Understanding the Impact of Dietary Cholesterol on Chronic Metabolic Diseases through Studies in Rodent Models


Abstract
1. Introduction
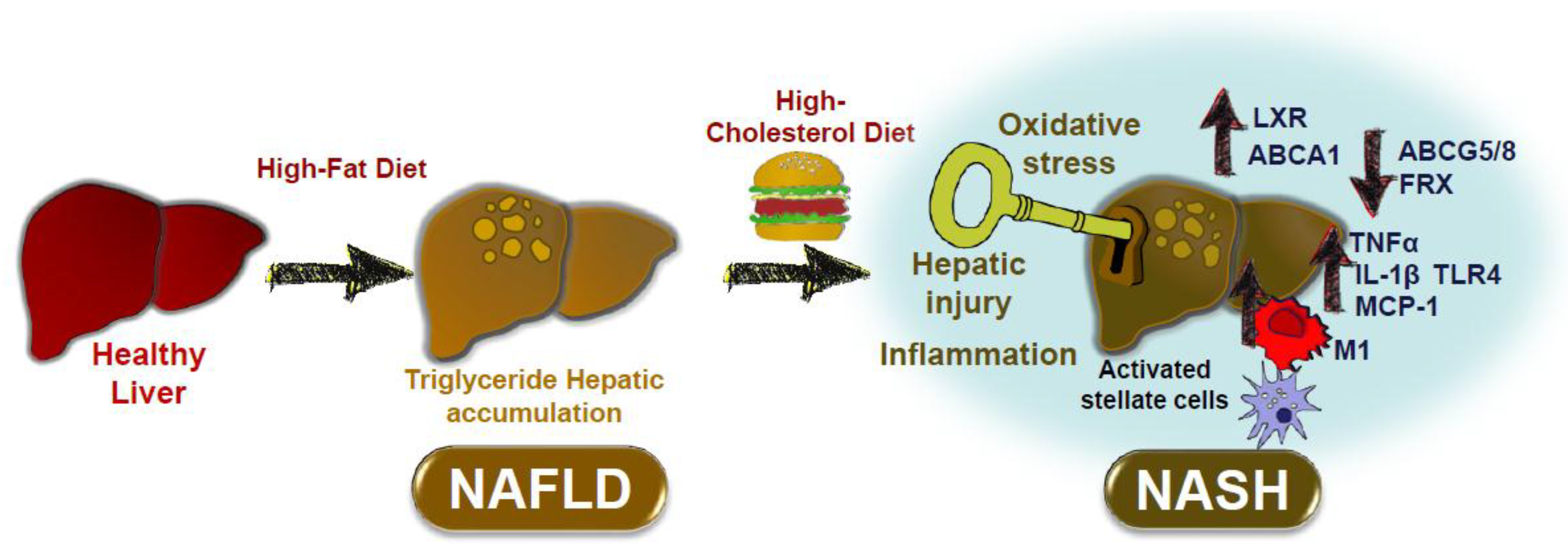
2. Dietary Cholesterol in Non-Alcoholic Fatty Liver Disease in Rodent Models
2.1. Studies in Rodent Models Using Different Cholesterol-Rich Diets
2.1.1. Studies in Rats
2.1.2. Studies in Mice
2.2. Studies in Mice Deficient in Lipid Metabolism Genes in Combination with Cholesterol-Rich Diets
2.2.1. Studies in Lipase-Deficient Mice in Combination with Cholesterol-Rich Diets
2.2.2. Studies in Genetically Modified Mice in Combination with Cholesterol-Rich Diets
Studies in LDLr-Deficient Mice
Studies in NOD.B10 Mice
Studies in apoE-Deficient Mice
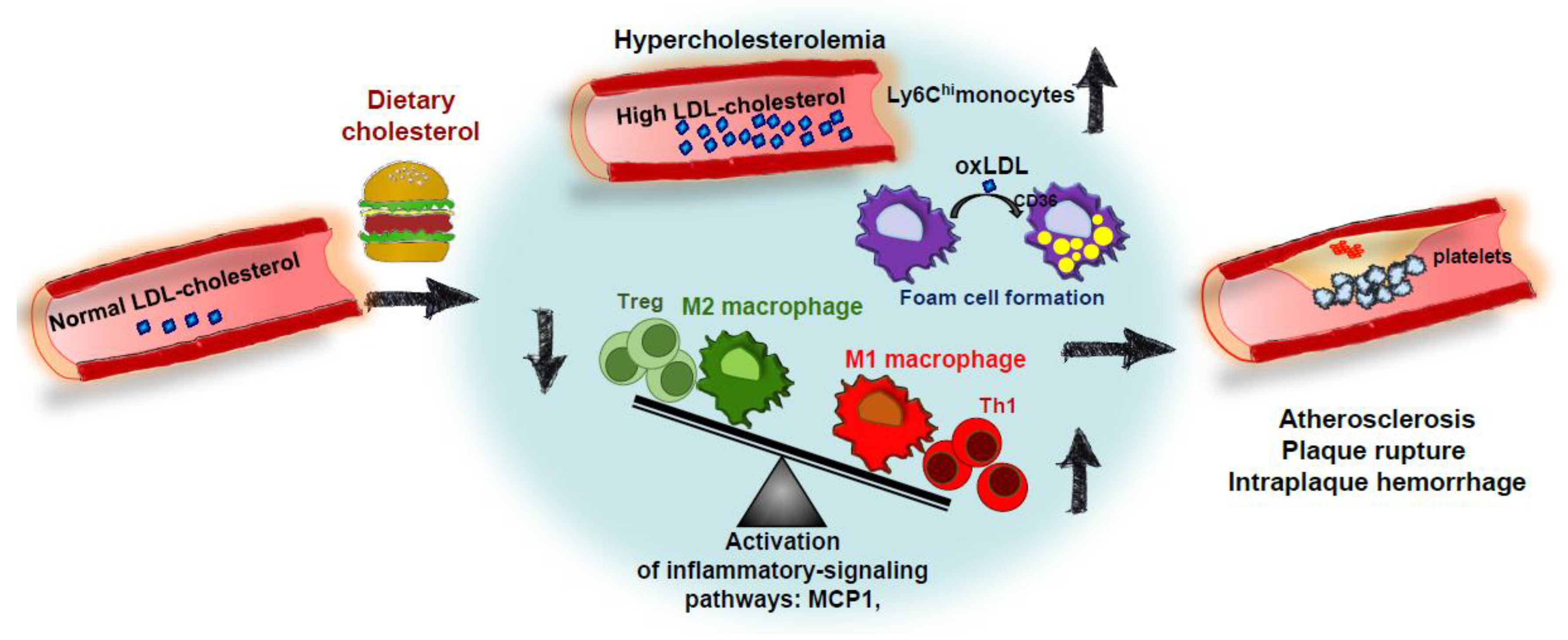
3. The Role of Dietary Cholesterol in the Development of Atherosclerosis in Animal Models
3.1. Impact of Dietary Cholesterol on Atherosclerosis in Mouse Models with an Historic Perspective
3.2. Impact of Dietary Cholesterol in Genetically-Modified Mouse Models of Atherosclerosis
3.2.1. Studies in LDLr-Deficient Mice
3.2.2. Studies in apoE-Deficient Mice
3.2.3. Studies in “Humanized” ApoB-100 Transgenic Mice
3.2.4. Studies in “Humanized” ApoE3*Leiden Transgenic Mice
4. Conclusions: Impact of Dietary Cholesterol on Inflammatory Processes Associated with NAFLD and ATHEROSCLEROSIS
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCG8: | adenosine triphosphate-binding cassette transporter G8 |
| ABCG5: | adenosine triphosphate-binding cassette transporter G5 |
| ApoE: | apolipoprotein E |
| ATGL: | adipose triglyceride lipase |
| CPT: | carnitine palmitoyltransferase, |
| FDFT1: | farnesyldiphosphate farnesyl-transferase 1 |
| FXR: | farnesoid X receptor |
| HMG-CoA-r: | 3-hydroxy-3-methylglutaryl coenzyme A reductase |
| Il2rγ: | interleukin-2 receptor γ |
| LDL: | low-density lipoprotein |
| LDL-C: | low-density lipoprotein cholesterol |
| LDLr: | low-density lipoprotein receptor |
| LXR: | liver X receptor |
| MCP1: | monocyte chemotactic protein 1 |
| MIP2: | macrophage inflammatory protein 2 |
| MTTP: | microsomal triglyceride transfer protein |
| NFkB: | nuclear factor kappa B |
| PDGF-B: | platelet-derived growth factor protein B |
| PPAR-α: | peroxisome proliferator-activated receptor |
| RANTES: | regulated on activation normal T cell expressed and secreted |
| ROS: | reactive oxygen species |
| SAA: | serum amyloid A |
References
- Fernandez, M.L. Rethinking dietary cholesterol. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Wouters, K.; van Gorp, P.J.; Bieghs, V.; Gijbels, M.J.; Duimel, H.; Lutjohann, D.; Kerksiek, A.; van Kruchten, R.; Maeda, N.; Staels, B.; et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 2008, 48, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Morrow, O.B.; Connole, M.L.; Lee, S.P. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology 2009, 50, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Yasutake, K.; Nakamuta, M.; Shima, Y.; Ohyama, A.; Masuda, K.; Haruta, N.; Fujino, T.; Aoyagi, Y.; Fukuizumi, K.; Yoshimoto, T.; et al. Nutritional investigation of non-obese patients with non-alcoholic fatty liver disease: The significance of dietary cholesterol. Scand. J. Gastroenterol. 2009, 44, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; De Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Faga, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, N.; Takamura, T.; Kurita, S.; Misu, H.; Ota, T.; Ando, H.; Yokoyama, M.; Honda, M.; Zen, Y.; Nakanuma, Y.; et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007, 46, 1392–1403. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, C.C. Lymphocytes in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Arbab-Zadeh, A.; Fuster, V. The myth of the “vulnerable plaque”: Transitioning from a focus on individual lesions to atherosclerotic disease burden for coronary artery disease risk assessment. J. Am. Coll. Cardiol. 2015, 65, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Lichtenstein, A.H.; Wu, J.H.Y.; Appel, L.J.; Creager, M.A.; Kris-Etherton, P.M.; Miller, M.; Rimm, E.B.; Rudel, L.L.; Robinson, J.G.; et al. Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association. Circulation 2017, 136, e1–e23. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Does Dietary Cholesterol Matter? Curr. Atheroscler. Rep. 2016, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- McNamara, D.J. Dietary cholesterol, heart disease risk and cognitive dissonance. Proc. Nutr. Soc. 2014, 73, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Barona, J.; Fernandez, M.L. Dietary cholesterol affects plasma lipid levels, the intravascular processing of lipoproteins and reverse cholesterol transport without increasing the risk for heart disease. Nutrients 2012, 4, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Millatt, L.J.; Bocher, V.; Fruchart, J.C.; Staels, B. Liver X receptors and the control of cholesterol homeostasis: Potential therapeutic targets for the treatment of atherosclerosis. Biochim. Biophys. Acta 2003, 1631, 107–118. [Google Scholar] [CrossRef]
- Zhao, C.; Dahlman-Wright, K. Liver X receptor in cholesterol metabolism. J. Endocrinol. 2010, 204, 233–240. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Cote, I.; Ngo Sock, E.T.; Levy, E.; Lavoie, J.M. An atherogenic diet decreases liver FXR gene expression and causes severe hepatic steatosis and hepatic cholesterol accumulation: Effect of endurance training. Eur. J. Nutr. 2013, 52, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, M.; Kawase, M.; Masuzumi, M.; Sakaki, M.; Nagata, Y.; Tanaka, K.; Suruga, K.; Tamaru, S.; Kato, S.; Tsuneyama, K.; et al. High-fat and high-cholesterol diet rapidly induces non-alcoholic steatohepatitis with advanced fibrosis in Sprague-Dawley rats. Hepatol. Res. 2015, 45, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Moriya, T.; Kitamori, K.; Naito, H.; Yanagiba, Y.; Ito, Y.; Yamagishi, N.; Tamada, H.; Jia, X.; Tsuchikura, S.; Ikeda, K.; et al. Simultaneous changes in high-fat and high-cholesterol diet-induced steatohepatitis and severe fibrosis and those underlying molecular mechanisms in novel SHRSP5/Dmcr rat. Environ. Health Prev. Med. 2012, 17, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Yetti, H.; Naito, H.; Jia, X.; Shindo, M.; Taki, H.; Tamada, H.; Kitamori, K.; Hayashi, Y.; Ikeda, K.; Yamori, Y.; et al. High-fat-cholesterol diet mainly induced necrosis in fibrotic steatohepatitis rat by suppressing caspase activity. Life Sci. 2013, 93, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Horai, Y.; Utsumi, H.; Ono, Y.; Kishimoto, T.; Ono, Y.; Fukunari, A. Pathological characterization and morphometric analysis of hepatic lesions in SHRSP5/Dmcr, an experimental non-alcoholic steatohepatitis model, induced by high-fat and high-cholesterol diet. Int. J. Exp. Pathol. 2016, 97, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Savard, C.; Tartaglione, E.V.; Kuver, R.; Haigh, W.G.; Farrell, G.C.; Subramanian, S.; Chait, A.; Yeh, M.M.; Quinn, L.S.; Ioannou, G.N. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013, 57, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Wang, D.Q. Excess cholesterol and fat in the diet: A dangerous liaison for energy expenditure and the liver. Hepatology 2013, 57, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Vergnes, L.; Phan, J.; Strauss, M.; Tafuri, S.; Reue, K. Cholesterol and cholate components of an atherogenic diet induce distinct stages of hepatic inflammatory gene expression. J. Biol. Chem. 2003, 278, 42774–42784. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.S.; Mariscalco, M.M.; Tawil, A.; Vallejo, J.G.; Smith, C.W. Atherogenic diet-induced hepatitis is partially dependent on murine TLR4. J. Leukoc. Biol. 2008, 83, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Sumiyoshi, M.; Sakanaka, M.; Kimura, Y. Chronic intake of a high-cholesterol diet resulted in hepatic steatosis, focal nodular hyperplasia and fibrosis in non-obese mice. Br. J. Nutr. 2010, 103, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Ganz, M.; Bukong, T.N.; Csak, T.; Saha, B.; Park, J.K.; Ambade, A.; Kodys, K.; Szabo, G. Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat-cholesterol-sugar diet model in mice. J. Transl. Med. 2015, 13, 193. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.N.; Showalter, M.R.; Cajka, T.; Fan, S.; Pillai, V.V.; Fiehn, O.; Selvaraj, V. Metabolomic characteristics of cholesterol-induced non-obese nonalcoholic fatty liver disease in mice. Sci. Rep. 2017, 7, 6120. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Wang, S.P.; Alvarez, F.; Casavant, S.; Gauthier, N.; Abed, L.; Soni, K.G.; Yang, G.; Mitchell, G.A. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 2011, 54, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Vegliante, R.; Di Leo, L.; Ciccarone, F.; Ciriolo, M.R. Hints on ATGL implications in cancer: Beyond bioenergetic clues. Cell Death Dis. 2018, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.; Claudel, T.; Baghdasaryan, A.; Mueller, M.; Halilbasic, E.; Das, S.K.; Lass, A.; Zimmermann, R.; Zechner, R.; Hoefler, G.; et al. Role of adipose triglyceride lipase (PNPLA2) in protection from hepatic inflammation in mouse models of steatohepatitis and endotoxemia. Hepatology 2014, 59, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Turpin, S.M.; Hoy, A.J.; Brown, R.D.; Rudaz, C.G.; Honeyman, J.; Matzaris, M.; Watt, M.J. Adipose triacylglycerol lipase is a major regulator of hepatic lipid metabolism but not insulin sensitivity in mice. Diabetologia 2011, 54, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Andres-Blasco, I.; Herrero-Cervera, A.; Vinue, A.; Martinez-Hervas, S.; Piqueras, L.; Sanz, M.J.; Burks, D.J.; Gonzalez-Navarro, H. Hepatic lipase deficiency produces glucose intolerance, inflammation and hepatic steatosis. J. Endocrinol. 2015, 227, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.K.; Qian, K.; Ogimoto, K.; Morton, G.J.; Wisse, B.E.; Agrawal, N.; McDonald, T.O.; Schwartz, M.W.; Dichek, H.L. Mice lacking hepatic lipase are lean and protected against diet-induced obesity and hepatic steatosis. Endocrinology 2010, 151, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Goodspeed, L.; Wang, S.; Kim, J.; Zeng, L.; Ioannou, G.N.; Haigh, W.G.; Yeh, M.M.; Kowdley, K.V.; O’Brien, K.D.; et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J. Lipid Res. 2011, 52, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Prieur, X.; Roszer, T.; Ricote, M. Lipotoxicity in macrophages: Evidence from diseases associated with the metabolic syndrome. Biochim. Biophys. Acta 2010, 1801, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Van Rooyen, D.M.; Larter, C.Z.; Haigh, W.G.; Yeh, M.M.; Ioannou, G.; Kuver, R.; Lee, S.P.; Teoh, N.C.; Farrell, G.C. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 2011, 141, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Schierwagen, R.; Maybuchen, L.; Zimmer, S.; Hittatiya, K.; Back, C.; Klein, S.; Uschner, F.E.; Reul, W.; Boor, P.; Nickenig, G.; et al. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci. Rep. 2015, 5, 12931. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sanabria, F.; Rull, A.; Aragones, G.; Beltran-Debon, R.; Alonso-Villaverde, C.; Camps, J.; Joven, J. Differential response of two models of genetically modified mice fed with high fat and cholesterol diets: Relationship to the study of non-alcoholic steatohepatitis. Mol. Cell. Biochem. 2010, 343, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Kampschulte, M.; Stockl, C.; Langheinrich, A.C.; Althohn, U.; Bohle, R.M.; Krombach, G.A.; Stieger, P.; Churin, Y.; Kremer, S.; Dierkes, C.; et al. Western diet in ApoE-LDLR double-deficient mouse model of atherosclerosis leads to hepatic steatosis, fibrosis, and tumorigenesis. Lab. Investig. 2014, 94, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Endo-Umeda, K.; Nakashima, H.; Komine-Aizawa, S.; Umeda, N.; Seki, S.; Makishima, M. Liver X receptors regulate hepatic F4/80 (+) CD11b(+) Kupffer cells/macrophages and innate immune responses in mice. Sci. Rep. 2018, 8, 9281. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.T.; Mashek, M.T.; Bu, S.Y.; Greenberg, A.S.; Mashek, D.G. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 2011, 53, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.L.; Teijeiro, A.; Buren, S.; Tummala, K.S.; Yilmaz, M.; Waisman, A.; Theurillat, J.P.; Perna, C.; Djouder, N. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016, 30, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Harley, I.T.; Stankiewicz, T.E.; Giles, D.A.; Softic, S.; Flick, L.M.; Cappelletti, M.; Sheridan, R.; Xanthakos, S.A.; Steinbrecher, K.A.; Sartor, R.B.; et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014, 59, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Singh, R.; Wang, Y.; Lefkowitch, J.H.; Rigoli, R.M.; Scherer, P.E.; Czaja, M.J. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology 2006, 43, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Wang, Y.; Xiang, Y.; Tanaka, K.E.; Gaarde, W.A.; Czaja, M.J. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology 2009, 49, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D. In celebration of the 100th anniversary of the lipid hypothesis of atherosclerosis. J. Lipid Res. 2013, 54, 2946–2949. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Lin, H.Y.; Chan, Y.W.; Li, K.H.; To, O.T.; Yan, B.P.; Liu, T.; Li, G.; Wong, W.T.; Keung, W.; et al. Mouse models of atherosclerosis: A historical perspective and recent advances. Lipids Health Dis. 2017, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Paigen, B.; Holmes, P.A.; Mitchell, D.; Albee, D. Comparison of atherosclerotic lesions and HDL-lipid levels in male, female, and testosterone-treated female mice from strains C57BL/6, BALB/c, and C3H. Atherosclerosis 1987, 64, 215–221. [Google Scholar] [CrossRef]
- Ishibashi, S.; Goldstein, J.L.; Brown, M.S.; Herz, J.; Burns, D.K. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J. Clin. Investig. 1994, 93, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, A.H.; Clinton, S.K.; Iiyama, K.; Connelly, P.W.; Libby, P.; Cybulsky, M.I. Hyperlipidemia and atherosclerotic lesion development in LDL receptor-deficient mice fed defined semipurified diets with and without cholate. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1938–1944. [Google Scholar] [CrossRef] [PubMed]
- Hartvigsen, K.; Binder, C.J.; Hansen, L.F.; Rafia, A.; Juliano, J.; Horkko, S.; Steinberg, D.; Palinski, W.; Witztum, J.L.; Li, A.C. A diet-induced hypercholesterolemic murine model to study atherogenesis without obesity and metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Han, C.Y.; Chiba, T.; McMillen, T.S.; Wang, S.A.; Haw, A., 3rd; Kirk, E.A.; O’Brien, K.D.; Chait, A. Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Teupser, D.; Persky, A.D.; Breslow, J.L. Induction of atherosclerosis by low-fat, semisynthetic diets in LDL receptor-deficient C57BL/6J and FVB/NJ mice: Comparison of lesions of the aortic root, brachiocephalic artery, and whole aorta (en face measurement). Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Vikramadithyan, R.; Yu, S.; Pau, C.; Hu, Y.; Goldberg, I.J.; Dansky, H.M. Addition of dietary fat to cholesterol in the diets of LDL receptor knockout mice: Effects on plasma insulin, lipoproteins, and atherosclerosis. J. Lipid Res. 2006, 47, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, W.; Zhang, J.; Lu, Y.; Wu, W.; Yan, H.; Wang, Y. Hyperlipidemia and atherosclerotic lesion development in Ldlr-deficient mice on a long-term high-fat diet. PLoS ONE 2012, 7, e35835. [Google Scholar] [CrossRef] [PubMed]
- Joyce, C.W.; Wagner, E.M.; Basso, F.; Amar, M.J.; Freeman, L.A.; Shamburek, R.D.; Knapper, C.L.; Syed, J.; Wu, J.; Vaisman, B.L.; et al. ABCA1 overexpression in the liver of LDLr-KO mice leads to accumulation of pro-atherogenic lipoproteins and enhanced atherosclerosis. J. Biol. Chem. 2006, 281, 33053–33065. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Kuchibhotla, S.D.; Guy, E.; Park, Y.M.; Nimako, G.; Vanegas, D.; Morton, R.E.; Febbraio, M. Dietary cholesterol plays a role in CD36-mediated atherogenesis in LDLR-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Dadoo, O.; Serkis, V.; Abutouk, D.; MacDonald, M.; Dhingani, N.; Macri, J.; Igdoura, S.A.; Trigatti, B.L. The effects of diet on occlusive coronary artery atherosclerosis and myocardial infarction in scavenger receptor class B, type 1/low-density lipoprotein receptor double knockout mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2394–2403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.H.; Reddick, R.L.; Burkey, B.; Maeda, N. Diet-induced atherosclerosis in mice heterozygous and homozygous for apolipoprotein E gene disruption. J. Clin. Investig. 1994, 94, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Plump, A.S.; Raines, E.W.; Breslow, J.L.; Ross, R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler. Thromb. 1994, 14, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Joyce, C.W.; Amar, M.J.; Lambert, G.; Vaisman, B.L.; Paigen, B.; Najib-Fruchart, J.; Hoyt, R.F., Jr.; Neufeld, E.D.; Remaley, A.T.; Fredrickson, D.S.; et al. The ATP binding cassette transporter A1 (ABCA1) modulates the development of aortic atherosclerosis in C57BL/6 and apoE-knockout mice. Proc. Natl. Acad. Sci. USA 2002, 99, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Jackson, C.L. Atherosclerotic plaque rupture in the apolipoprotein E knockout mouse. Atherosclerosis 2001, 154, 399–406. [Google Scholar] [CrossRef]
- Bond, A.R.; Jackson, C.L. The fat-fed apolipoprotein E knockout mouse brachiocephalic artery in the study of atherosclerotic plaque rupture. J. Biomed. Biotechnol. 2011, 2011, 379069. [Google Scholar] [CrossRef] [PubMed]
- Acin, S.; Navarro, M.A.; Carnicer, R.; Arbones-Mainar, J.M.; Guzman, M.A.; Arnal, C.; Beltran, G.; Uceda, M.; Maeda, N.; Osada, J. Dietary cholesterol suppresses the ability of olive oil to delay the development of atherosclerotic lesions in apolipoprotein E knockout mice. Atherosclerosis 2005, 182, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Mailer, R.K.W.; Gistera, A.; Polyzos, K.A.; Ketelhuth, D.F.J.; Hansson, G.K. Hypercholesterolemia Enhances T Cell Receptor Signaling and Increases the Regulatory T Cell Population. Sci. Rep. 2017, 7, 15655. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Hong, C.; Oka, K.; Salazar, J.V.; Diehl, C.; Witztum, J.L.; Diaz, M.; Castrillo, A.; Bensinger, S.J.; Chan, L.; et al. Cholesterol Accumulation in CD11c(+) Immune Cells Is a Causal and Targetable Factor in Autoimmune Disease. Immunity 2016, 45, 1311–1326. [Google Scholar] [CrossRef] [PubMed]
- Purcell-Huynh, D.A.; Farese, R.V., Jr.; Johnson, D.F.; Flynn, L.M.; Pierotti, V.; Newland, D.L.; Linton, M.F.; Sanan, D.A.; Young, S.G. Transgenic mice expressing high levels of human apolipoprotein B develop severe atherosclerotic lesions in response to a high-fat diet. J. Clin. Investig. 1995, 95, 2246–2257. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.A.; Charbonneau, A.; Avramoglu, R.K.; Pelletier, P.; Fang, X.; Bachelard, H.; Yla-Herttuala, S.; Laakso, M.; Despres, J.P.; Deshaies, Y.; et al. Distinct metabolic and vascular effects of dietary triglycerides and cholesterol in atherosclerotic and diabetic mouse models. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E573–E584. [Google Scholar] [CrossRef] [PubMed]
- van Vlijmen, B.J.; van den Maagdenberg, A.M.; Gijbels, M.J.; van der Boom, H.; HogenEsch, H.; Frants, R.R.; Hofker, M.H.; Havekes, L.M. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J. Clin. Investig. 1994, 93, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, R.; Verschuren, L.; van Erk, M.J.; Nikolsky, Y.; Cnubben, N.H.; Verheij, E.R.; Smilde, A.K.; Hendriks, H.F.; Zadelaar, S.; Smith, G.J.; et al. Atherosclerosis and liver inflammation induced by increased dietary cholesterol intake: A combined transcriptomics and metabolomics analysis. Genome Biol. 2007, 8, R200. [Google Scholar] [CrossRef] [PubMed]
- Paigen, B.; Mitchell, D.; Reue, K.; Morrow, A.; Lusis, A.J.; LeBoeuf, R.C. Ath-1, a gene determining atherosclerosis susceptibility and high density lipoprotein levels in mice. Proc. Natl. Acad. Sci. USA 1987, 84, 3763–3767. [Google Scholar] [CrossRef] [PubMed]
- van der Wulp, M.Y.; Verkade, H.J.; Groen, A.K. Regulation of cholesterol homeostasis. Mol. Cell. Endocrinol. 2013, 368, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R. Functions of cholesterol ester transfer protein and relationship to coronary artery disease risk. J. Clin. Lipidol. 2010, 4, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Plump, A.S.; Smith, J.D.; Hayek, T.; Aalto-Setala, K.; Walsh, A.; Verstuyft, J.G.; Rubin, E.M.; Breslow, J.L. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 1992, 71, 343–353. [Google Scholar] [CrossRef]
- Gijbels, M.J.; van der Cammen, M.; van der Laan, L.J.; Emeis, J.J.; Havekes, L.M.; Hofker, M.H.; Kraal, G. Progression and regression of atherosclerosis in APOE3-Leiden transgenic mice: An immunohistochemical study. Atherosclerosis 1999, 143, 15–25. [Google Scholar] [CrossRef]
- von Scheidt, M.; Zhao, Y.; Kurt, Z.; Pan, C.; Zeng, L.; Yang, X.; Schunkert, H.; Lusis, A.J. Applications and Limitations of Mouse Models for Understanding Human Atherosclerosis. Cell Metab. 2016, 25, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Linton, M.F.; Farese, R.V., Jr.; Chiesa, G.; Grass, D.S.; Chin, P.; Hammer, R.E.; Hobbs, H.H.; Young, S.G. Transgenic mice expressing high plasma concentrations of human apolipoprotein B100 and lipoprotein(a). J. Clin. Investig. 1993, 92, 3029–3037. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; Reardon, C.A. Diet and murine atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Peters, W.; Charo, I.F. Involvement of chemokine receptor 2 and its ligand, monocyte chemoattractant protein-1, in the development of atherosclerosis: Lessons from knockout mice. Curr. Opin. Lipidol. 2001, 12, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S.; Tedgui, A.; Mallat, Z. Adaptive T cell immune responses and atherogenesis. Curr. Opin. Pharmacol. 2010, 10, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Luscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Foks, A.C.; Lichtman, A.H.; Kuiper, J. Treating atherosclerosis with regulatory T cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 280–287. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study/Animal Model | Diet | Age | Stage Disease | Mechanism |
|---|---|---|---|---|
| Cote el al., 2013 [19] Sprague–Dawley rats | 40% fat and 1.25% cholesterol | 8-week-old female | Fatty liver disease | Hepatic accumulation triglycerides and cholesterol Decreased FXRs Lower expression of HMG-CoA-r, FDFT1, and ABCG8 |
| Ichimura et al., 2015 [20] Sprague–Dawley rats | Fat alone or in combination with 1.25% or 2.5% cholesterol | 9-week-old male | Hepatic steatosis | Diminished CPT activity and ABCG5 |
| Moriya et al., 2012 [21] SHRSP5/Dmcr rats | High-fat diet | 10-week-old male | Hepatic fibrotic and inflammatory status of NASH | Altered TNFα proinflammatory cytokine and NFkB pathways |
| Yeti et al., 2013 [22] SHRSP5/Dmcr rats | Fat and cholesterol High-fat high-cholesterol diet | Male SHRSP5/Dmcr rats at 10 weeks old | Phenotype similar to NASH in humans Inflammatory fibrotic liver disease Hepatocyte necrosis | Downregulation of caspase activity |
| Horai et al., 2016 [23] SHRSP5/Dmcr rats | High cholesterol | 6-week-old male rats | Hepatic steatosis, inflammation, and fibrosis | Eosinophilic inclusion bodies and mega-mitochondria |
| Study/Mouse Model | Diet | Age | Stage Disease | Mechanism |
|---|---|---|---|---|
| C57BL/6J | ||||
| Matsuzawa et al., 2007 [8] C57BL/6J mice | 1.25% cholesterol and two different amounts fat (7.5% and 60%) | Males at 6 weeks of age | Insulin resistance Fibrosis Steatohepatitis | Down-regulation of antioxidant enzymes |
| Savard et al., 2013 [24] C57BL/6J | 15% fat or 1% cholesterol | 30 weeks | Hepatic fat accumulation | |
| Neuschwander-Tetri et al., 2013 [25] C57BL/6J mice | 15% fat and 1% cholesterol | 30 weeks | Severe steatosis Inflammation Fibrosis | Inappropriate suppression of fatty acid β-oxidation |
| Vergnes et al., 2003 [26] C57BL/6J | Fat, cholate, and/or cholesterol | At 3 months of age, mice were fed with the specified diet for 3 weeks | Hepatic steatohepatitis | Activation of hepatic stellate cells, SAA family genes, histocompatibility antigens, Il2rγ, Scyb9, Samhd1 |
| Desai et al., 2008 [27] C57BL/6J | 1.25% cholesterol, 0.5% cholic acid, and 16% fat | Males at 8–10 weeks of age were fed for 3 weeks with the diet | Hepatic steatohepatitis | Mononuclear leukocyte infiltration in liver Enhanced MCP1, RANTES, MIP2 |
| Sumiyoshi et al., 2010 [28] C57BL/6 mice | 15% milk fat, 1.5% cholesterol and 0.1% cholic acid | Males at 4 weeks old were fed with diet for 25 or 55 weeks | Hepatic steatosis Fibrosis Tumor formation (focal nodular hyperplasia) | Elevated levels of MCP1 levels and PDGF-B protein |
| Ganz et al., 2015 [29] C57BL/6 | High fat, high cholesterol and high sugar supplement | Males aged 8–10 weeks old were fed with diet for 8, 27, or 49 weeks | Hepatic steatosis at early stage Fibrosis and characteristics of NASH at a late stage | Enhanced levels of MCP1, TNFα, and IL-1β Macrophage polarization toward an M1 |
| Tu et al., 2017 [30] C57BL/6J | High-fat high-cholesterol and cholate diet | Males and females at 8 weeks of age fed their respective diets for 3 weeks | Hepatic pathology similar to NAFLD and NASH | Elevated free cholesterol, cholesterol esters, and cholic acid Changes to metabolism of sphingomyelins and phosphatidylcholines |
| Studies in lipase-deficient mice | ||||
| ATGL-/- [31,32,33,34] | High-fat high-cholesterol diet | 2–12 months old | Severe hepatic steatosis | Activation of inflammatory pathways |
| Andres-Blasco et al., 2015 [35] HL-/- | High-fat high-cholesterol diet | At two months of age, mice were fed for 16 weeks with diet | Hepatic steatosis and liver inflammation | Dyslipidemia Increased NEFA Enhanced macrophages Circulating levels of MCP1 and Th17 T-cell subset |
| Chiu et al., HL-/-, 2010 [36] | High-fat 21% diet and 0.15% cholesterol | Females 21–23 weeks old | Decreased hepatic steatosis | No dyslipidemia and IR |
| Studies in low-density lipoprotein receptor and apolipoprotein E-deficient mice | ||||
| Wouters et al., 2008 [3] LDLr-deficient and apoE2 knock-in | High-fat diet with cholesterol | Males or/and females were fed for 2, 4, 7, and 21 days or for 7 days according to experiments | NASH Hepatic steatosis with inflammation | |
| Subramanian et al., 2011 [37] LDLr-deficient | Fat, carbohydrate and cholesterol | 10-week-old males were fed for 24 weeks with diet | NASH | Macrovesicular steatosis, inflammatory cell foci and fibrosis |
| Prieur et al., 2010 [38] LDLr-deficient mice | Diet enriched in fat, carbohydrate and cholesterol | Males at 10 weeks of age were fed for 24 weeks with diet | Hepatic inflammation | Hepatic macrophage infiltration, apoptosis, and oxidative stress. |
| Van Rooyen et al., 2011 [39]. Alms1 mutant (foz/foz) and wild-type diabetes NOD.B10, | Dietary cholesterol | Females at 8 weeks of age were fed for 12 or 24 weeks with diet | Hepatic free cholesterol accumulation | Increased macrophage, liver apoptosis and fibrosis |
| Schierwagen et al., 2015 [40]. apoE-/- | Western-type diet containing 1.25% of cholesterol | 12 weeks age + 7 weeks diet | Phenotype resembling that of human NASH. | Hepatic fibrosis Upregulation of TGFβ Increased hepatic collagen Activation of hepatic stellate cells |
| Rodríguez Sanabria et al., 2010 [41] apoE-/- vs LDLr-/- | 20% fat and 0.25% cholesterol | Males 10 weeks age + 6 weeks diet | Inflammation vs. fatty liver | Increased macrophage and inflammatory nodules (apolipoprotein E, apoE-/-) vs. hepatic steatosis (LDLr-/-) |
| Kampschulte et al., 2014 [42] apoE-/-LDLr-/- | Western diet containing 5% cholesterol and 21% or regular chow control diet | Males at 4 weeks of age were fed for 35 weeks with diet | Hepatic steatosis Fibrosis Hepatocellular injury | Macrophage and T cell infiltration, hepatic ROS accumulation, JNK activation Induction of PPAR-α |
| Study/Mouse Model | Diet | Age | Stage of Disease |
|---|---|---|---|
| C57BL/6J | |||
| Lee et al., 2017 [51] | 30% fat, 5% cholesterol and 2% cholic acid | Increased serum cholesterol levels Atheroma lesions and extreme hepatic damage | |
| Paigen et al., 1987 [52] C57BL/6 mouse | 1.25% cholesterol, 15% fat, and 0.5% cholic acid (toxic atherogenic diet) | Diet for 14 weeks | Atheroma lesions Discrete hypercholesterolemia |
| Vergnes et al., 2003 [26]. C57BL mice | Different diets (1.25% cholesterol, 0.5% sodium cholate, and 7.5% cocoa butter, and three other diets which omitted one of the three components of the atherogenic diet) | Males at 3 months of age were fed with the diet for 3 weeks | Dietary cholesterol induces the expression of inflammatory genes Cholate induces the expression of extracellular matrix deposition genes such as collagen |
| Studies in LDLr-deficient mice | |||
| Ishibashi et al., 1994 [53]. | 1.25% cholesterol, 7.5% cocoa butter, 7.5% casein, and 0.5% cholic acid | Diet for 6, 7, or 8 months | Severe hypercholesterolemia Atheroma lesions in the vascular wall |
| Lichtman et al., 1999 [54] | 0.5% and 1.25% of cholesterol to a high-fat diet | Males at 8 to 12 weeks of age were fed with the diet for 12 weeks | Lesion plaque formation in a dose-dependent manner |
| Hartvigsen et al., 2007 [55] | Western-type diet, 0.06% cholesterol/21% milk fat, or a cholesterol-enriched diet, 1% cholesterol/4.4% fat | Males were fed with the diet for 28 weeks | Atherosclerotic lesions |
| Subramanian et al., 2008 [56] | Carbohydrate-rich diet with 0.15% cholesterol | Males at 8 weeks old were fed with diet for 24 weeks | Accumulation of macrophages in adipose tissue Acceleration of atherosclerosis |
| Teupser et al. 2003 [57] LDLr-/- mice in C57BL/6J | 4.3% fat in combination with 0.02% 0.15%, 0.30%, or 0.50% cholesterol | Mice at 28 days of age were fed the diet for 16 weeks (20 weeks of age) | Atherosclerosis in aortic root, brachiocephalic artery and whole aorta |
| Wu et al., 2006 [58] | 0.15% cholesterol versus 0.03% cholesterol with high fat in each diet | Diet 20 or 40 weeks | Lesion size was bigger in mice fed 0.15% cholesterol versus 0.03% cholesterol Addition of fat to a cholesterol-rich diet did not increase atherosclerotic lesion hypertriglyceridemia |
| Ma et al., 2012 [59] | 21% fat, 0.15% cholesterol | Males at 8 weeks were fed with the diet for 1, 3, 6, 9, 12 months | Before 3 months: slight atherosclerotic lesions in aortic roots and innominate artery At 3 months: advanced lesions in the aortic sinus At 6–9 months: advanced lesions in the innominate artery |
| Joyce et al., 2006 [60] | 0.02% cholesterol and 4% fat or 0.2% cholesterol and 21.2% fat | Diet 4, 9, or 12 weeks prior to sacrifice | Increased hepatic content of cholesterol and aggravated aortic root atherosclerosis in LDLr-/- mice that overexpressed ABCA1 in the liver |
| Kennedy et al., 2009 [61] LDLr-/-CD36-/- mice | High-cholesterol diet | Diet for 12 weeks | Atherosclerotic lesions |
| Fuller et al., 2014 [62]. LDLr-/- SRBI-/- | High-cholesterol high-fat high-cholesterol high-fat high-cholesterol cholate diet | Females at 10–12 weeks of age were fed 12 weeks with the atherogenic diet | Occluded coronary arteries Accumulation of platelets in atherosclerotic plaque in coronary arteries (thrombosis) Myocardial fibrosis Increased circulating Ly6Chi monocytes |
| Studies in apoE-deficient mice | |||
| Zhang et al., 1994 [63] Partially deficient in apoE | 15.8% fat, 1.25% cholesterol, and 0.5% cholate | Beginning at 8 weeks of age, mice were fed 6 or 12 weeks with the diet | Atheroma lesions |
| Nakashima et al., 1994 [64] | Western-type diet, containing 21% fat, 0.15% cholesterol and 19.5% casein without sodium cholate | Males at 5 weeks of age were fed the diet until 6, 8, 10, 15, 20, 30, and 40 weeks of age | Advanced plaques Development of fibrous cap, presence of smooth muscle cell and adhesion of monocytes |
| Joyce et al., 2002 [65] ABCA1-Tg apoE-/- | 1.25% cholesterol and 0.5% cholic acid | Mice at 2–3 months of age were maintained on the diet for 15 weeks before sacrifice | Overexpression of ABCA1 increased apoB-lipoprotein and HDL levels in plasma and reduced atherosclerosis in vivo |
| Johnson et al., 2001 [66] | 0.15% cholesterol into a high-fat diet | Seven-week-old mice were fed the diet for 14 months | Plaque rupture in the brachiocephalic artery Lesions with intraplaque hemorrhage |
| Bond et al., 2011 [67]. | Cholesterol diet | Lesions in the brachiocephalic artery | |
| Acin et al., 2005 [68] | Different fat-enriched diet and cholesterol with or no unsaturated sources such as extra virgin olive oil (EVOO) | 2-month-old mice were fed with different diets for 10 weeks | Reduced lesion formation with EVOO Cholesterol prevented the beneficial effects of unsaturated fat from EVOO |
| Swirski et al., 2007 [69] | Cholesterol diet | Beginning at 10 weeks of age, mice were fed 20–25 weeks with the diet | Accelerated atherosclerosis by increasing Ly6Chi |
| Mailer et al., 2017 [70] | Cholesterol diet | Mice at 8–10 weeks of age were fed for 4, 12, or 24 weeks with the diet | Accelerated atherosclerosis by activating T cell receptor signaling |
| Ito et al., 2016 (in combination with LXRβ-/-) [71] | Cholesterol diet | Diet for 8, 12, or 16 weeks | Accelerated atherosclerosis and production of autoantibodies and B cell expansion |
| Studies in “humanized” ApoB-100 and ApoE3*Leiden transgenic mice | |||
| Purcell-Huynh et al., 1998 [72] | 16% fat and 1.25% cholesterol | Males and females at 5 weeks of age were fed the diet for 5 or 8 weeks | Severe hypercholesterolemia and atherosclerosis |
| Laplante et al. 2013 [73] LDLr-/-TgApoB100 and LDLr-/TgApoB100IGFII | Standard chow, 0.2% cholesterol diet, high-fat diet or high-fat 0.2% cholesterol diet | Males aged 6 weeks were fed the diet for 6 months | Cholesterol accelerates lesion formation in both LDLr-/-TgApoB100 and LDLr-/-TgApoB100IGFII. |
| Van Vlijmen et al., 1994 [74] ApoE*3Leiden transgenic mice | High cholesterol | At 8–10 weeks old age, mice were fed for 6 weeks with the diet | Hypercholesterolemia and atherosclerotic lesions in the whole aorta and carotid arteries |
| Kleemann et al., 2007 [75] ApoE*3Leiden transgenic mice | 0%, 0.25%, and 1.25% cholesterol | Female E3L mice at 12 weeks old were treated with diet for 10 weeks | Hypercholesterolemia and atherosclerosis |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinué, Á.; Herrero-Cervera, A.; González-Navarro, H. Understanding the Impact of Dietary Cholesterol on Chronic Metabolic Diseases through Studies in Rodent Models. Nutrients 2018, 10, 939. https://doi.org/10.3390/nu10070939
Vinué Á, Herrero-Cervera A, González-Navarro H. Understanding the Impact of Dietary Cholesterol on Chronic Metabolic Diseases through Studies in Rodent Models. Nutrients. 2018; 10(7):939. https://doi.org/10.3390/nu10070939
Chicago/Turabian StyleVinué, Ángela, Andrea Herrero-Cervera, and Herminia González-Navarro. 2018. "Understanding the Impact of Dietary Cholesterol on Chronic Metabolic Diseases through Studies in Rodent Models" Nutrients 10, no. 7: 939. https://doi.org/10.3390/nu10070939
APA StyleVinué, Á., Herrero-Cervera, A., & González-Navarro, H. (2018). Understanding the Impact of Dietary Cholesterol on Chronic Metabolic Diseases through Studies in Rodent Models. Nutrients, 10(7), 939. https://doi.org/10.3390/nu10070939