Pectolinarigenin Induced Cell Cycle Arrest, Autophagy, and Apoptosis in Gastric Cancer Cell via PI3K/AKT/mTOR Signaling Pathway

 , ,
, , 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Chemicals and Antibodies
2.2. Cell Culture and Cell Viability Assay
2.3. Cell Cycle Distribution and Measurement of Cell Apoptosis
2.4. Annexin V Propidium Iodide Apoptosis Detection
2.5. Morphological Change and Hoechst 33342 Fluorescent Staining
2.6. Analysis of Acidic Vesicular Organelles (AVOs) with Acridine Orange Staining
2.7. Western Blotting Analysis
2.8. RNA Purification and Quantification of mRNAs by Real-Time PCR
2.9. Statistical Analysis
3. Results
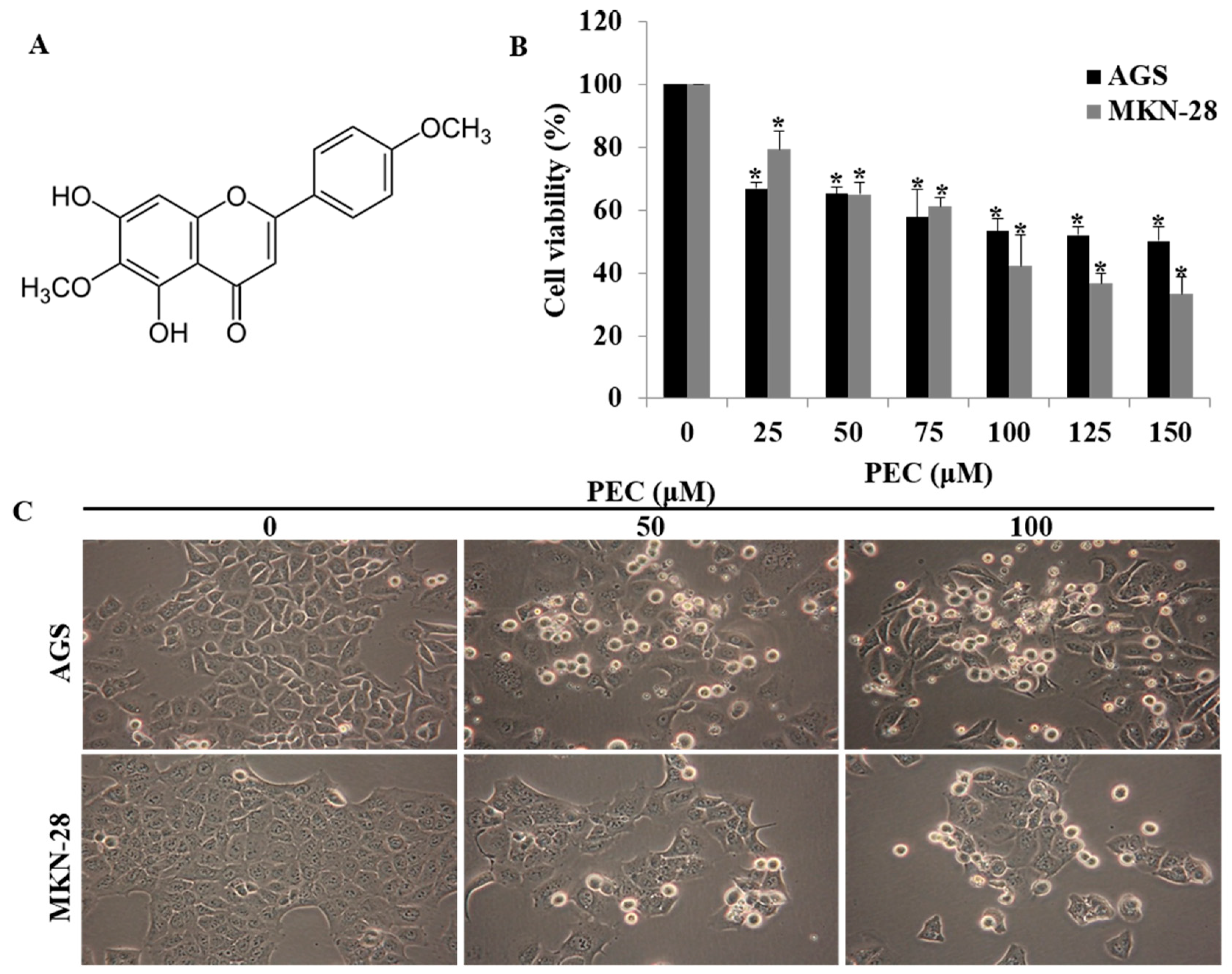
3.1. Pectolinarigenin (PEC) Inhibits Gastric Cancer Cell Growth in a Dose-Dependent Manner
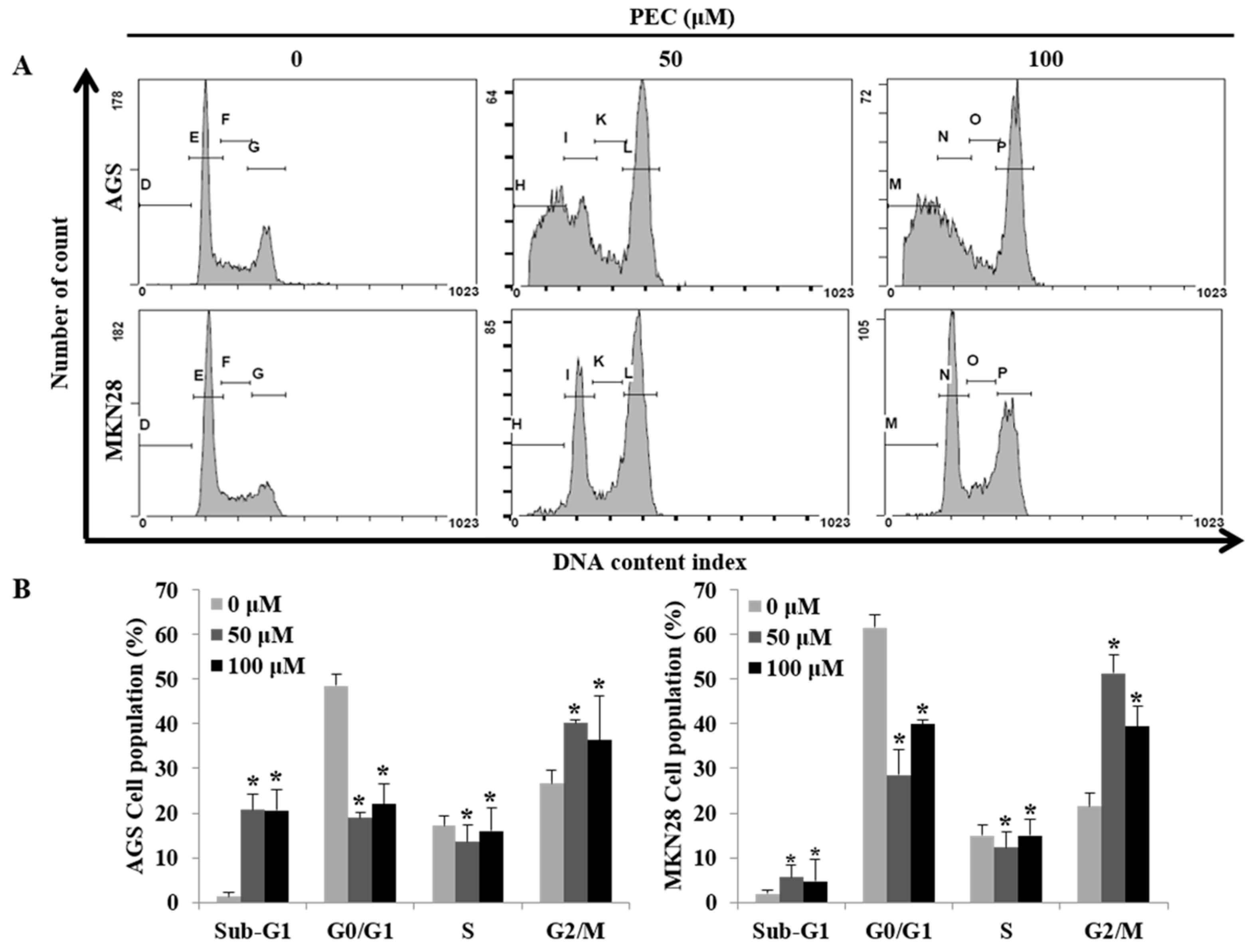
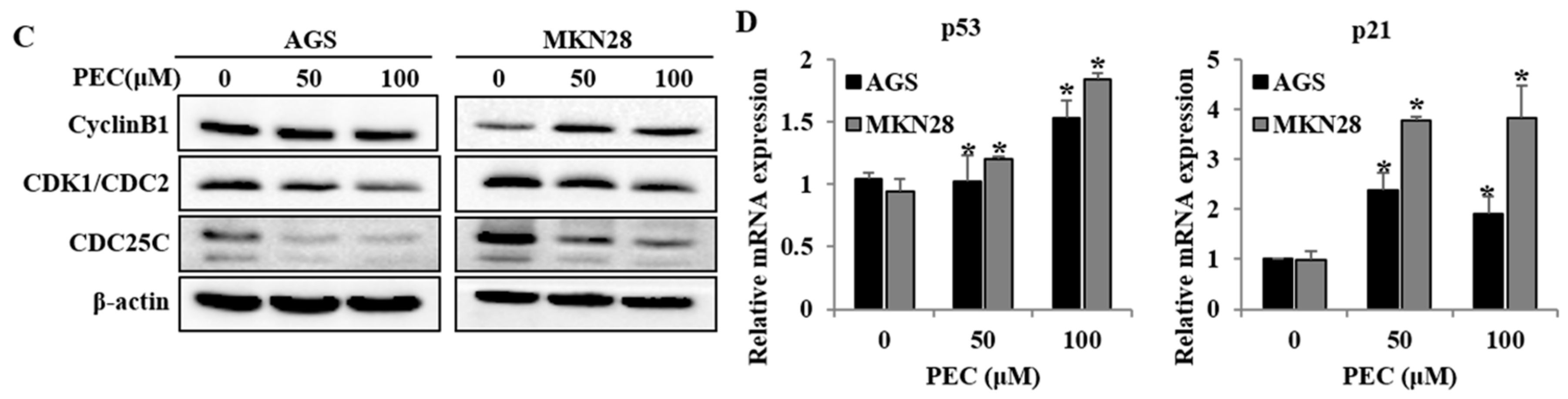
3.2. PEC Induced G2/M Phase Cell Cycle Arrest in AGS and MKN28 Cells and Involved Modulation of the Expression of Cell Cycle Regulators
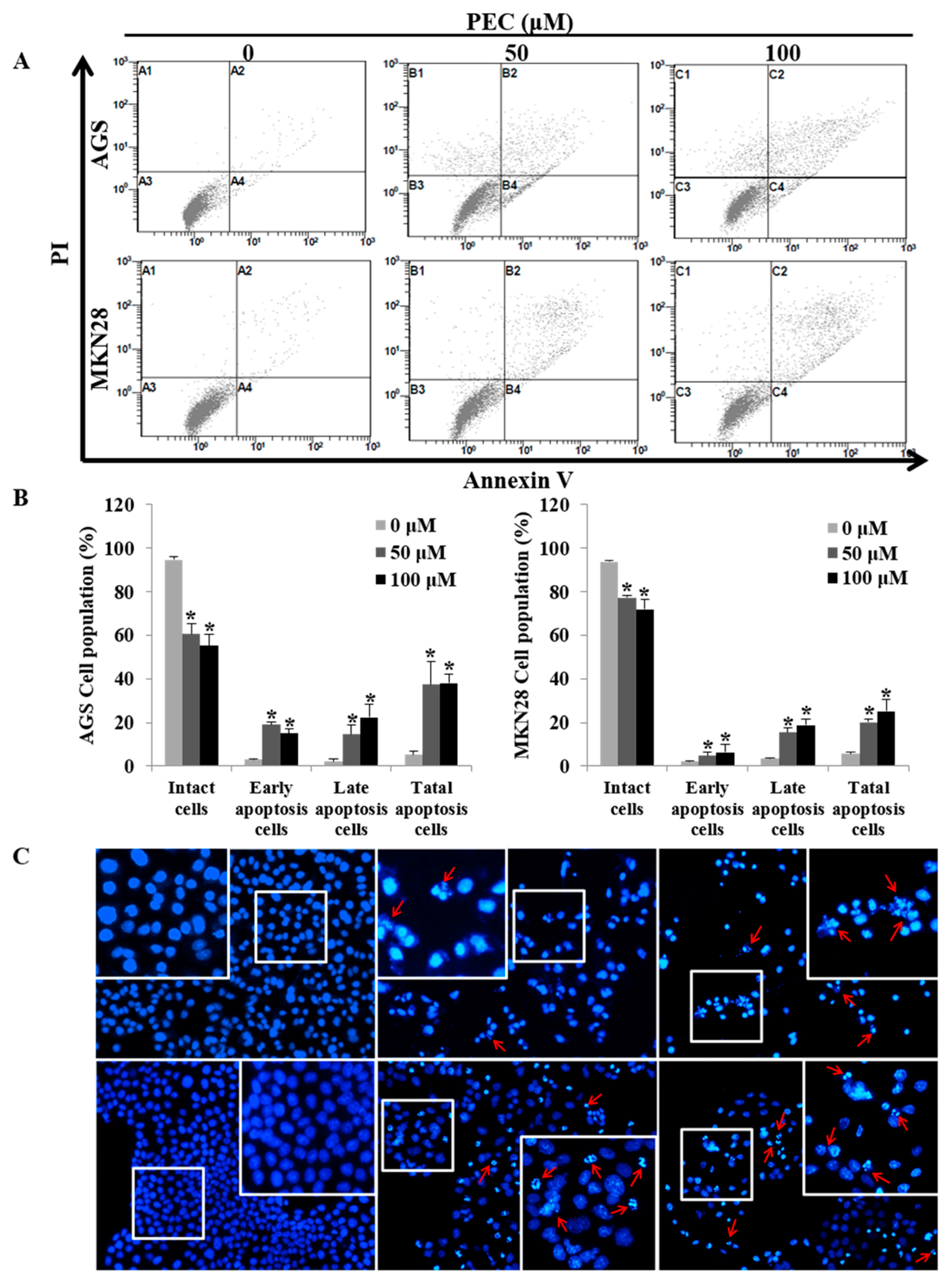
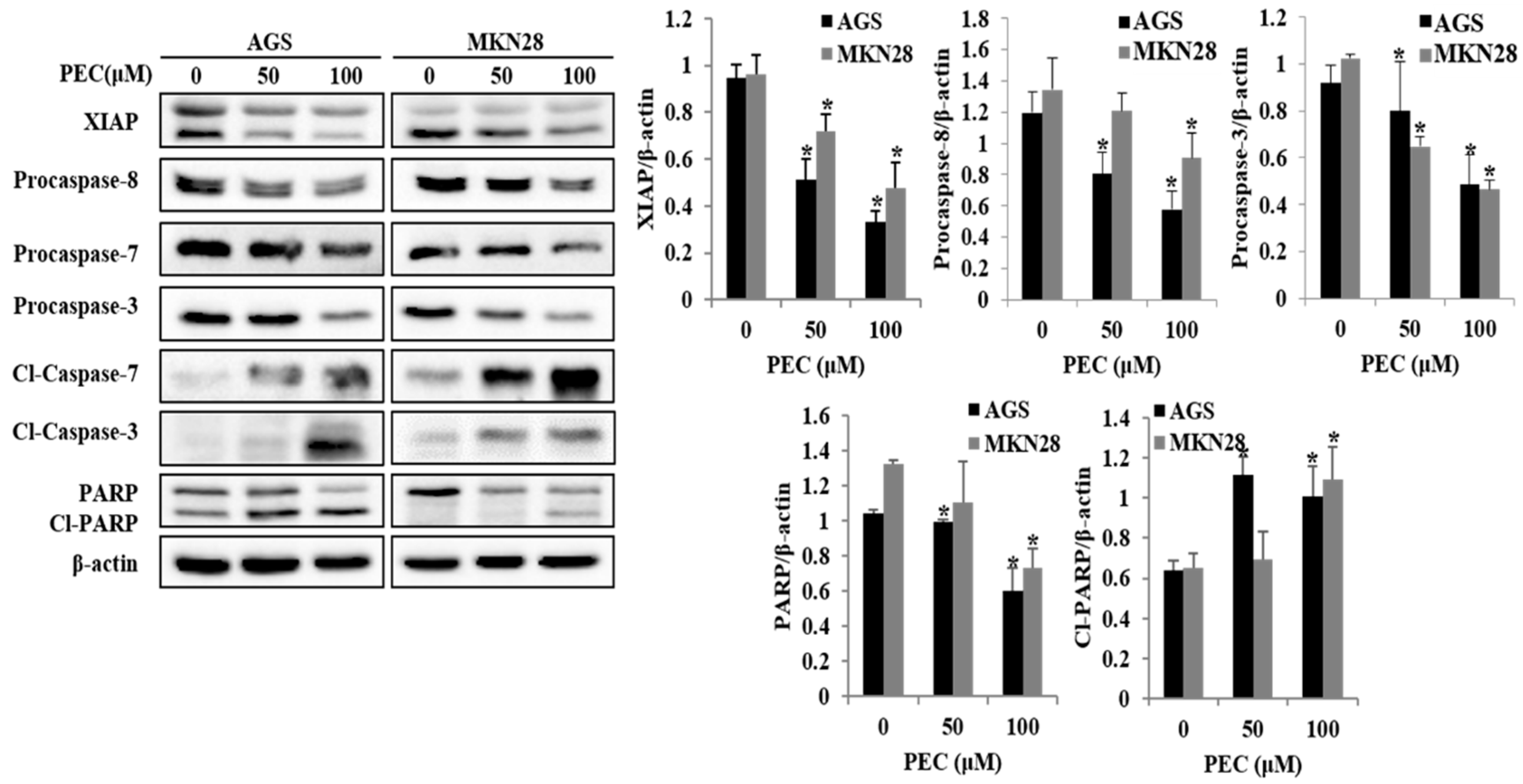
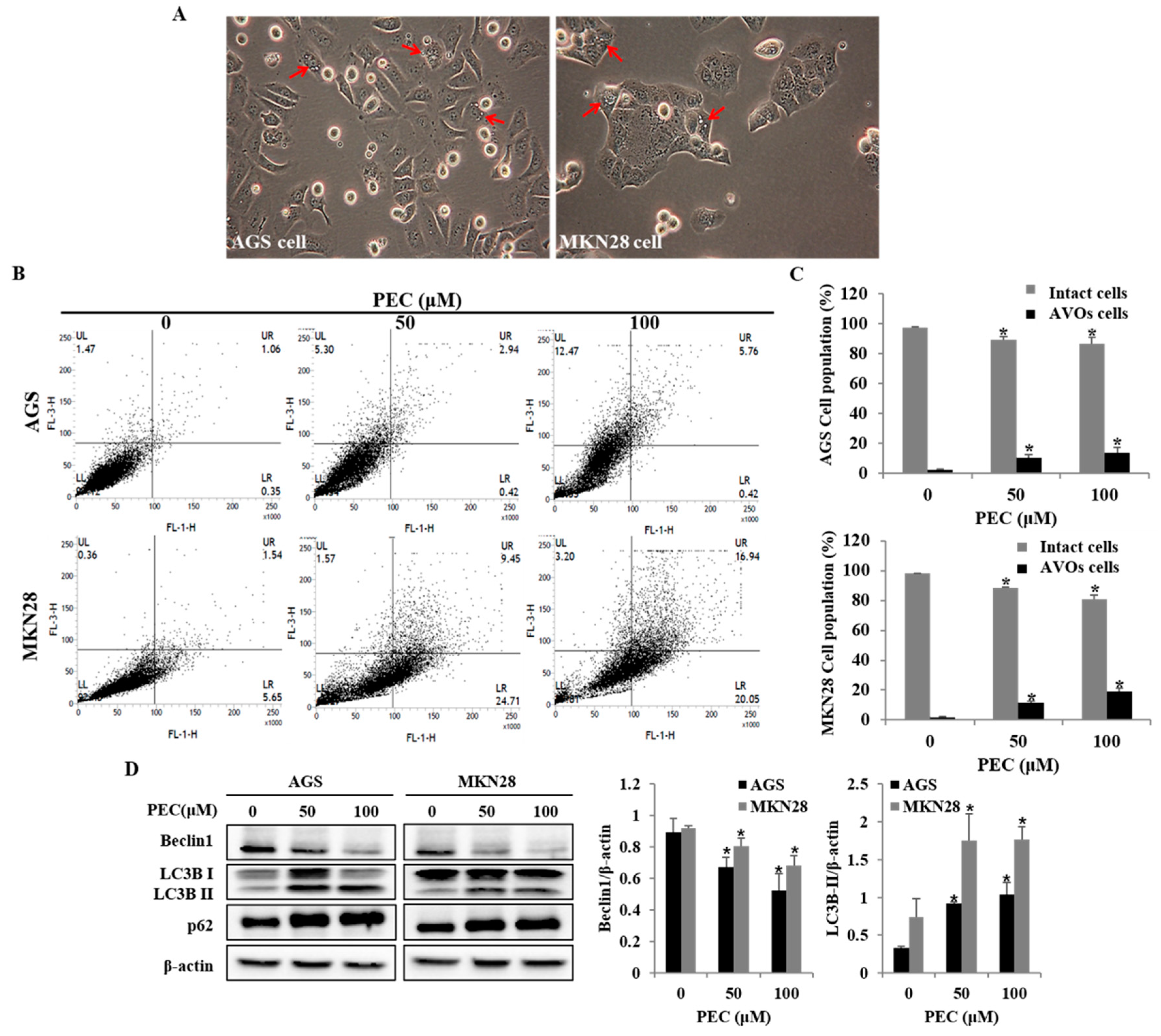
3.3. PEC Induces Apoptosis in AGS and MKN28 Cells
3.4. PEC Induces Autophagy in AGS and MKN28 Cells
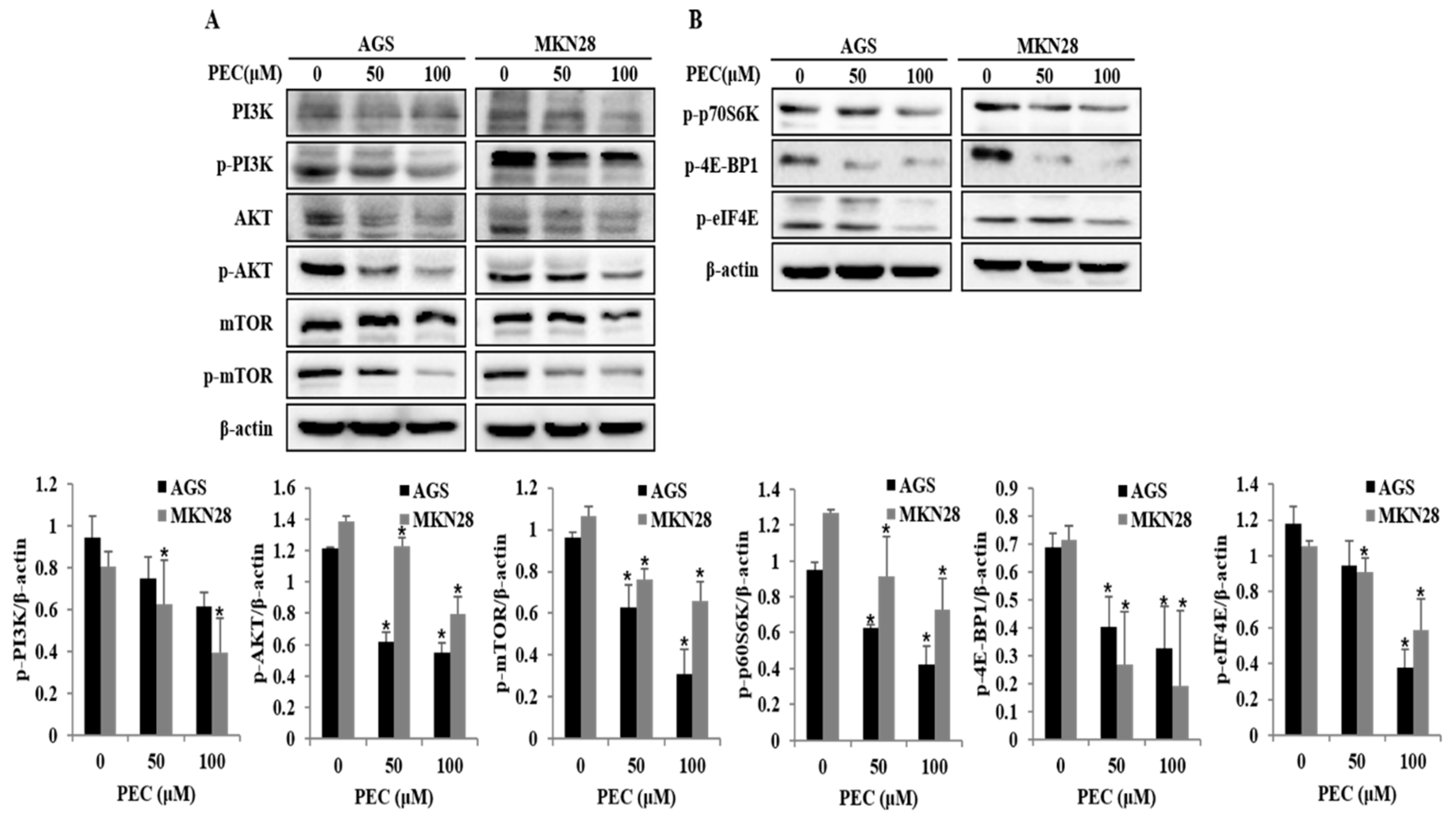
3.5. Effects of PEC on PI3K/AKT/mTOR Signaling Pathway for the Induction of Cell Cycle Arrest, Autophagy, and Apoptosis in AGS and MKN28 Cells
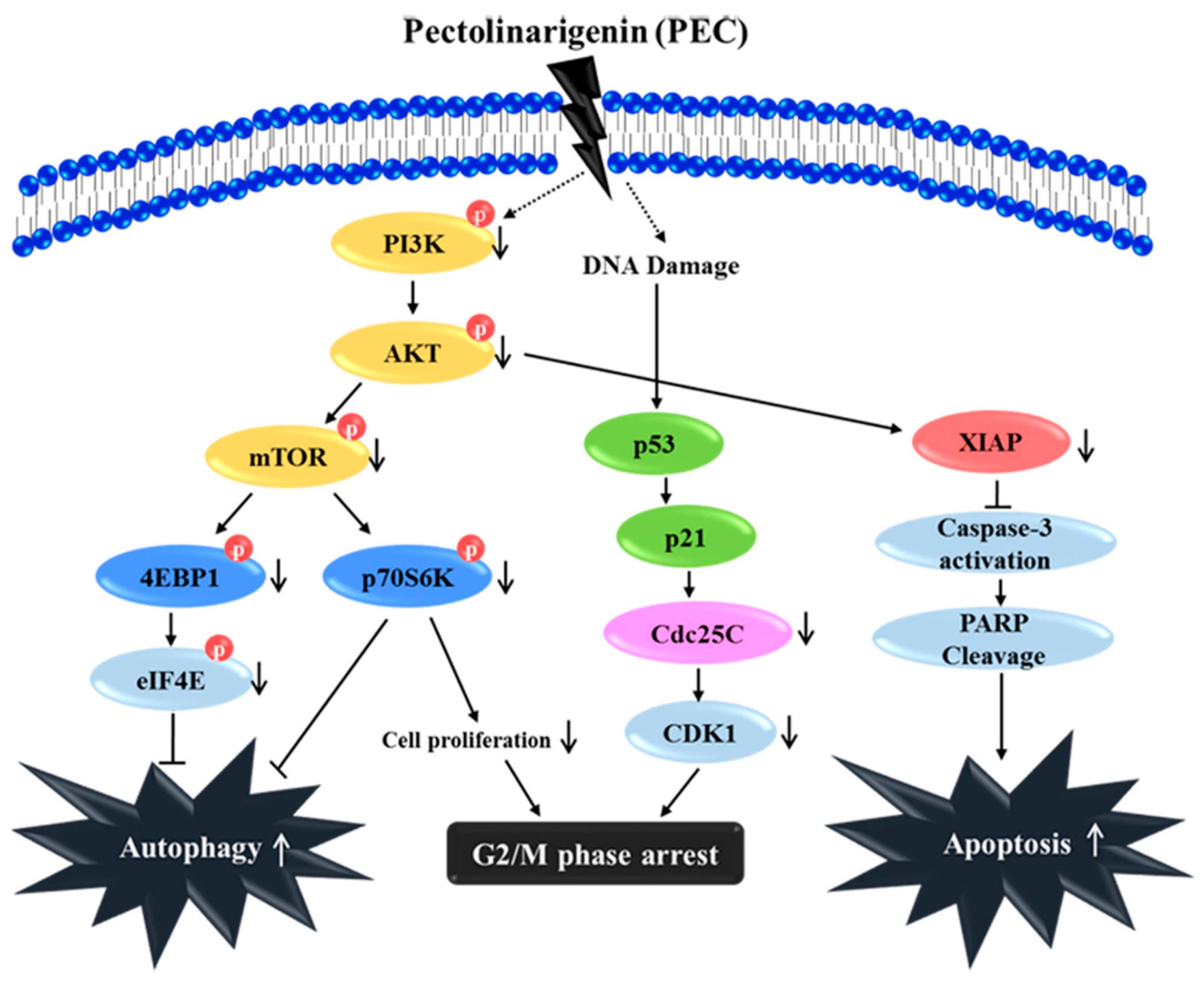
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crew, K.D.; Neugut, A.I. Epidemiology of gastric cancer. World J. Gastroenterol. 2006, 12, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Rocco, A.; Nardone, G.; Diet, H. Pylori infection and gastric cancer: Evidence and controversies. World J. Gastroenterol. 2007, 13, 2901–2912. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Mizumura, K.; Choi, A.M. The impact of autophagy on cell death modalities. Int. J. Cell Biol. 2014, 2014, 502676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chang, J.T.; Guo, B.; Hansen, M.; Jia, K.; Kovacs, A.L.; Kumsta, C.; Lapierre, L.R.; Legouis, R.; Lin, L.; et al. Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy 2015, 11, 9–27. [Google Scholar] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting apoptosis pathways in cancer therapy. Cancer J. Clin. 2005, 55, 178–194. [Google Scholar] [CrossRef]
- Dan, H.C.; Sun, M.; Kaneko, S.; Feldman, R.I.; Nicosia, S.V.; Wang, H.G.; Tsang, B.K.; Cheng, J.Q. Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP). J. Biol. Chem. 2004, 279, 5405–5412. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, K.S.; Siveen, K.S.; Kuttikrishnan, S.; Iskandarani, A.; Tsakou, M.; Achkar, I.W.; Therachiyil, L.; Krishnankutty, R.; Parray, A.; Kulinski, M.; et al. Targeting of X-linked inhibitor of apoptosis protein and PI3-kinase/AKT signaling by embelin suppresses growth of leukemic cells. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Milella, M.; Tsao, T.; McQueen, T.; Schober, W.D.; Hu, W.; Dean, N.M.; Steelman, L.; McCubrey, J.A.; Andreeff, M. Regulation and targeting of antiapoptotic XIAP in acute myeloid leukemia. Leukemia 2003, 17, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Edinger, A.L.; Thompson, C.B. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell 2002, 13, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Garrett, M.D.; Walton, M.I.; Raynaud, F.; de Bono, J.S.; Workman, P. Targeting the PI3K-AKT-mTOR pathway: Progress, pitfalls, and promises. Curr. Opin. Pharmacol. 2008, 8, 393–412. [Google Scholar] [CrossRef] [PubMed]
- Tapia, O.; Riquelme, I.; Leal, P.; Sandoval, A.; Aedo, S.; Weber, H.; Letelier, P.; Bellolio, E.; Villaseca, M.; Garcia, P.; et al. The PI3K/AKT/mTOR pathway is activated in gastric cancer with potential prognostic and predictive significance. Int. J. Pathol. 2014, 465, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Kumar, D.; Shankar, S.; Srivastava, R.K. Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochem. Pharmacol. 2012, 84, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Gontijo, V.S.; Dos Santos, M.H.; Viegas, C., Jr. Biological and chemical aspects of natural biflavonoids from plants: A brief review. Mini Rev. Med. Chem. 2016, 17, 834–862. [Google Scholar] [CrossRef] [PubMed]
- Uzoigwe, J.; Sauter, E.R. Cancer prevention and treatment using combination therapy with plant- and animal-derived compounds. Expert Rev. Clin. Pharmacol. 2012, 5, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.U.; Ryu, S.Y.; Yoon, S.K.; Jung, N.P.; Park, S.H.; Kim, K.H.; Choi, E.J.; Lee, C.O. Effects of flavonoids on the growth and cell cycle of cancer cells. Anticancer Res. 1999, 19, 5229–5233. [Google Scholar] [PubMed]
- Lim, H.; Son, K.H.; Chang, H.W.; Bae, K.; Kang, S.S.; Kim, H.P. Anti-inflammatory activity of pectolinarigenin and pectolinarin isolated from Cirsium chanroenicum. Biol. Pharm. Bull. 2008, 31, 2063–2067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, S.; Li, J.; Yin, F.; Hua, Y.; Wang, Z.; Lin, B.; Wang, H.; Zou, D.; Zhou, Z.; et al. Natural product pectolinarigenin inhibits osteosarcoma growth and metastasis via SHP-1-mediated STAT3 signaling inhibition. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Bonesi, M.; Tundis, R.; Deguin, B.; Loizzo, M.R.; Menichini, F.; Tillequin, F.; Menichini, F. In vitro biological evaluation of novel 7-O-dialkylaminoalkyl cytotoxic pectolinarigenin derivatives against a panel of human cancer cell lines. Bioorganic Med. Chem. Lett. 2008, 18, 5431–5434. [Google Scholar] [CrossRef] [PubMed]
- Erukainure, O.L.; Mesaik, M.A.; Atolani, O.; Muhammad, A.; Chukwuma, C.I.; Islam, M.S. Pectolinarigenin from the leaves of Clerodendrum volubile shows potent immunomodulatory activity by inhibiting T-cell proliferation and modulating respiratory oxidative burst in phagocytes. Biomed. Pharm. 2017, 93, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Yumnam, S.; Hong, G.E.; Lee, H.J.; Saralamma, V.V.; Park, H.S.; Heo, J.D.; Lee, S.J.; Kim, E.H.; Kim, J.A.; et al. Naringin induces autophagy-mediated growth inhibition by downregulating the PI3K/Akt/mTOR cascade via activation of MAPK pathways in AGS cancer cells. Int. J. Oncol. 2015, 47, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Saralamma, V.V.; Nagappan, A.; Hong, G.E.; Lee, H.J.; Yumnam, S.; Raha, S.; Heo, J.D.; Lee, S.J.; Lee, W.S.; Kim, E.H.; et al. Poncirin induces apoptosis in AGS human gastric cancer cells through extrinsic apoptotic pathway by up-regulation of fas ligand. Int. J. Mol. Sci. 2015, 16, 22676–22691. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Nagappan, A.; Park, H.S.; Hong, G.E.; Yumnam, S.; Raha, S.; Saralamma, V.V.; Lee, W.S.; Kim, E.H.; Kim, G.S. Flavonoids isolated from Citrus platymamma induce mitochondrial-dependent apoptosis in AGS cells by modulation of the PI3K/AKT and MAPK pathways. Oncol. Rep. 2015, 34, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Park, K.I.; Park, H.S.; Kang, S.R.; Nagappan, A.; Kim, J.A.; Kim, E.H.; Lee, W.S.; Hah, Y.S.; Chung, H.J.; et al. Flavonoids Isolated from Korea Citrus aurantium L. Induce G2/M Phase arrest and apoptosis in human gastric cancer AGS Cells. Evid. Based Complement. Altern. Med. 2012, 2012. [Google Scholar] [CrossRef]
- Gowda Saralamma, V.V.; Lee, H.J.; Raha, S.; Lee, W.S.; Kim, E.H.; Lee, S.J.; Heo, J.D.; Won, C.; Kang, C.K.; Kim, G.S. Inhibition of IAP’s and activation of p53 leads to caspase-dependent apoptosis in gastric cancer cells treated with Scutellarein. Oncotarget 2018, 9, 5993–6006. [Google Scholar] [PubMed]
- Fan, S.; Ramalingam, S.S.; Kauh, J.; Xu, Z.; Khuri, F.R.; Sun, S.Y. Phosphorylated eukaryotic translation initiation factor 4 (eIF4E) is elevated in human cancer tissues. Cancer Biol. Ther. 2009, 8, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Green, D.; Ponce de Leon, S.; Leon-Rodriguez, E.; Sosa-Sanchez, R. Adenocarcinoma of the stomach: Univariate and multivariate analysis of factors associated with survival. Am. J. Clin. Oncol. 2002, 25, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Sooro, M.A.; Zhang, N.; Zhang, P. Targeting EGFR-mediated autophagy as a potential strategy for cancer therapy. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.J.; Venkatarame Gowda Saralamma, V.; Kim, S.M.; Ha, S.E.; Raha, S.; Lee, W.S.; Kim, E.H.; Lee, S.J.; Heo, J.D.; Kim, G.S. Pectolinarigenin Induced Cell Cycle Arrest, Autophagy, and Apoptosis in Gastric Cancer Cell via PI3K/AKT/mTOR Signaling Pathway. Nutrients 2018, 10, 1043. https://doi.org/10.3390/nu10081043
Lee HJ, Venkatarame Gowda Saralamma V, Kim SM, Ha SE, Raha S, Lee WS, Kim EH, Lee SJ, Heo JD, Kim GS. Pectolinarigenin Induced Cell Cycle Arrest, Autophagy, and Apoptosis in Gastric Cancer Cell via PI3K/AKT/mTOR Signaling Pathway. Nutrients. 2018; 10(8):1043. https://doi.org/10.3390/nu10081043
Chicago/Turabian StyleLee, Ho Jeong, Venu Venkatarame Gowda Saralamma, Seong Min Kim, Sang Eun Ha, Suchismita Raha, Won Sup Lee, Eun Hee Kim, Sang Joon Lee, Jeong Doo Heo, and Gon Sup Kim. 2018. "Pectolinarigenin Induced Cell Cycle Arrest, Autophagy, and Apoptosis in Gastric Cancer Cell via PI3K/AKT/mTOR Signaling Pathway" Nutrients 10, no. 8: 1043. https://doi.org/10.3390/nu10081043
APA StyleLee, H. J., Venkatarame Gowda Saralamma, V., Kim, S. M., Ha, S. E., Raha, S., Lee, W. S., Kim, E. H., Lee, S. J., Heo, J. D., & Kim, G. S. (2018). Pectolinarigenin Induced Cell Cycle Arrest, Autophagy, and Apoptosis in Gastric Cancer Cell via PI3K/AKT/mTOR Signaling Pathway. Nutrients, 10(8), 1043. https://doi.org/10.3390/nu10081043