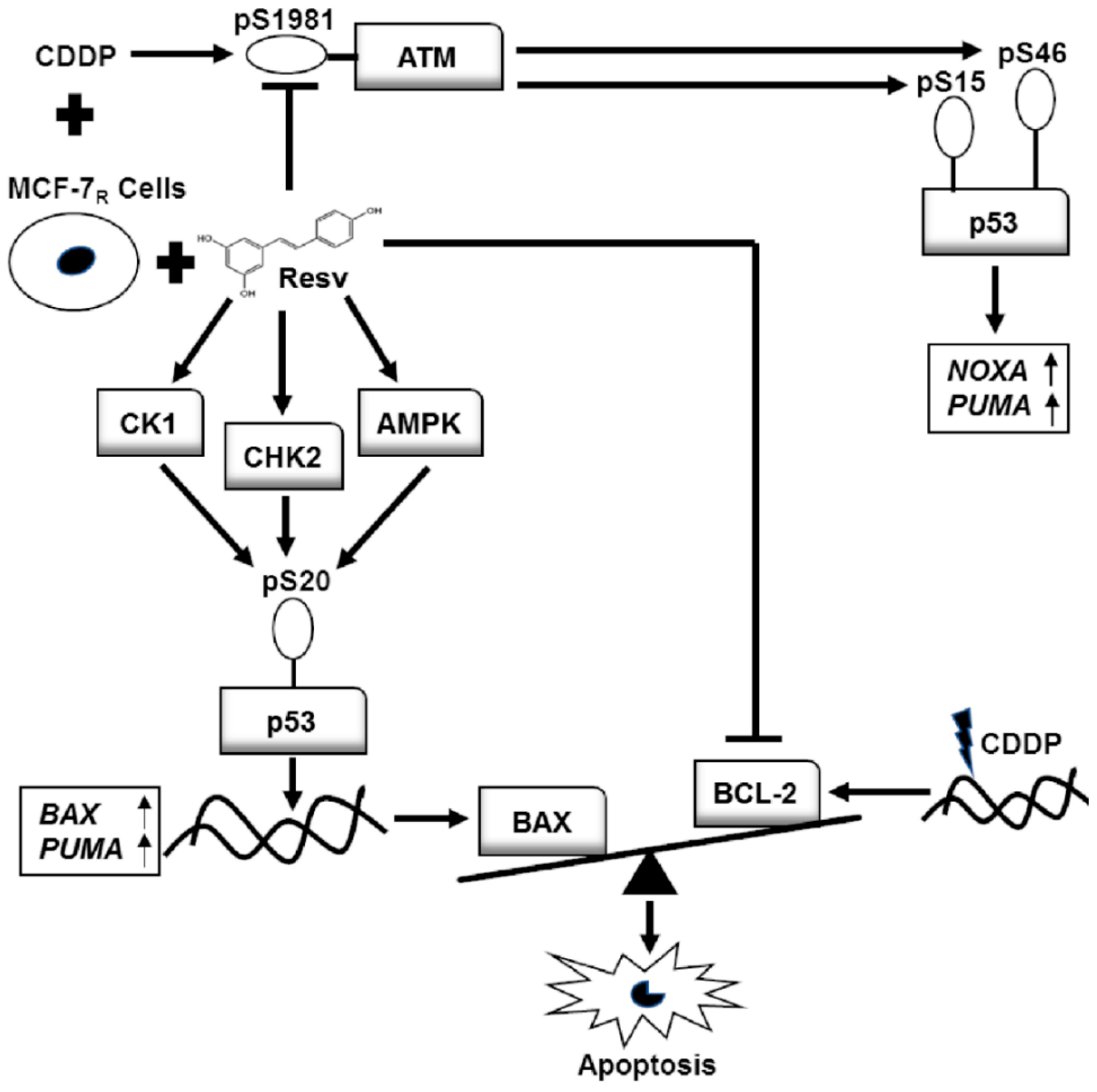
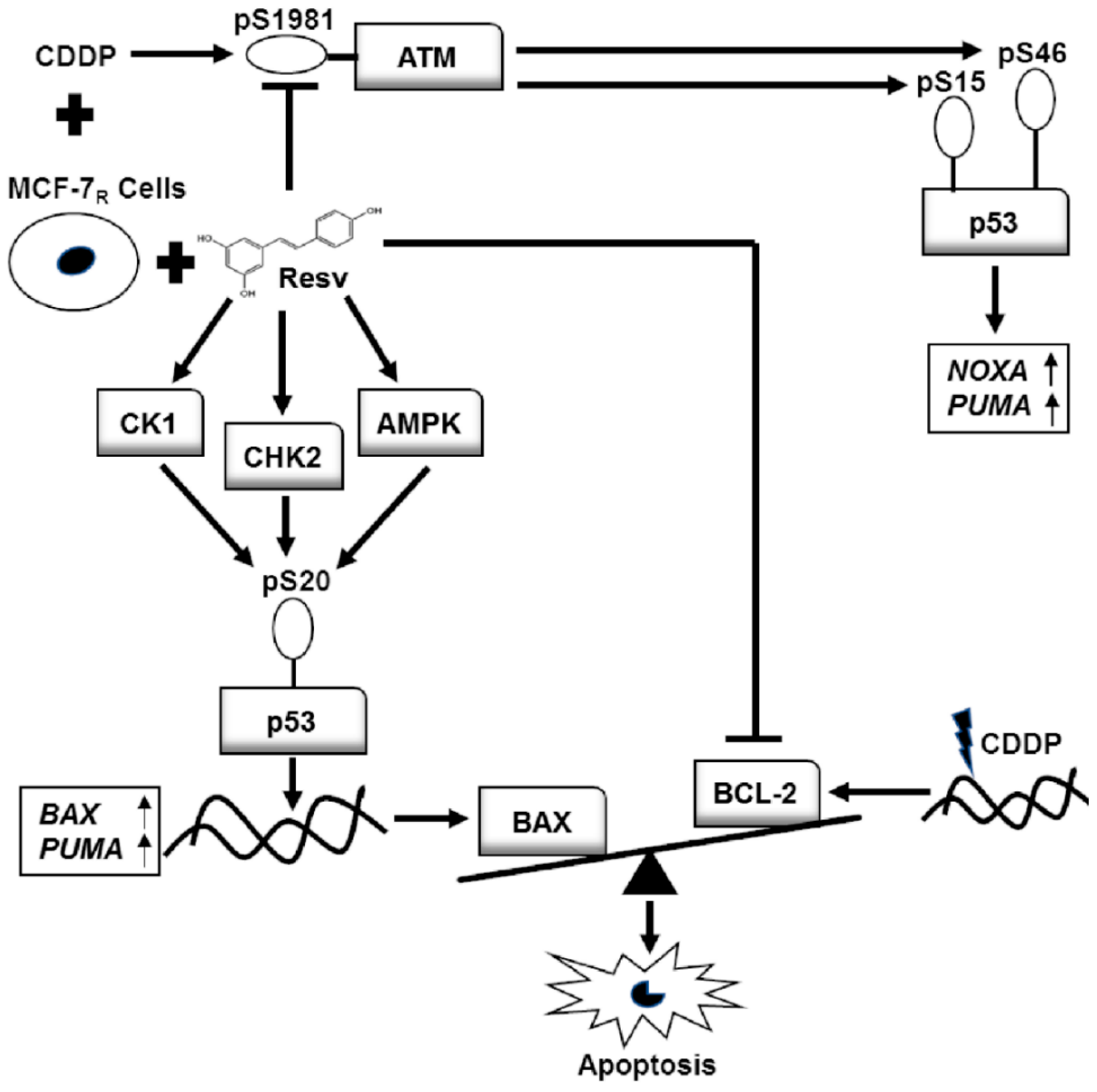
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brabec, V.; Kasparkova, J. Modifications of DNA by platinum complexes. Relation to resistance of tumors to platinum antitumor drugs. Drug Resist. Updat. 2005, 8, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Verma, N. Nature curing cancer—Review on structural modification studies with natural active compounds having anti-tumor efficiency. Biotechnol. Rep. (Amst.) 2015, 6, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Resveratrol and derivatives for the prevention and treatment of cancer. Drug Discov. Today 2010, 15, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Fabris, S.; Momo, F.; Ravagnan, G.; Stevanato, R. Antioxidant properties of resveratrol and piceid on lipid peroxidation in micelles and monolamellar liposomes. Biophys. Chem. 2008, 135, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, F.; Tanski, J.; Villegas-Estrada, A.; Rossi, M. Structural basis for antioxidant activity of trans-resveratrol: Ab initio calculations and crystal and molecular structure. J. Agric. Food Chem. 2004, 52, 7279–7285. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Bhardwaj, A.; Aggarwal, R.S.; Seeram, N.P.; Shishodia, S.; Takada, Y. Role of resveratrol in prevention and therapy of cancer: Preclinical and clinical studies. Anticancer Res. 2004, 24, 2783–2840. [Google Scholar] [PubMed]
- Gupta, S.C.; Kannappan, R.; Reuter, S.; Kim, J.H.; Aggarwal, B.B. Chemosensitization of tumors by resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 150–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; Liebermann, D.A.; Hoffman, B.; Reed, J.C. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar] [PubMed]
- Ferraz da Costa, D.C.; Casanova, F.A.; Quarti, J.; Malheiros, M.S.; Sanches, D.; Dos Santos, P.S.; Fialho, E.; Silva, J.L. Transient transfection of a wild-type p53 gene triggers resveratrol-induced apoptosis in cancer cells. PLoS ONE 2012, 7, e48746. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ma, W.Y.; Goranson, A.; Dong, Z. Resveratrol suppresses cell transformation and induces apoptosis through a p53-dependent pathway. Carcinogenesis 1999, 20, 237–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogada, R.; Prabhu, V.; Amadori, M.; Scott, R.; Hashmi, S.; Chandra, D. Resveratrol induces p53-independent, x-linked inhibitor of apoptosis protein (xiap)-mediated bax protein oligomerization on mitochondria to initiate cytochrome c release and caspase activation. J. Biol. Chem. 2011, 286, 28749–28760. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, V.; Srivastava, P.; Yadav, N.; Amadori, M.; Schneider, A.; Seshadri, A.; Pitarresi, J.; Scott, R.; Zhang, H.; Koochekpour, S.; et al. Resveratrol depletes mitochondrial DNA and inhibition of autophagy enhances resveratrol-induced caspase activation. Mitochondrion 2013, 13, 493–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taira, N.; Nihira, K.; Yamaguchi, T.; Miki, Y.; Yoshida, K. Dyrk2 is targeted to the nucleus and controls p53 via ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell 2007, 25, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by mdm2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Kodama, M.; Otsubo, C.; Hirota, T.; Yokota, J.; Enari, M.; Taya, Y. Requirement of ATM for rapid p53 phosphorylation at Ser46 without ser/thr-gln sequences. Mol. Cell. Biol. 2010, 30, 1620–1633. [Google Scholar] [CrossRef] [PubMed]
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; Del Sal, G.; et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 2002, 4, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Mullauer, F.; Bock, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. P53- and drug-induced apoptotic responses mediated by bh3-only proteins puma and noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Jiang, X.; Mercer, W.E. Downregulation of wip-1 phosphatase expression in mcf-7 breast cancer cells enhances doxorubicin-induced apoptosis through p53-mediated transcriptional activation of bax. Cancer Biol. Ther. 2009, 8, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Luo, J.; Chen, F.; Yang, F.; Song, W.; Zhu, A.; Guan, X. Brca1 regulates pig3-mediated apoptosis in a p53-dependent manner. Oncotarget 2015, 6, 7608–7618. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.C.; Wong, C.; John Bennett, D.; Wu, J.M. Regulation of p53 and cell proliferation by resveratrol and its derivatives in breast cancer cells: An in silico and biochemical approach targeting integrin alphavbeta3. Int. J. Cancer 2011, 129, 2732–2743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cao, H.J.; Davis, F.B.; Tang, H.Y.; Davis, P.J.; Lin, H.Y. Oestrogen inhibits resveratrol-induced post-translational modification of p53 and apoptosis in breast cancer cells. Br. J. Cancer 2004, 91, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon-Galicia, I.; Diaz-Chavez, J.; Garcia-Villa, E.; Uribe-Figueroa, L.; Hidalgo-Miranda, A.; Herrera, L.A.; Alvarez-Rios, E.; Garcia-Mena, J.; Gariglio, P. Resveratrol induces downregulation of DNA repair genes in mcf-7 human breast cancer cells. Eur. J. Cancer Prev. 2013, 22, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Leon-Galicia, I.; Diaz-Chavez, J.; Albino-Sanchez, M.E.; Garcia-Villa, E.; Bermudez-Cruz, R.; Garcia-Mena, J.; Herrera, L.A.; Garcia-Carranca, A.; Gariglio, P. Resveratrol decreases rad51 expression and sensitizes cisplatinresistant mcf7 breast cancer cells. Oncol. Rep. 2018, 39, 3025–3033. [Google Scholar] [PubMed]
- Fraser, M.; Bai, T.; Tsang, B.K. Akt promotes cisplatin resistance in human ovarian cancer cells through inhibition of p53 phosphorylation and nuclear function. Int. J. Cancer 2008, 122, 534–546. [Google Scholar] [CrossRef] [PubMed]
- MacLaine, N.J.; Oster, B.; Bundgaard, B.; Fraser, J.A.; Buckner, C.; Lazo, P.A.; Meek, D.W.; Hollsberg, P.; Hupp, T.R. A central role for ck1 in catalyzing phosphorylation of the p53 transactivation domain at serine 20 after hhv-6b viral infection. J. Biol. Chem. 2008, 283, 28563–28573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, A.; Scott, M.; Burch, L.; Smith, G.; Ball, K.; Hupp, T. Allosteric effects mediate chk2 phosphorylation of the p53 transactivation domain. EMBO Rep. 2003, 4, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Makela, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the lkb1 tumor suppressor, strad alpha/beta and mo25 alpha/beta are upstream kinases in the amp-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, L.; Xing, Z.; Liu, D.; Sun, J.; Li, X.; Zhang, Y. Status of bi- and multi-nuclear platinum anticancer drug development. Anticancer Agents Med. Chem. 2010, 10, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Tsang, R.Y.; Al-Fayea, T.; Au, H.J. Cisplatin overdose: Toxicities and management. Drug Saf. 2009, 32, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Maubant, S.; Staedel, C.; Gauduchon, P. Integrins, cell response to anti-tumor agents and chemoresistance. Bull. Cancer 2002, 89, 923–934. [Google Scholar] [PubMed]
- Nista, A.; Leonetti, C.; Bernardini, G.; Mattioni, M.; Santoni, A. Functional role of alpha4beta1 and alpha5beta1 integrin fibronectin receptors expressed on adriamycin-resistant mcf-7 human mammary carcinoma cells. Int. J. Cancer 1997, 72, 133–141. [Google Scholar] [CrossRef]
- Nadkarni, A.; Rajesh, P.; Ruch, R.J.; Pittman, D.L. Cisplatin resistance conferred by the rad51d (e233g) genetic variant is dependent upon p53 status in human breast carcinoma cell lines. Mol. Carcinog. 2009, 48, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Yi, X.; Hsu, S.; Wang, C.Y.; Dong, Z. Role of p53 in cisplatin-induced tubular cell apoptosis: Dependence on p53 transcriptional activity. Am. J. Physiol. Ren. Physiol. 2004, 287, F1140–F1147. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Luong, K.V.; Wang, L.; Roberts, B.J.; Wahl, J.K., 3rd; Peng, A. Cell fate determination in cisplatin resistance and chemosensitization. Oncotarget 2016, 7, 23383–23394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, T.; Horiguchi, H.; Oguma, E.; Kayama, F. Effects of diverse dietary phytoestrogens on cell growth, cell cycle and apoptosis in estrogen-receptor-positive breast cancer cells. J. Nutr. Biochem. 2010, 21, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Chen, Q.; Siddiqui, I.; Sarva, K.; Srivastava, R.K. Sensitization of trail-resistant lncap cells by resveratrol (3, 4′, 5 tri-hydroxystilbene): Molecular mechanisms and therapeutic potential. J. Mol. Signal. 2007, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Hwang, J.T.; Yun, H.; Chi, S.G.; Lee, S.J.; Kang, I.; Yoon, K.S.; Choe, W.J.; Kim, S.S.; Ha, J. Inhibition of amp-activated protein kinase sensitizes cancer cells to cisplatin-induced apoptosis via hyper-induction of p53. J. Biol. Chem. 2008, 283, 3731–3742. [Google Scholar] [CrossRef] [PubMed]
- Harhaji-Trajkovic, L.; Vilimanovich, U.; Kravic-Stevovic, T.; Bumbasirevic, V.; Trajkovic, V. Ampk-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J. Cell. Mol. Med. 2009, 13, 3644–3654. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Smith, M.L.; Rivet, D.J., 2nd; Duba, D.; Zhan, Q.; Kohn, K.W.; Fornace, A.J., Jr.; O’Connor, P.M. Disruption of p53 function sensitizes breast cancer mcf-7 cells to cisplatin and pentoxifylline. Cancer Res. 1995, 55, 1649–1654. [Google Scholar] [PubMed]
- Menendez, J.A.; Lupu, R. RNA interference-mediated silencing of the p53 tumor-suppressor protein drastically increases apoptosis after inhibition of endogenous fatty acid metabolism in breast cancer cells. Int. J. Mol. Med. 2005, 15, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Li, W.; Qian, C.; Zhou, Z.; Xu, W.; Wu, J. Down-regulated p53 by sirna increases smad4′s activity in promoting cell apoptosis in mcf-7 cells. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 1243–1248. [Google Scholar] [PubMed]
- Struckhoff, A.P.; Patel, B.; Beckman, B.S. Inhibition of p53 sensitizes mcf-7 cells to ceramide treatment. Int. J. Oncol. 2010, 37, 21–30. [Google Scholar] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of bcl-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. P53 modulates homologous recombination by transcriptional regulation of the rad51 gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandez-Valencia, J.; Garcia-Villa, E.; Arenas-Hernandez, A.; Garcia-Mena, J.; Diaz-Chavez, J.; Gariglio, P. Induction of p53 Phosphorylation at Serine 20 by Resveratrol Is Required to Activate p53 Target Genes, Restoring Apoptosis in MCF-7 Cells Resistant to Cisplatin. Nutrients 2018, 10, 1148. https://doi.org/10.3390/nu10091148
Hernandez-Valencia J, Garcia-Villa E, Arenas-Hernandez A, Garcia-Mena J, Diaz-Chavez J, Gariglio P. Induction of p53 Phosphorylation at Serine 20 by Resveratrol Is Required to Activate p53 Target Genes, Restoring Apoptosis in MCF-7 Cells Resistant to Cisplatin. Nutrients. 2018; 10(9):1148. https://doi.org/10.3390/nu10091148
Chicago/Turabian StyleHernandez-Valencia, Jorge, Enrique Garcia-Villa, Aquetzalli Arenas-Hernandez, Jaime Garcia-Mena, Jose Diaz-Chavez, and Patricio Gariglio. 2018. "Induction of p53 Phosphorylation at Serine 20 by Resveratrol Is Required to Activate p53 Target Genes, Restoring Apoptosis in MCF-7 Cells Resistant to Cisplatin" Nutrients 10, no. 9: 1148. https://doi.org/10.3390/nu10091148
APA StyleHernandez-Valencia, J., Garcia-Villa, E., Arenas-Hernandez, A., Garcia-Mena, J., Diaz-Chavez, J., & Gariglio, P. (2018). Induction of p53 Phosphorylation at Serine 20 by Resveratrol Is Required to Activate p53 Target Genes, Restoring Apoptosis in MCF-7 Cells Resistant to Cisplatin. Nutrients, 10(9), 1148. https://doi.org/10.3390/nu10091148