Fatty Acids and Calcium Regulation in Prostate Cancer


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Dietary Intake and Intracellular Signal Conversion
2.1. Co-Consumption of Fatty Acids and Calcium
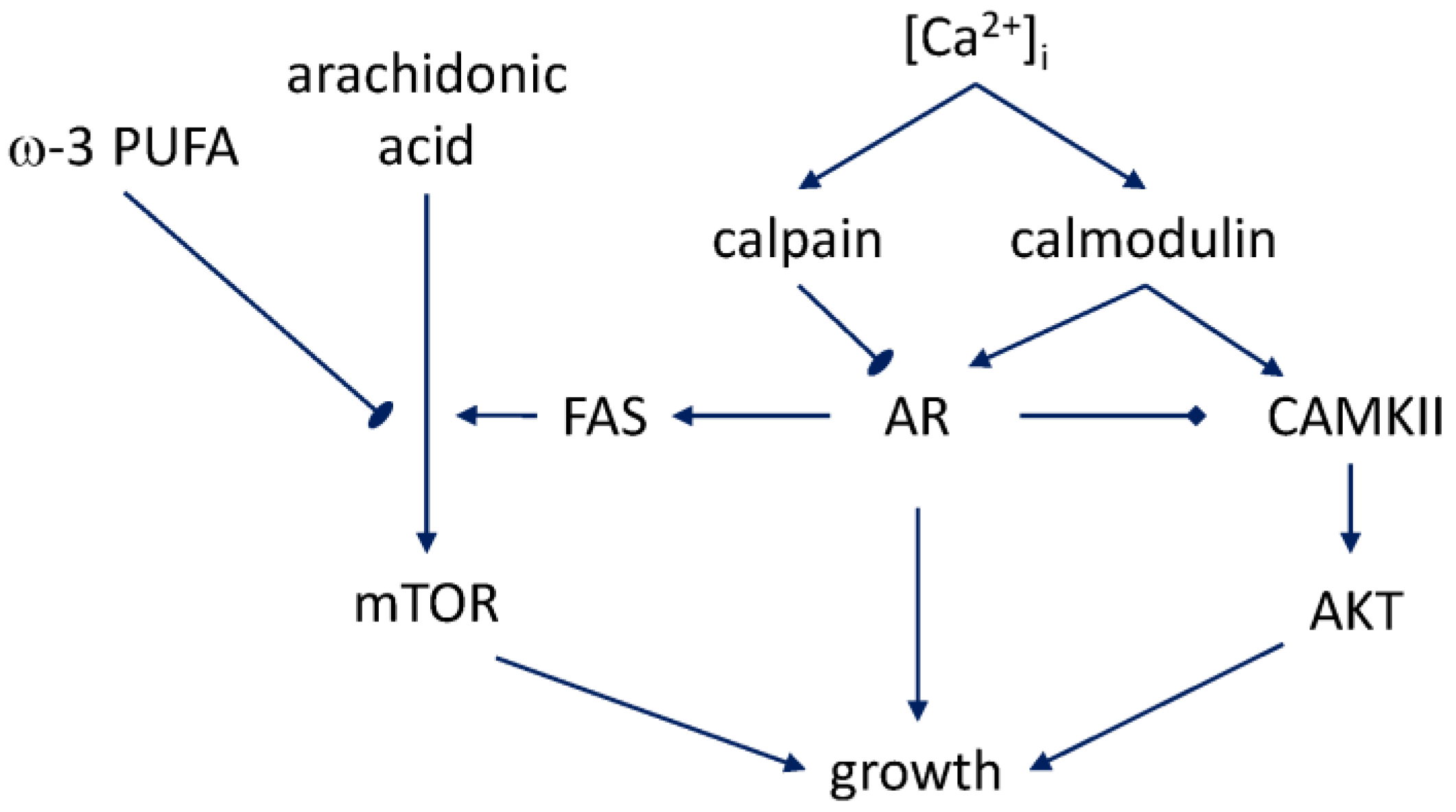
2.2. Conversion to Intracellular Calcium Signaling
3. Mechanisms of Castration Resistance
3.1. First-Line Therapy and the Escape Mechanisms
3.2. Calcium Regulation
3.3. The Feedback Model
3.4. Bidirectional Connection with Fatty Acid Metabolism
4. Resistance to Growth Inhibition
5. Angiogenesis and Hypoxia
5.1. Calcium-Mediated and Fatty Acid-Regulated Hypoxia Responses
5.2. Involvement of Exosomes
5.3. Related HIF-Dependent Mechanisms
5.4. Connection to Castration Resistance
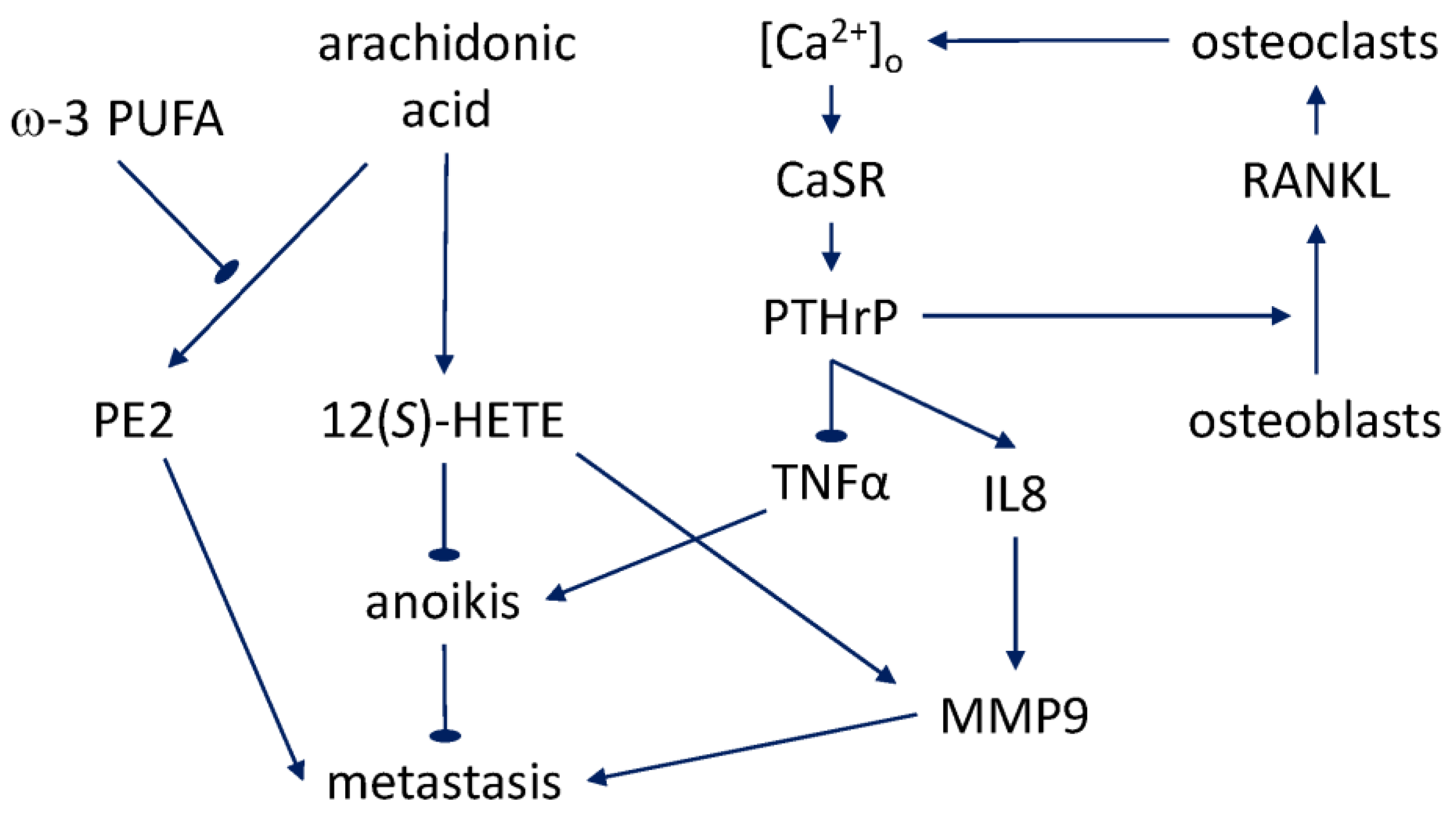
6. Metastasis and Bone Tropism
6.1. Connection with Obesity and Fatty Acid Transfer
6.2. Calcium Sensitivity
6.3. PTHrP Signaling
7. Summary and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AR | Androgen receptor |
| CAMK | Calcium-calmodulin dependent kinase |
| CaSR | Calcium-sensing receptor |
| DHT | Dihydrotestosterone |
| EGF | Epidermal growth factor |
| EGFR | EGF receptor |
| EMT | Epithelial-mesenchymal transition |
| ERK | Extracellular signal-regulated kinase |
| ETE | Eicosatetraenoic (acid) |
| FABP | Fatty acid-binding protein |
| FAS | Fatty acid synthase |
| HETE | Hydroxyeicosatetraenoic (acid) |
| HIF | Hypoxia-induced (transcription) factor |
| HSP | Heat shock protein |
| LDL | Low density lipoprotein |
| LDLR | LDL receptor |
| MMP | Matrix metalloproteases |
| NLS | Nuclear localization signal (sequence) |
| PE2 | Prostaglandin E2 |
| PSA | Prostate-specific antigen |
| PTHrP | Parathyroid hormone-related peptide |
| PUFA | Polyunsaturated fatty acid |
| RANK | Receptor activator of nuclear factor κB |
| RANKL | RANK ligand |
| ROS | Reactive oxygen species |
| SOCE | Store-operated calcium entry |
| TGF | Transforming growth factor |
| TRAMP | Transgenic adenocarcinoma of the mouse prostate |
| VEGF | Vascular endothelial growth factor |
References
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.; Vickers, A. Spotlight on prostate cancer: The latest evidence and current controversies. BMC Med. 2015, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [PubMed]
- Di Sebastiano, K.M.; Mourtzakis, M. The role of dietary fat throughout the prostate cancer trajectory. Nutrients 2014, 6, 6095–6109. [Google Scholar] [CrossRef] [PubMed]
- Berquin, I.M.; Edwards, I.J.; Kridel, S.J.; Chen, Y.Q. Polyunsaturated fatty acid metabolism in prostate cancer. Cancer Metastasis Rev. 2011, 30, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Maly, I.V.; Hofmann, W.A. Calcium and Nuclear Signaling in Prostate Cancer. Int. J. Mol. Sci. 2018, 19, 1237. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, N.; Inoue, M.; Iwasaki, M.; Sasazuki, S.; Tsugane, A.S.; Japan Public Health Center-Based Prospective Study Group. Dairy product, saturated fatty acid, and calcium intake and prostate cancer in a prospective cohort of Japanese men. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, T.; Nagata, Y.; Mori, M.; Miyanaga, N.; Takashima, N.; Okumura, K.; Goto, K.; Naito, S.; Fujimoto, K.; Hirao, Y.; et al. A case-control study of diet and prostate cancer in Japan: Possible protective effect of traditional Japanese diet. Cancer Sci. 2004, 95, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Tat, D.; Kenfield, S.A.; Cowan, J.E.; Broering, J.M.; Carroll, P.R.; Van Blarigan, E.L.; Chan, J.M. Milk and other dairy foods in relation to prostate cancer recurrence: Data from the cancer of the prostate strategic urologic research endeavor (CaPSURE). Prostate 2018, 78, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Kenfield, S.A.; Van Blarigan, E.L.; Wilson, K.M.; Batista, J.L.; Sesso, H.D.; Ma, J.; Stampfer, M.J.; Chavarro, J.E. Dairy intake after prostate cancer diagnosis in relation to disease-specific and total mortality. Int. J. Cancer 2015, 137, 2462–2469. [Google Scholar] [CrossRef] [PubMed]
- Downer, M.K.; Batista, J.L.; Mucci, L.A.; Stampfer, M.J.; Epstein, M.M.; Hakansson, N.; Wolk, A.; Johansson, J.E.; Andren, O.; Fall, K.; et al. Dairy intake in relation to prostate cancer survival. Int. J. Cancer 2017, 140, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Schwedhelm, C.; Galbete, C.; Hoffmann, G. Adherence to Mediterranean Diet and Risk of Cancer: An Updated Systematic Review and Meta-Analysis. Nutrients 2017, 9, 1063. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Hoffmann, G. Mediterranean dietary pattern, inflammation and endothelial function: A systematic review and meta-analysis of intervention trials. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Willett, W.C.; Sacks, F.; Trichopoulou, A.; Drescher, G.; Ferro-Luzzi, A.; Helsing, E.; Trichopoulos, D. Mediterranean diet pyramid: A cultural model for healthy eating. Am. J. Clin. Nutr. 1995, 61, 1402S–1406S. [Google Scholar] [CrossRef] [PubMed]
- Kenfield, S.A.; DuPre, N.; Richman, E.L.; Stampfer, M.J.; Chan, J.M.; Giovannucci, E.L. Mediterranean diet and prostate cancer risk and mortality in the Health Professionals Follow-up Study. Eur. Urol. 2014, 65, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Capurso, C.; Vendemiale, G. The Mediterranean Diet Reduces the Risk and Mortality of the Prostate Cancer: A Narrative Review. Front. Nutr. 2017, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Aune, D.; Navarro Rosenblatt, D.A.; Chan, D.S.; Vieira, A.R.; Vieira, R.; Greenwood, D.C.; Vatten, L.J.; Norat, T. Dairy products, calcium, and prostate cancer risk: A systematic review and meta-analysis of cohort studies. Am. J. Clin. Nutr. 2015, 101, 87–117. [Google Scholar] [CrossRef] [PubMed]
- Niclis, C.; Roman, M.D.; Osella, A.R.; Eynard, A.R.; Diaz Mdel, P. Traditional Dietary Pattern Increases Risk of Prostate Cancer in Argentina: Results of a Multilevel Modeling and Bias Analysis from a Case-Control Study. J. Cancer Epidemiol. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Niclis, C.; Diaz Mdel, P.; Eynard, A.R.; Roman, M.D.; La Vecchia, C. Dietary habits and prostate cancer prevention: A review of observational studies by focusing on South America. Nutr. Cancer 2012, 64, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Diaz, M.P.; Munoz, S.E.; Lantieri, M.J.; Eynard, A.R. Characterization of meat consumption and risk of colorectal cancer in Cordoba, Argentina. Nutrition 2003, 19, 7–10. [Google Scholar] [CrossRef]
- Riserus, U.; Smedman, A.; Basu, S.; Vessby, B. Metabolic effects of conjugated linoleic acid in humans: The Swedish experience. Am. J. Clin. Nutr. 2004, 79, 1146S–1148S. [Google Scholar] [CrossRef] [PubMed]
- Riserus, U.; Vessby, B.; Arnlov, J.; Basu, S. Effects of cis-9,trans-11 conjugated linoleic acid supplementation on insulin sensitivity, lipid peroxidation, and proinflammatory markers in obese men. Am. J. Clin. Nutr. 2004, 80, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Colombel, M.; Filleur, S.; Fournier, P.; Merle, C.; Guglielmi, J.; Courtin, A.; Degeorges, A.; Serre, C.M.; Bouvier, R.; Clezardin, P.; et al. Androgens repress the expression of the angiogenesis inhibitor thrombospondin-1 in normal and neoplastic prostate. Cancer Res. 2005, 65, 300–308. [Google Scholar] [PubMed]
- Firlej, V.; Mathieu, J.R.; Gilbert, C.; Lemonnier, L.; Nakhle, J.; Gallou-Kabani, C.; Guarmit, B.; Morin, A.; Prevarskaya, N.; Delongchamps, N.B.; et al. Thrombospondin-1 triggers cell migration and development of advanced prostate tumors. Cancer Res. 2011, 71, 7649–7658. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 2015, 5, 14752. [Google Scholar] [CrossRef] [PubMed]
- Dramane, G.; Akpona, S.; Besnard, P.; Khan, N.A. Cell mechanisms of gustatory lipids perception and modulation of the dietary fat preference. Biochimie 2014, 107, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Kuda, O.; Jenkins, C.M.; Skinner, J.R.; Moon, S.H.; Su, X.; Gross, R.W.; Abumrad, N.A. CD36 protein is involved in store-operated calcium flux, phospholipase A2 activation, and production of prostaglandin E2. J. Biol. Chem. 2011, 286, 17785–17795. [Google Scholar] [CrossRef] [PubMed]
- Pihlajamaa, P.; Sahu, B.; Janne, O.A. Determinants of Receptor- and Tissue-Specific Actions in Androgen Signaling. Endocr. Rev. 2015, 36, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, P.E.; Tindall, D.J. Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 2011, 10, 20. [Google Scholar] [PubMed]
- Wilson, J.D.; Leihy, M.W.; Shaw, G.; Renfree, M.B. Androgen physiology: Unsolved problems at the millennium. Mol. Cell. Endocrinol. 2002, 198, 1–5. [Google Scholar] [CrossRef]
- Turcu, A.; Smith, J.M.; Auchus, R.; Rainey, W.E. Adrenal androgens and androgen precursors-definition, synthesis, regulation and physiologic actions. Compr. Physiol. 2014, 4, 1369–1381. [Google Scholar] [PubMed]
- Stanbrough, M.; Leav, I.; Kwan, P.W.; Bubley, G.J.; Balk, S.P. Prostatic intraepithelial neoplasia in mice expressing an androgen receptor transgene in prostate epithelium. Proc. Natl. Acad. Sci. USA 2001, 98, 10823–10828. [Google Scholar] [CrossRef] [PubMed]
- Rove, K.O.; Crawford, E.D. Traditional androgen ablation approaches to advanced prostate cancer: New insights. Can. J. Urol. 2014, 21, 14–21. [Google Scholar] [PubMed]
- Watson, P.A.; Ellwood-Yen, K.; King, J.C.; Wongvipat, J.; Lebeau, M.M.; Sawyers, C.L. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 2005, 65, 11565–11571. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Ouyang, X.; Banach-Petrosky, W.A.; Gerald, W.L.; Shen, M.M.; Abate-Shen, C. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 14477–14482. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Altuwaijri, S.; Yeh, S.; Lai, K.P.; Yu, S.; Chuang, K.H.; Huang, S.P.; Lardy, H.; Chang, C. Targeting the stromal androgen receptor in primary prostate tumors at earlier stages. Proc. Natl. Acad. Sci. USA 2008, 105, 12188–12193. [Google Scholar] [CrossRef] [PubMed]
- Mercader, M.; Sengupta, S.; Bodner, B.K.; Manecke, R.G.; Cosar, E.F.; Moser, M.T.; Ballman, K.V.; Wojcik, E.M.; Kwon, E.D. Early effects of pharmacological androgen deprivation in human prostate cancer. BJU Int. 2007, 99, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Dai, B.; Jiang, T.; Xu, K.; Xie, Y.; Kim, O.; Nesheiwat, I.; Kong, X.; Melamed, J.; Handratta, V.D.; et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 2006, 10, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Wang, S.; Qiao, R.; Vivanco, I.; Watson, P.A.; Sawyers, C.L.; Wu, H. Murine cell lines derived from Pten null prostate cancer show the critical role of PTEN in hormone refractory prostate cancer development. Cancer Res. 2007, 67, 6083–6091. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Shimelis, H.; Linn, D.E.; Jiang, R.; Yang, X.; Sun, F.; Guo, Z.; Chen, H.; Li, W.; Chen, H.; et al. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell 2009, 15, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Baek, S.H.; Bourk, E.M.; Ohgi, K.A.; Garcia-Bassets, I.; Sanjo, H.; Akira, S.; Kotol, P.F.; Glass, C.K.; Rosenfeld, M.G.; et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell 2006, 124, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.L.; Tan, W.; Ricono, J.M.; Korchynskyi, O.; Zhang, M.; Gonias, S.L.; Cheresh, D.A.; Karin, M. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature 2007, 446, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Sivanandam, A.; Murthy, S.; Chinnakannu, K.; Bai, V.U.; Kim, S.H.; Barrack, E.R.; Menon, M.; Reddy, G.P. Calmodulin protects androgen receptor from calpain-mediated breakdown in prostate cancer cells. J. Cell. Physiol. 2011, 226, 1889–1896. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, E.; Mataraza, J.M.; Yoshida, B.A.; Menon, M.; Sacks, D.B.; Barrack, E.R.; Reddy, G.P. Physical and functional interaction of androgen receptor with calmodulin in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2004, 101, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Pelley, R.P.; Chinnakannu, K.; Murthy, S.; Strickland, F.M.; Menon, M.; Dou, Q.P.; Barrack, E.R.; Reddy, G.P. Calmodulin-androgen receptor (AR) interaction: Calcium-dependent, calpain-mediated breakdown of AR in LNCaP prostate cancer cells. Cancer Res. 2006, 66, 11754–11762. [Google Scholar] [CrossRef] [PubMed]
- Black, B.E.; Paschal, B.M. Intranuclear organization and function of the androgen receptor. Trends Endocrinol. Metab. 2004, 15, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Johnson, M.; Le, K.H.; Sato, M.; Ilagan, R.; Iyer, M.; Gambhir, S.S.; Wu, L.; Carey, M. Interrogating androgen receptor function in recurrent prostate cancer. Cancer Res. 2003, 63, 4552–4560. [Google Scholar] [PubMed]
- Wu, M.; Kim, S.H.; Datta, I.; Levin, A.; Dyson, G.; Li, J.; Kaypee, S.; Swamy, M.M.; Gupta, N.; Kwon, H.J.; et al. Hydrazinobenzoylcurcumin inhibits androgen receptor activity and growth of castration-resistant prostate cancer in mice. Oncotarget 2015, 6, 6136–6150. [Google Scholar] [CrossRef] [PubMed]
- Rokhlin, O.W.; Taghiyev, A.F.; Bayer, K.U.; Bumcrot, D.; Koteliansk, V.E.; Glover, R.A.; Cohen, M.B. Calcium/calmodulin-dependent kinase II plays an important role in prostate cancer cell survival. Cancer Biol. Ther. 2007, 6, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Logsdon, C.D. S100P: A novel therapeutic target for cancer. Amino Acids 2011, 41, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Mousses, S.; Bubendorf, L.; Wagner, U.; Hostetter, G.; Kononen, J.; Cornelison, R.; Goldberger, N.; Elkahloun, A.G.; Willi, N.; Koivisto, P.; et al. Clinical validation of candidate genes associated with prostate cancer progression in the CWR22 model system using tissue microarrays. Cancer Res. 2002, 62, 1256–1260. [Google Scholar] [PubMed]
- Basu, G.D.; Azorsa, D.O.; Kiefer, J.A.; Rojas, A.M.; Tuzmen, S.; Barrett, M.T.; Trent, J.M.; Kallioniemi, O.; Mousses, S. Functional evidence implicating S100P in prostate cancer progression. Int. J. Cancer 2008, 123, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Averboukh, L.; Liang, P.; Kantoff, P.W.; Pardee, A.B. Regulation of S100P expression by androgen. Prostate 1996, 29, 350–355. [Google Scholar] [CrossRef]
- Amler, L.C.; Agus, D.B.; LeDuc, C.; Sapinoso, M.L.; Fox, W.D.; Kern, S.; Lee, D.; Wang, V.; Leysens, M.; Higgins, B.; et al. Dysregulated expression of androgen-responsive and nonresponsive genes in the androgen-independent prostate cancer xenograft model CWR22-R1. Cancer Res. 2000, 60, 6134–6141. [Google Scholar] [PubMed]
- Wang, Q.; Zhang, J.G.; Wang, W. Expression and significance of S100P, CD147, and OCT4 in different prostate cancer tissue TNM stages. Genet. Mol. Res. 2015, 14, 6844–6851. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.P.; Kaygusuz, G.; Wang, L.; Montgomery, K.; Mason, V.; Zhu, S.X.; Marinelli, R.J.; Presti, J.C., Jr.; van de Rijn, M.; Brooks, J.D. Placental S100 (S100P) and GATA3: Markers for transitional epithelium and urothelial carcinoma discovered by complementary DNA microarray. Am. J. Surg. Pathol. 2007, 31, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.Y.; Fang, Y.; Zhen, L.; Zhu, X.J.; Chen, H.; Liu, H.; Jiang, B.; Li, G.X.; Deng, H.J. Analysis of the predictive efficiency of S100P on adverse prognosis and the pathogenesis of S100P-mediated invasion and metastasis of colon adenocarcinoma. Cancer Genet. 2016, 209, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Dakhel, S.; Padilla, L.; Adan, J.; Masa, M.; Martinez, J.M.; Roque, L.; Coll, T.; Hervas, R.; Calvis, C.; Messeguer, R.; et al. S100P antibody-mediated therapy as a new promising strategy for the treatment of pancreatic cancer. Oncogenesis 2014, 3, e92. [Google Scholar] [CrossRef] [PubMed]
- Soekmadji, C.; Rockstroh, A.; Ramm, G.A.; Nelson, C.C.; Russell, P.J. Extracellular Vesicles in the Adaptive Process of Prostate Cancer during Inhibition of Androgen Receptor Signaling by Enzalutamide. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Fedele, C.; Lu, H.; Nevalainen, M.T.; Keen, J.H.; Languino, L.R. Exosome-mediated Transfer of alphavbeta3 Integrin from Tumorigenic to Nontumorigenic Cells Promotes a Migratory Phenotype. Mol. Cancer Res. 2016, 14, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, C.; Rani, S.; O’Brien, K.; O’Neill, A.; Prencipe, M.; Sheikh, R.; Webb, G.; McDermott, R.; Watson, W.; Crown, J.; et al. Docetaxel-resistance in prostate cancer: Evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PLoS ONE 2012, 7, e50999. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, X.; Guan, H.; Mizokami, A.; Keller, E.T.; Xu, X.; Liu, X.; Tan, J.; Hu, L.; Lu, Y.; et al. Exosome-derived microRNAs contribute to prostate cancer chemoresistance. Int. J. Oncol. 2016, 49, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Soekmadji, C.; Riches, J.D.; Russell, P.J.; Ruelcke, J.E.; McPherson, S.; Wang, C.; Hovens, C.M.; Corcoran, N.M.; Australian Prostate Cancer Collaboration BioResource; Hill, M.M.; et al. Modulation of paracrine signaling by CD9 positive small extracellular vesicles mediates cellular growth of androgen deprived prostate cancer. Oncotarget 2017, 8, 52237–52255. [Google Scholar] [CrossRef] [PubMed]
- Karacosta, L.G.; Foster, B.A.; Azabdaftari, G.; Feliciano, D.M.; Edelman, A.M. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J. Biol. Chem. 2012, 287, 24832–24843. [Google Scholar] [CrossRef] [PubMed]
- Frigo, D.E.; Howe, M.K.; Wittmann, B.M.; Brunner, A.M.; Cushman, I.; Wang, Q.; Brown, M.; Means, A.R.; McDonnell, D.P. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011, 71, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Carmichael, M.; Partin, A.W. OA-519 (fatty acid synthase) as an independent predictor of pathologic state in adenocarcinoma of the prostate. Urology 1995, 45, 81–86. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Esquenet, M.; Goossens, K.; Heyns, W.; Verhoeven, G. Androgens stimulate fatty acid synthase in the human prostate cancer cell line LNCaP. Cancer Res. 1997, 57, 1086–1090. [Google Scholar] [PubMed]
- Swinnen, J.V.; Van Veldhoven, P.P.; Esquenet, M.; Heyns, W.; Verhoeven, G. Androgens markedly stimulate the accumulation of neutral lipids in the human prostatic adenocarcinoma cell line LNCaP. Endocrinology 1996, 137, 4468–4474. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, W.; Ruparel, S.B.; Marciniak, R.A.; deGraffenried, L. Omega-3 fatty acid inhibition of prostate cancer progression to hormone independence is associated with suppression of mTOR signaling and androgen receptor expression. Nutr. Cancer 2011, 63, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Webber, J.P.; Gurney, M.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget 2015, 6, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Ramteke, A.; Ting, H.; Agarwal, C.; Mateen, S.; Somasagara, R.; Hussain, A.; Graner, M.; Frederick, B.; Agarwal, R.; Deep, G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol. Carcinog. 2015, 54, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Bijnsdorp, I.V.; Geldof, A.A.; Lavaei, M.; Piersma, S.R.; van Moorselaar, R.J.; Jimenez, C.R. Exosomal ITGA3 interferes with non-cancerous prostate cell functions and is increased in urine exosomes of metastatic prostate cancer patients. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.Y.; Chang, C.C.; Ohgami, N.; Yamauchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 2006, 22, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Maly, I.V.; Domaradzki, T.M.; Gosy, V.A.; Hofmann, W.A. Myosin isoform expressed in metastatic prostate cancer stimulates cell invasion. Sci. Rep. 2017, 7, 8476. [Google Scholar] [CrossRef] [PubMed]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-beta—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.; Pu, H.; Kyprianou, N. Targeting TGF-beta in prostate cancer: Therapeutic possibilities during tumor progression. Expert Opin. Ther. Targets 2009, 13, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.Y.; Ahn, H.J.; Zelner, D.J.; Shaw, J.W.; Lang, S.; Kato, M.; Oefelein, M.G.; Miyazono, K.; Nemeth, J.A.; Kozlowski, J.M.; et al. Loss of expression of transforming growth factor beta type I and type II receptors correlates with tumor grade in human prostate cancer tissues. Clin. Cancer Res. 1996, 2, 1255–1261. [Google Scholar] [PubMed]
- Guo, Y.; Jacobs, S.C.; Kyprianou, N. Down-regulation of protein and mRNA expression for transforming growth factor-beta (TGF-beta1) type I and type II receptors in human prostate cancer. Int. J. Cancer 1997, 71, 573–579. [Google Scholar] [CrossRef]
- Tu, W.H.; Thomas, T.Z.; Masumori, N.; Bhowmick, N.A.; Gorska, A.E.; Shyr, Y.; Kasper, S.; Case, T.; Roberts, R.L.; Shappell, S.B.; et al. The loss of TGF-beta signaling promotes prostate cancer metastasis. Neoplasia 2003, 5, 267–277. [Google Scholar] [CrossRef]
- Yang, J.; Wahdan-Alaswad, R.; Danielpour, D. Critical role of Smad2 in tumor suppression and transforming growth factor-beta-induced apoptosis of prostate epithelial cells. Cancer Res. 2009, 69, 2185–2190. [Google Scholar] [CrossRef] [PubMed]
- Latil, A.; Pesche, S.; Valeri, A.; Fournier, G.; Cussenot, O.; Lidereau, R. Expression and mutational analysis of the MADR2/Smad2 gene in human prostate cancer. Prostate 1999, 40, 225–231. [Google Scholar] [CrossRef]
- Gizatullina, Z.Z.; Grapengiesser, E.; Shabalina, I.G.; Nedergaard, J.; Heldin, C.H.; Aspenstrom, P. Effect of transforming growth factor-beta on calcium homeostasis in prostate carcinoma cells. Biochem. Biophys. Res. Commun. 2003, 304, 643–649. [Google Scholar] [CrossRef]
- Hahm, S.H.; Cooper, R.H. Transforming growth factor-beta 1 rapidly activates phosphorylase in a calcium-dependent manner in rat hepatocytes. FEBS Lett. 1992, 311, 37–40. [Google Scholar] [CrossRef]
- McGowan, T.A.; Madesh, M.; Zhu, Y.; Wang, L.; Russo, M.; Deelman, L.; Henning, R.; Joseph, S.; Hajnoczky, G.; Sharma, K. TGF-beta-induced Ca2+ influx involves the type III IP3 receptor and regulates actin cytoskeleton. Am. J. Physiol.-Ren. Physiol. 2002, 282, F910–F920. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Kolb, M.R.; Duan, F.; Janssen, L.J. Transforming growth factor-beta evokes Ca2+ waves and enhances gene expression in human pulmonary fibroblasts. Am. J. Respir. Cell Mol. Biol. 2012, 46, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Nesti, L.J.; Caterson, E.J.; Li, W.J.; Chang, R.; McCann, T.D.; Hoek, J.B.; Tuan, R.S. TGF-beta1 calcium signaling in osteoblasts. J. Cell. Biochem. 2007, 101, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Miyazaki, M.; Sonegawa, H.; Kashiwagi, M.; Ohba, M.; Kuroki, T.; Namba, M.; Huh, N.H. PKCalpha mediates TGFbeta-induced growth inhibition of human keratinocytes via phosphorylation of S100C/A11. J. Cell Biol. 2004, 164, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Miyazaki, M.; Takaishi, M.; Sakaguchi, Y.; Makino, E.; Kataoka, N.; Yamada, H.; Namba, M.; Huh, N.H. S100C/A11 is a key mediator of Ca2+-induced growth inhibition of human epidermal keratinocytes. J. Cell Biol. 2003, 163, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Sonegawa, H.; Nukui, T.; Sakaguchi, Y.; Miyazaki, M.; Namba, M.; Huh, N.H. Bifurcated converging pathways for high Ca2+- and TGFbeta-induced inhibition of growth of normal human keratinocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 13921–13926. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Schveigert, D.; Valuckas, K.P.; Kovalcis, V.; Ulys, A.; Chvatovic, G.; Didziapetriene, J. Significance of MMP-9 expression and MMP-9 polymorphism in prostate cancer. Tumori 2013, 99, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Maly, I.V.; Hofmann, W.A. Calcium-regulated import of myosin IC into the nucleus. Cytoskeleton 2016, 73, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Wikstrom, P.; Stattin, P.; Franck-Lissbrant, I.; Damber, J.E.; Bergh, A. Transforming growth factor beta1 is associated with angiogenesis, metastasis, and poor clinical outcome in prostate cancer. Prostate 1998, 37, 19–29. [Google Scholar] [CrossRef]
- Yu, N.; Kozlowski, J.M.; Park, I.I.; Chen, L.; Zhang, Q.; Xu, D.; Doll, J.A.; Crawford, S.E.; Brendler, C.B.; Lee, C. Overexpression of transforming growth factor beta1 in malignant prostate cells is partly caused by a runaway of TGF-beta1 auto-induction mediated through a defective recruitment of protein phosphatase 2A by TGF-beta type I receptor. Urology 2010, 76, 1519.e8–1519.e13. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Cornelius, S.C.; Pultz, N.J.; Jorgensen, J.S.; Bonham, M.J.; Kim, S.J.; Danielpour, D. The androgen receptor represses transforming growth factor-beta signaling through interaction with Smad3. J. Biol. Chem. 2002, 277, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.A.; Zarnegar, M.; Sharma, M.; Yang, F.; Peehl, D.M.; ten Dijke, P.; Sun, Z. SMAD3 represses androgen receptor-mediated transcription. Cancer Res. 2001, 61, 2112–2118. [Google Scholar] [PubMed]
- Kang, H.Y.; Huang, K.E.; Chang, S.Y.; Ma, W.L.; Lin, W.J.; Chang, C. Differential modulation of androgen receptor-mediated transactivation by Smad3 and tumor suppressor Smad4. J. Biol. Chem. 2002, 277, 43749–43756. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Wang, H.; Krebs, T.L.; Kim, S.J.; Danielpour, D. Androgenic control of transforming growth factor-beta signaling in prostate epithelial cells through transcriptional suppression of transforming growth factor-beta receptor II. Cancer Res. 2008, 68, 8173–8182. [Google Scholar] [CrossRef] [PubMed]
- Pu, H.; Begemann, D.E.; Kyprianou, N. Aberrant TGF-beta Signaling Drives Castration-Resistant Prostate Cancer in a Male Mouse Model of Prostate Tumorigenesis. Endocrinology 2017, 158, 1612–1622. [Google Scholar] [CrossRef] [PubMed]
- Miles, F.L.; Tung, N.S.; Aguiar, A.A.; Kurtoglu, S.; Sikes, R.A. Increased TGF-beta1-mediated suppression of growth and motility in castrate-resistant prostate cancer cells is consistent with Smad2/3 signaling. Prostate 2012, 72, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Lekas, A.; Lazaris, A.C.; Deliveliotis, C.; Chrisofos, M.; Zoubouli, C.; Lapas, D.; Papathomas, T.; Fokitis, I.; Nakopoulou, L. The expression of hypoxia-inducible factor-1alpha (HIF-1alpha) and angiogenesis markers in hyperplastic and malignant prostate tissue. Anticancer Res. 2006, 26, 2989–2993. [Google Scholar] [PubMed]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Mottet, D.; Michel, G.; Renard, P.; Ninane, N.; Raes, M.; Michiels, C. Role of ERK and calcium in the hypoxia-induced activation of HIF-1. J. Cell. Physiol. 2003, 194, 30–44. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, A.P.; De Vries, G.W. Role of PLCgamma and Ca2+ in VEGF- and FGF-induced choroidal endothelial cell proliferation. Am. J. Physiol. Cell Physiol. 2001, 281, C1448–C1456. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, M.P.; Takhar, M.K.; Nason, R.; Santacruz, S.; Tam, K.J.; Massah, S.; Haegert, A.; Bell, R.H.; Altamirano-Dimas, M.; Collins, C.C.; et al. The retinoblastoma protein regulates hypoxia-inducible genetic programs, tumor cell invasiveness and neuroendocrine differentiation in prostate cancer cells. Oncotarget 2016, 7, 24284–24302. [Google Scholar] [CrossRef] [PubMed]
- Calviello, G.; Di Nicuolo, F.; Gragnoli, S.; Piccioni, E.; Serini, S.; Maggiano, N.; Tringali, G.; Navarra, P.; Ranelletti, F.O.; Palozza, P. n-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE2 induced ERK-1 and -2 and HIF-1alpha induction pathway. Carcinogenesis 2004, 25, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Ambring, A.; Johansson, M.; Axelsen, M.; Gan, L.; Strandvik, B.; Friberg, P. Mediterranean-inspired diet lowers the ratio of serum phospholipid n-6 to n-3 fatty acids, the number of leukocytes and platelets, and vascular endothelial growth factor in healthy subjects. Am. J. Clin. Nutr. 2006, 83, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, M.; Murota, S.I.; Morita, I. Docosapentaenoic acid (22:5, n-3) suppressed tube-forming activity in endothelial cells induced by vascular endothelial growth factor. Prostagland. Leuk. Essent. Fat. Acids 2003, 68, 337–342. [Google Scholar] [CrossRef]
- Tsuzuki, T.; Shibata, A.; Kawakami, Y.; Nakagawa, K.; Miyazawa, T. Conjugated eicosapentaenoic acid inhibits vascular endothelial growth factor-induced angiogenesis by suppressing the migration of human umbilical vein endothelial cells. J. Nutr. 2007, 137, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Panigrahi, G.K. Hypoxia-Induced Signaling Promotes Prostate Cancer Progression: Exosomes Role as Messenger of Hypoxic Response in Tumor Microenvironment. Crit. Rev. Oncog. 2015, 20, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, G.K.; Deep, G. Exosomes-based biomarker discovery for diagnosis and prognosis of prostate cancer. Front. Biosci. 2017, 22, 1682–1696. [Google Scholar]
- Miyake, H.; Hara, I.; Arakawa, S.; Kamidono, S. Stress protein GRP78 prevents apoptosis induced by calcium ionophore, ionomycin, but not by glycosylation inhibitor, tunicamycin, in human prostate cancer cells. J. Cell. Biochem. 2000, 77, 396–408. [Google Scholar] [CrossRef]
- Dai, Y.; Bae, K.; Siemann, D.W. Impact of hypoxia on the metastatic potential of human prostate cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zahringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Gorlach, A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Chadderton, N.; Cowen, R.L.; Sheppard, F.C.; Robinson, S.; Greco, O.; Scott, S.D.; Stratford, I.J.; Patterson, A.V.; Williams, K.J. Dual responsive promoters to target therapeutic gene expression to radiation-resistant hypoxic tumor cells. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Dayal, R.; Singh, A.; Pandey, A.; Mishra, K.P. Reactive oxygen species as mediator of tumor radiosensitivity. J. Cancer Res. Ther. 2014, 10, 811–818. [Google Scholar] [PubMed]
- Jayadevappa, R.; Chhatre, S.; Wong, Y.N.; Wittink, M.N.; Cook, R.; Morales, K.H.; Vapiwala, N.; Newman, D.K.; Guzzo, T.; Wein, A.J.; et al. Comparative effectiveness of prostate cancer treatments for patient-centered outcomes: A systematic review and meta-analysis (PRISMA Compliant). Medicine 2017, 96, e6790. [Google Scholar] [CrossRef] [PubMed]
- Jamieson-Gladney, W.L.; Zhang, Y.; Fong, A.M.; Meucci, O.; Fatatis, A. The chemokine receptor CX(3)CR1 is directly involved in the arrest of breast cancer cells to the skeleton. Breast Cancer Res. 2011, 13, R91. [Google Scholar] [CrossRef] [PubMed]
- Shulby, S.A.; Dolloff, N.G.; Stearns, M.E.; Meucci, O.; Fatatis, A. CX3CR1-fractalkine expression regulates cellular mechanisms involved in adhesion, migration, and survival of human prostate cancer cells. Cancer Res. 2004, 64, 4693–4698. [Google Scholar] [CrossRef] [PubMed]
- Nevo, I.; Sagi-Assif, O.; Meshel, T.; Ben-Baruch, A.; Johrer, K.; Greil, R.; Trejo, L.E.; Kharenko, O.; Feinmesser, M.; Yron, I.; et al. The involvement of the fractalkine receptor in the transmigration of neuroblastoma cells through bone-marrow endothelial cells. Cancer Lett. 2009, 273, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.J.; Chen, Y.Y.; Lin, P.; Zou, H.F.; Lin, F.; Zhao, L.N.; Li, D.; Guo, L.; Tang, J.B.; Zheng, X.L.; et al. Hypoxia increases CX3CR1 expression via HIF-1 and NFkappaB in androgen-independent prostate cancer cells. Int. J. Oncol. 2012, 41, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Salnikow, K.; Costa, M.; Figg, W.D.; Blagosklonny, M.V. Hyperinducibility of hypoxia-responsive genes without p53/p21-dependent checkpoint in aggressive prostate cancer. Cancer Res. 2000, 60, 5630–5634. [Google Scholar] [PubMed]
- Eliasson, P.; Jonsson, J.I. The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J. Cell. Physiol. 2010, 222, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.C.; Sadek, H.A. Hypoxia and metabolic properties of hematopoietic stem cells. Antioxid Redox Signal. 2014, 20, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Peng, L.; Tang, Y.; Zhang, L.; Guo, W.; Zou, X.; Peng, X. Hypoxia of PC-3 prostate cancer cells enhances migration and vasculogenesis in vitro of bone marrow-derived endothelial progenitor cells by secretion of cytokines. Oncol. Rep. 2013, 29, 2369–2377. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.P.; Wu, K.J. Hypoxia-regulated target genes implicated in tumor metastasis. J. Biomed. Sci. 2012, 19, 102. [Google Scholar] [CrossRef] [PubMed]
- Hermani, A.; Hess, J.; De Servi, B.; Medunjanin, S.; Grobholz, R.; Trojan, L.; Angel, P.; Mayer, D. Calcium-binding proteins S100A8 and S100A9 as novel diagnostic markers in human prostate cancer. Clin. Cancer Res. 2005, 11, 5146–5152. [Google Scholar] [CrossRef] [PubMed]
- Grebhardt, S.; Veltkamp, C.; Strobel, P.; Mayer, D. Hypoxia and HIF-1 increase S100A8 and S100A9 expression in prostate cancer. Int. J. Cancer 2012, 131, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Tenbrock, K.; Ludwig, S.; Leukert, N.; Ehrhardt, C.; van Zoelen, M.A.; Nacken, W.; Foell, D.; van der Poll, T.; Sorg, C.; et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 2007, 13, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, G. S100A8 and S100A9: New insights into their roles in malignancy. J. Innate Immun. 2012, 4, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, J.; Stein, I.; Haag, D.; Riehl, A.; Longerich, T.; Horwitz, E.; Breuhahn, K.; Gebhardt, C.; Schirmacher, P.; Hahn, M.; et al. S100A8 and S100A9 are novel nuclear factor kappa B target genes during malignant progression of murine and human liver carcinogenesis. Hepatology 2009, 50, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Byrne, N.M.; Nesbitt, H.; Ming, L.; McKeown, S.R.; Worthington, J.; McKenna, D.J. Androgen deprivation in LNCaP prostate tumour xenografts induces vascular changes and hypoxic stress, resulting in promotion of epithelial-to-mesenchymal transition. Br. J. Cancer 2016, 114, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Halin, S.; Hammarsten, P.; Wikstrom, P.; Bergh, A. Androgen-insensitive prostate cancer cells transiently respond to castration treatment when growing in an androgen-dependent prostate environment. Prostate 2007, 67, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Shabsigh, A.; Ghafar, M.A.; de la Taille, A.; Burchardt, M.; Kaplan, S.A.; Anastasiadis, A.G.; Buttyan, R. Biomarker analysis demonstrates a hypoxic environment in the castrated rat ventral prostate gland. J. Cell. Biochem. 2001, 81, 437–444. [Google Scholar] [CrossRef]
- Mitani, T.; Yamaji, R.; Higashimura, Y.; Harada, N.; Nakano, Y.; Inui, H. Hypoxia enhances transcriptional activity of androgen receptor through hypoxia-inducible factor-1alpha in a low androgen environment. J. Steroid Biochem. Mol. Biol. 2011, 123, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Horii, K.; Suzuki, Y.; Kondo, Y.; Akimoto, M.; Nishimura, T.; Yamabe, Y.; Sakaue, M.; Sano, T.; Kitagawa, T.; Himeno, S.; et al. Androgen-dependent gene expression of prostate-specific antigen is enhanced synergistically by hypoxia in human prostate cancer cells. Mol. Cancer Res. 2007, 5, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, W.K.; Xiao, L.; Kovac, S.; Chang, M.; Michiels, C.; Bolton, D.; Shulkes, A.; Baldwin, G.S.; Patel, O. The role of hypoxia-inducible factor 1alpha in determining the properties of castrate-resistant prostate cancers. PLoS ONE 2013, 8, e54251. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.C.; Tille, J.C.; Combescure, C.; Egger, J.F.; Laouiti, M.; Hammad, K.; Granger, P.; Rubbia-Brandt, L.; Miralbell, R. The prognostic value of expression of HIF1alpha, EGFR and VEGF-A, in localized prostate cancer for intermediate- and high-risk patients treated with radiation therapy with or without androgen deprivation therapy. Radiat. Oncol. 2012, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, W.K.; Baldwin, G.S.; Bolton, D.; Shulkes, A.; Ischia, J.; Patel, O. HIF1alpha expression under normoxia in prostate cancer—Which pathways to target? J. Urol. 2015, 193, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Marin-Hernandez, A.; Gallardo-Perez, J.C.; Ralph, S.J.; Rodriguez-Enriquez, S.; Moreno-Sanchez, R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, W.K.; Sengupta, S.; Williams, S.; Chang, M.; Shulkes, A.; Bolton, D.M.; Baldwin, G.; Patel, O. The effects of nonspecific HIF1alpha inhibitors on development of castrate resistance and metastases in prostate cancer. Cancer Med. 2014, 3, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Freedland, S.J.; Banez, L.L.; Sun, L.L.; Fitzsimons, N.J.; Moul, J.W. Obese men have higher-grade and larger tumors: An analysis of the duke prostate center database. Prostate Cancer Prostatic Dis. 2009, 12, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Hardaway, A.L.; Herroon, M.K.; Rajagurubandara, E.; Podgorski, I. Bone marrow fat: Linking adipocyte-induced inflammation with skeletal metastases. Cancer Metastasis Rev. 2014, 33, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Agalliu, I.; Lin, D.W.; Stanford, J.L.; Kristal, A.R. Obesity is associated with increased risks of prostate cancer metastasis and death after initial cancer diagnosis in middle-aged men. Cancer 2007, 109, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Keto, C.J.; Aronson, W.J.; Terris, M.K.; Presti, J.C.; Kane, C.J.; Amling, C.L.; Freedland, S.J. Obesity is associated with castration-resistant disease and metastasis in men treated with androgen deprivation therapy after radical prostatectomy: Results from the SEARCH database. BJU Int. 2012, 110, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, A.; Satoh, Y.; Tokuda, Y.; Fujiyama, C.; Udo, K.; Uozumi, J. Effects of adipocytes on the proliferation and differentiation of prostate cancer cells in a 3-D culture model. Int. J. Urol. 2010, 17, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Gazi, E.; Gardner, P.; Lockyer, N.P.; Hart, C.A.; Brown, M.D.; Clarke, N.W. Direct evidence of lipid translocation between adipocytes and prostate cancer cells with imaging FTIR microspectroscopy. J. Lipid Res. 2007, 48, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Hart, C.A.; Gazi, E.; Bagley, S.; Clarke, N.W. Promotion of prostatic metastatic migration towards human bone marrow stoma by Omega 6 and its inhibition by Omega 3 PUFAs. Br. J. Cancer 2006, 94, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Karmali, R.A.; Reichel, P.; Cohen, L.A.; Terano, T.; Hirai, A.; Tamura, Y.; Yoshida, S. The effects of dietary omega-3 fatty acids on the DU-145 transplantable human prostatic tumor. Anticancer Res. 1987, 7, 1173–1179. [Google Scholar] [PubMed]
- Rose, D.P. Effects of dietary fatty acids on breast and prostate cancers: Evidence from in vitro experiments and animal studies. Am. J. Clin. Nutr. 1997, 66, 1513S–1522S. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J. Rapid induction of apoptosis in prostate cancer cells by selenium: Reversal by metabolites of arachidonate 5-lipoxygenase. Biochem. Biophys. Res. Commun. 2004, 315, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Myers, C.E. Inhibition of arachidonate 5-lipoxygenase triggers massive apoptosis in human prostate cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 13182–13187. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Myers, C.E. Arachidonic acid stimulates prostate cancer cell growth: Critical role of 5-lipoxygenase. Biochem. Biophys. Res. Commun. 1997, 235, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, S.; Ghosh, J. Expression of 5-oxoETE receptor in prostate cancer cells: Critical role in survival. Biochem. Biophys. Res. Commun. 2006, 339, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Sarveswaran, S.; Ghosh, J. OXER1, a G protein-coupled oxoeicosatetraenoid receptor, mediates the survival-promoting effects of arachidonate 5-lipoxygenase in prostate cancer cells. Cancer Lett. 2013, 336, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Sarveswaran, S.; Thamilselvan, V.; Brodie, C.; Ghosh, J. Inhibition of 5-lipoxygenase triggers apoptosis in prostate cancer cells via down-regulation of protein kinase C-epsilon. Biochim. Biophys. Acta 2011, 1813, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Kalyvianaki, K.; Gebhart, V.; Peroulis, N.; Panagiotopoulou, C.; Kiagiadaki, F.; Pediaditakis, I.; Aivaliotis, M.; Moustou, E.; Tzardi, M.; Notas, G.; et al. Antagonizing effects of membrane-acting androgens on the eicosanoid receptor OXER1 in prostate cancer. Sci. Rep. 2017, 7, 44418. [Google Scholar] [CrossRef] [PubMed]
- Dilly, A.K.; Ekambaram, P.; Guo, Y.; Cai, Y.; Tucker, S.C.; Fridman, R.; Kandouz, M.; Honn, K.V. Platelet-type 12-lipoxygenase induces MMP9 expression and cellular invasion via activation of PI3K/Akt/NF-kappaB. Int. J. Cancer 2013, 133, 1784–1791. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.P.; Selman, S.H.; Jankun, J. Vascular endothelial growth factor production in human prostate cancer cells is stimulated by overexpression of platelet 12-lipoxygenase. Prostate 2006, 66, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Nie, D.; Krishnamoorthy, S.; Jin, R.; Tang, K.; Chen, Y.; Qiao, Y.; Zacharek, A.; Guo, Y.; Milanini, J.; Pages, G.; et al. Mechanisms regulating tumor angiogenesis by 12-lipoxygenase in prostate cancer cells. J. Biol. Chem. 2006, 281, 18601–18609. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kim, J.H. Activation of the leukotriene B4 receptor 2-reactive oxygen species (BLT2-ROS) cascade following detachment confers anoikis resistance in prostate cancer cells. J. Biol. Chem. 2013, 288, 30054–30063. [Google Scholar] [CrossRef] [PubMed]
- Herroon, M.K.; Rajagurubandara, E.; Hardaway, A.L.; Powell, K.; Turchick, A.; Feldmann, D.; Podgorski, I. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget 2013, 4, 2108–2123. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Narita, S.; Inoue, T.; Koizumi, A.; Saito, M.; Tsuruta, H.; Numakura, K.; Satoh, S.; Nanjo, H.; Sasaki, T.; et al. Fatty acid binding protein 4 enhances prostate cancer progression by upregulating matrix metalloproteinases and stromal cell cytokine production. Oncotarget 2017, 8, 111780–111794. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G. Prostate cancer, serum parathyroid hormone, and the progression of skeletal metastases. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Schneider, A.; Datta, N.S.; McCauley, L.K. Extracellular calcium as a candidate mediator of prostate cancer skeletal metastasis. Cancer Res. 2006, 66, 9065–9073. [Google Scholar] [CrossRef] [PubMed]
- Ahearn, T.U.; Tchrakian, N.; Wilson, K.M.; Lis, R.; Nuttall, E.; Sesso, H.D.; Loda, M.; Giovannucci, E.; Mucci, L.A.; Finn, S.; et al. Calcium-Sensing Receptor Tumor Expression and Lethal Prostate Cancer Progression. J. Clin. Endocrinol. Metab. 2016, 101, 2520–2527. [Google Scholar] [CrossRef] [PubMed]
- Gladden, A.B.; Diehl, J.A. Location, location, location: The role of cyclin D1 nuclear localization in cancer. J. Cell. Biochem. 2005, 96, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Tennakoon, S.; Aggarwal, A.; Kallay, E. The calcium-sensing receptor and the hallmarks of cancer. Biochim. Biophys. Acta 2016, 1863, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Binder, M.; Shui, I.M.; Wilson, K.M.; Penney, K.L.; Consortium, P.E.; Mucci, L.A.; Kibel, A.S. Calcium intake, polymorphisms of the calcium-sensing receptor, and recurrent/aggressive prostate cancer. Cancer Causes Control 2015, 26, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Shui, I.M.; Mucci, L.A.; Wilson, K.M.; Kraft, P.; Penney, K.L.; Stampfer, M.J.; Giovannucci, E. Common genetic variation of the calcium-sensing receptor and lethal prostate cancer risk. Cancer Epidemiol. Biomarkers Prev. 2013, 22, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Xu, X.; Li, B.; Brown, E.; Farris, A.B.; Sun, S.Y.; Yang, J.J. Prostate cancer metastatic to bone has higher expression of the calcium-sensing receptor (CaSR) than primary prostate cancer. Recept. Clin. Investig. 2014, 1, e270. [Google Scholar]
- Sanders, J.L.; Chattopadhyay, N.; Kifor, O.; Yamaguchi, T.; Brown, E.M. Ca2+-sensing receptor expression and PTHrP secretion in PC-3 human prostate cancer cells. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1267–E1274. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Macleod, R.J.; Chattopadhyay, N.; Tfelt-Hansen, J.; Kifor, O.; Butters, R.R.; Brown, E.M. Calcium-sensing receptor activation stimulates parathyroid hormone-related protein secretion in prostate cancer cells: Role of epidermal growth factor receptor transactivation. Bone 2004, 35, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Asadi, F.; Faraj, M.; Malakouti, S.; Kukreja, S.C. Effect of parathyroid hormone related protein, and dihydrotestosterone on proliferation and ornithine decarboxylase mRNA in human prostate cancer cell lines. Int. Urol. Nephrol. 2001, 33, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Jans, D.A.; Thomas, R.J.; Gillespie, M.T. Parathyroid hormone-related protein (PTHrP): A nucleocytoplasmic shuttling protein with distinct paracrine and intracrine roles. Vitam. Horm. 2003, 66, 345–384. [Google Scholar] [PubMed]
- Ongkeko, W.M.; Burton, D.; Kiang, A.; Abhold, E.; Kuo, S.Z.; Rahimy, E.; Yang, M.; Hoffman, R.M.; Wang-Rodriguez, J.; Deftos, L.J. Parathyroid hormone related-protein promotes epithelial-to-mesenchymal transition in prostate cancer. PLoS ONE 2014, 9, e85803. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Kyprianou, N. Targeting anoikis resistance in prostate cancer metastasis. Mol. Asp. Med. 2010, 31, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Park, S.I.; McCauley, L.K. Nuclear localization of parathyroid hormone-related peptide confers resistance to anoikis in prostate cancer cells. Endocr. Relat. Cancer 2012, 19, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Gujral, A.; Burton, D.W.; Terkeltaub, R.; Deftos, L.J. Parathyroid hormone-related protein induces interleukin 8 production by prostate cancer cells via a novel intracrine mechanism not mediated by its classical nuclear localization sequence. Cancer Res. 2001, 61, 2282–2288. [Google Scholar] [PubMed]
- Ferrer, F.A.; Miller, L.J.; Andrawis, R.I.; Kurtzman, S.H.; Albertsen, P.C.; Laudone, V.P.; Kreutzer, D.L. Angiogenesis and prostate cancer: In vivo and in vitro expression of angiogenesis factors by prostate cancer cells. Urology 1998, 51, 161–167. [Google Scholar] [CrossRef]
- Inoue, K.; Slaton, J.W.; Eve, B.Y.; Kim, S.J.; Perrotte, P.; Balbay, M.D.; Yano, S.; Bar-Eli, M.; Radinsky, R.; Pettaway, C.A.; et al. Interleukin 8 expression regulates tumorigenicity and metastases in androgen-independent prostate cancer. Clin. Cancer Res. 2000, 6, 2104–2119. [Google Scholar] [PubMed]
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Dushyanthen, S.; Cossigny, D.A.; Quan, G.M. The osteoblastic and osteoclastic interactions in spinal metastases secondary to prostate cancer. Cancer Growth Metastasis 2013, 6, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Li, X.; Koh, A.J.; Berry, J.E.; Thudi, N.; Rosol, T.J.; Pienta, K.J.; McCauley, L.K. Tumor expressed PTHrP facilitates prostate cancer-induced osteoblastic lesions. Int. J. Cancer 2008, 123, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, L.A.; Fournier, P.G.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.P.; Miller, R.E.; Jones, J.C.; Zhang, J.; Keller, E.T.; Dougall, W.C. RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate 2008, 68, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Sterling, J.A.; Edwards, J.R.; DeGraff, D.J.; Lee, C.; Park, S.I.; Matusik, R.J. Activation of NF-kappa B signaling promotes growth of prostate cancer cells in bone. PLoS ONE 2013, 8, e60983. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maly, I.V.; Hofmann, W.A. Fatty Acids and Calcium Regulation in Prostate Cancer. Nutrients 2018, 10, 788. https://doi.org/10.3390/nu10060788
Maly IV, Hofmann WA. Fatty Acids and Calcium Regulation in Prostate Cancer. Nutrients. 2018; 10(6):788. https://doi.org/10.3390/nu10060788
Chicago/Turabian StyleMaly, Ivan V., and Wilma A. Hofmann. 2018. "Fatty Acids and Calcium Regulation in Prostate Cancer" Nutrients 10, no. 6: 788. https://doi.org/10.3390/nu10060788
APA StyleMaly, I. V., & Hofmann, W. A. (2018). Fatty Acids and Calcium Regulation in Prostate Cancer. Nutrients, 10(6), 788. https://doi.org/10.3390/nu10060788