Saccharin Increases Fasting Blood Glucose but Not Liver Insulin Resistance in Comparison to a High Fructose-Fed Rat Model


 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Study Protocol
2.3. Diets
Metabolic Parameters
2.4. Liver Histological Studies
2.5. Real-Time Quantitative Reverse Transcription PCR
2.6. Statistical Analysis
3. Results
3.1. The Effects of Saccharin Versus a High Fructose Diet on Metabolic Parameters
3.2. The Effects of Saccharin Versus a High Fructose Diet on Fatty Changes in the Liver
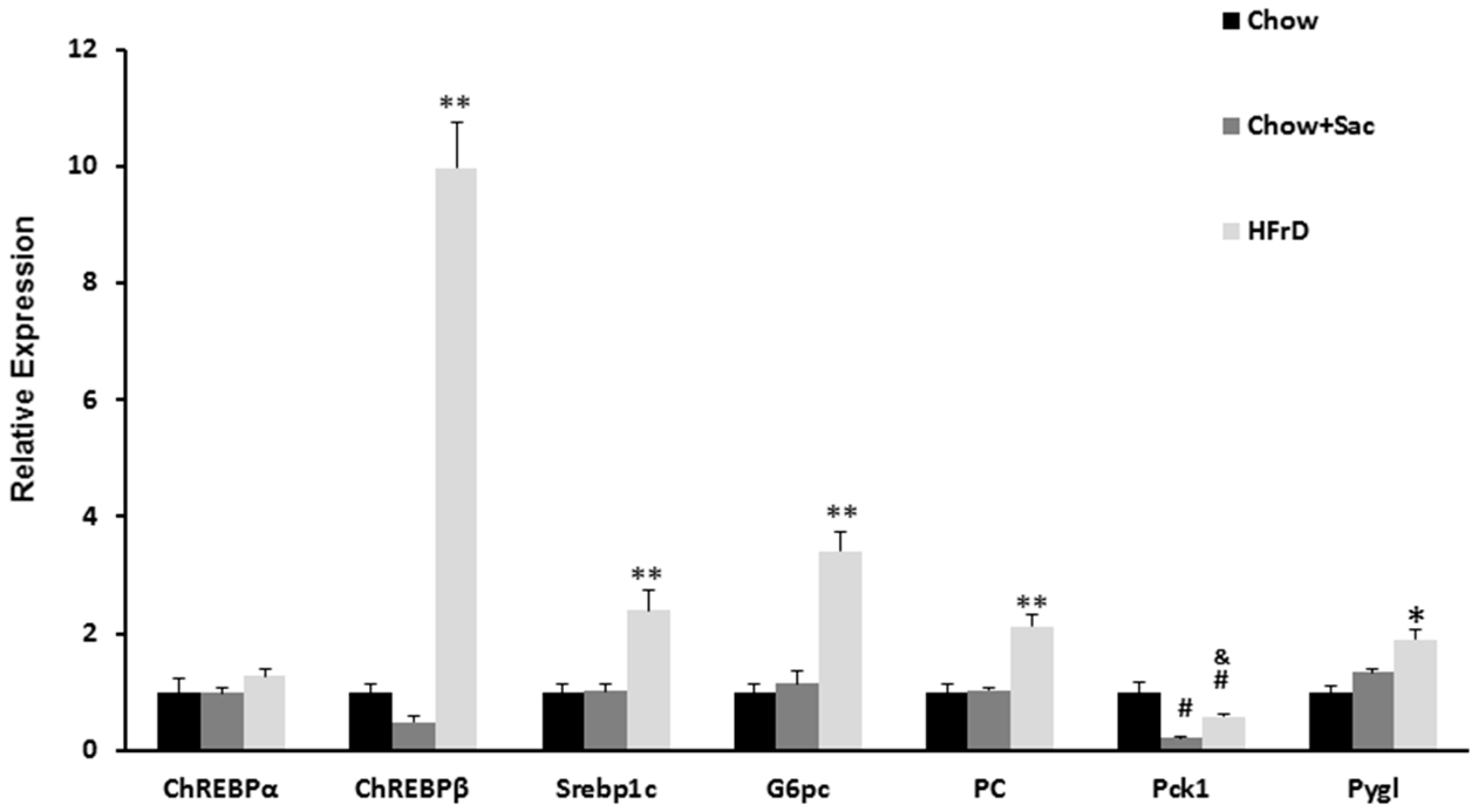
3.3. The Effects of Saccharin Versus a High Fructose Diet on the Expression of Genes Linked to Insulin Resistance in the Liver
4. Discussion
Study Limitations
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- The GBD 2015 Obesity Collaborators. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report from the American Heart Association. Circulation 2017, 35, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, D.; Roy, A.; Praveen, P.A.; Ramakrishnan, L.; Gupta, R.; Amarchand, R.; Kondal, D.; Singh, K.; Sharma, M.; Shukla, D.K.; et al. 20-Year Trend of cardiovascular disease risk factors: Urban and rural national capital region of Delhi, India. Glob. Heart 2017, 12, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Trogdon, J.G.; Khavjou, O.A.; Butler, J.; Dracup, K.; Ezekowitz, M.D.; Finkelstein, E.A.; Hong, Y.; Johnston, S.C.; Khera, A.; et al. Forecasting the future of cardiovascular disease in the United States: A policy statement from the American Heart Association. Circulation 2011, 123, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.; Tommy, N. Non-alcoholic fatty liver disease-a chronic disease of the 21st century. J. Biomed. Res. 2017, 1–9. [Google Scholar] [CrossRef]
- Ekstedt, M.; Hagstrom, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Arulanandan, A.; Ang, B.; Bettencourt, R.; Hooker, J.; Behling, C.; Lin, G.Y.; Valasek, M.A.; Ix, J.H.; Schnabl, B.; Sirlin, C.B.; et al. Association between quantity of liver fat and cardiovascular risk in patients with nonalcoholic fatty liver disease independent of nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2015, 13, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, M.; Rafiq, N.; Makhlouf, H.; Agrawal, R.; Kaur, I.; Younoszai, Z.; Valasek, M.A.; Ix, J.H.; Schnabl, B.; Sirlin, C.B.; et al. Predictors of all-cause mortality and liver-related mortality in patients with non-alcoholic fatty liver disease (NAFLD). Dig. Dis. Sci. 2013, 58, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic fatty liver disease and insulin resistance: New insights and potential new treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H.; Schmidt, L.A.; Brindis, C.D. Public health: The toxic truth about sugar. Nature 2012, 482, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Swithers, S.E. Artificial sweeteners produce the counterintuitive effect of inducing metabolic derangements. Trends Endocrinol. Metab. 2013, 24, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Abou-Setta, A.M.; Chauhan, B.F.; Rabbani, R.; Lys, J.; Copstein, L.; Mann, A.; Jeyaraman, M.M.; Reid, A.E.; Fiander, M.; et al. Nonnutritive sweeteners and cardiometabolic health: A systematic review and meta-analysis of randomized controlled trials and prospective cohort studies. Can. Med. Assoc. J. 2017, 189, E929–E939. [Google Scholar] [CrossRef] [PubMed]
- Suez, J.; Korem, T.; Zeevi, D.; Zilberman-Schapira, G.; Thaiss, C.A.; Maza, O.; Israeli, D.; Zmora, N.; Gilad, S.; Weinberger, A.; et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 2014, 514, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Kamari, Y.; Shaish, A.; Vax, E.; Shemesh, S.; Kandel-Kfir, M.; Arbel, Y.; Olteanu, S.; Barshack, I.; Dotan, S.; Voronov, E.; et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J. Hepatol. 2011, 55, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Lutsey, P.L.; Steffen, L.M.; Stevens, J. Dietary intake and the development of the metabolic syndrome: The Atherosclerosis Risk in Communities study. Circulation 2008, 117, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Nettleton, J.A.; Lutsey, P.L.; Wang, Y.; Lima, J.A.; Michos, E.D.; Jacobs, D.R., Jr. Diet soda intake and risk of incident metabolic syndrome and type 2 diabetes in the Multi-Ethnic Study of Atherosclerosis (MESA). Diabetes Care 2009, 32, 688–694. [Google Scholar] [CrossRef] [PubMed]
- De Koning, L.; Malik, V.S.; Rimm, E.B.; Willett, W.C.; Hu, F.B. Sugar-sweetened and artificially sweetened beverage consumption and risk of type 2 diabetes in men. Am. J. Clin. Nutr. 2011, 93, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Tate, D.F.; Turner-McGrievy, G.; Lyons, E.; Stevens, J.; Erickson, K.; Polzien, K.; Diamond, M.; Wang, X.; Popkin, B. Replacing caloric beverages with water or diet beverages for weight loss in adults: Main results of the Choose Healthy Options Consciously Everyday (CHOICE) randomized clinical trial. Am. J. Clin. Nutr. 2012, 95, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.A.; AlMuzafar, H.M. Alterations in lipid profile, oxidative stress and hepatic function in rat fed with saccharin and methyl-salicylates. Int. J. Clin. Exp. Med. 2015, 8, 6133–6144. [Google Scholar] [PubMed]
- Alkafafy Mel, S.; Ibrahim, Z.S.; Ahmed, M.M.; El-Shazly, S.A. Impact of aspartame and saccharin on the rat liver: Biochemical, molecular, and histological approach. Int. J. Immunopathol. Pharmacol. 2015, 28, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Andrejic, B.M.; Mijatovic, V.M.; Samojlik, I.N.; Horvat, O.J.; Calasan, J.D.; Dolai, M.A. The influence of chronic intake of saccharin on rat hepatic and pancreatic function and morphology: Gender differences. Bosn. J. Basic Med. Sci. 2013, 13, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Parlee, S.D.; Simon, B.R.; Scheller, E.L.; Alejandro, E.U.; Learman, B.S.; Krishnan, V.; Bernal-Mizrachi, E.; MacDougald, O.A. Administration of saccharin to neonatal mice influences body composition of adult males and reduces body weight of females. Endocrinology 2014, 155, 1313–1326. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, Z.; Oron-Herman, M.; Grozovski, M.; Rosenthal, T.; Pappo, O.; Link, G.; Sela, B.A. Fructose-induced fatty liver disease: Hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension 2005, 45, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Kramer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schon, M.R.; Abumrad, N.A.; Bluher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Krawczyk, S.A.; Doridot, L.; Fowler, A.J.; Wang, J.X.; Trauger, S.A.; Noh, H.L.; Kang, H.J.; Meissen, J.K.; Blatnik, M.; et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Investig. 2016, 126, 4372–4386. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Steatosis parenchymal involvement | <5% | 0 |
| 5–33% | 1 | |
| 33–66% | 2 | |
| >66% | 3 | |
| Location | Zone 3 | 0 |
| Zone 1 | 1 | |
| Azonal | 2 | |
| Panacinar | 3 | |
| Microvesicular steatosis | Not present | 0 |
| Present | 1 | |
| Fibrosis | None | 0 |
| Perisinusoidal/periportal | 1 | |
| Perisinusoidal and periportal | 2 | |
| Bridging fibrosis | 3 | |
| Cirrhosis | 4 |
| Gene Name | Forward | Reverse |
|---|---|---|
| Rplp0 * | GAACATCTCCCCCTTCTCCTTC | ATTGCGGACACCCTCTAGGAA |
| PC | CCAAGCAGGTTGGCTATGAGAA | GATGTTTTCCTGCCGCAGCC |
| Pck1 | GGATGTGGCCAGGATCGAAA | ATACATGGTGCGGCCTTTCA |
| ChREBP-α | TGCATCGATCACAGGTCATT | AGGCTCAAGCATTCGAAGAG |
| ChREBP-β | TCTGCAGATCGCGCGGAG | CTTGTCCCGGCATAGCAAC |
| G6pc | CGTCACCTGTGAGACTGGAC | ACGACATTCAAGCACCGGAA |
| Pygl | ATAATTGGTGGGAAAGCTGCC | GCCAGCAGTGGAGATCTGTT |
| Srebp-1c | CATGGATTGCACATTTGAAGAC | GCAGGAGAAGAGAAGCTCTCAG |
| Type Diet | Ctrl | Saccharin | HFrD |
|---|---|---|---|
| Body weight (g) | 408 ± 9 | 407 ± 9 | 415 ± 8 |
| Blood pressure (mmHg) | 135 ± 1 | 138 ± 1 | 156 ± 2 * |
| Triglycerides (mg/dL) | 152 ± 20 | 127 ± 13 | 420 ± 43 * |
| Glucose (mg/dL) | 118 ± 3 | 137 ± 6 # | 140 ± 5 # |
| Insulin (ug/L) | 0.7 ± 0.1 | 0.6 ± 0.1 | 0.99 ± 0.12 * |
| HOMA2-IR | 2.3 ± 0.36 | 2.13 ± 0.3 | 3.4 ± 0.4 * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leibowitz, A.; Bier, A.; Gilboa, M.; Peleg, E.; Barshack, I.; Grossman, E. Saccharin Increases Fasting Blood Glucose but Not Liver Insulin Resistance in Comparison to a High Fructose-Fed Rat Model. Nutrients 2018, 10, 341. https://doi.org/10.3390/nu10030341
Leibowitz A, Bier A, Gilboa M, Peleg E, Barshack I, Grossman E. Saccharin Increases Fasting Blood Glucose but Not Liver Insulin Resistance in Comparison to a High Fructose-Fed Rat Model. Nutrients. 2018; 10(3):341. https://doi.org/10.3390/nu10030341
Chicago/Turabian StyleLeibowitz, Avshalom, Ariel Bier, Mayan Gilboa, Edna Peleg, Iris Barshack, and Ehud Grossman. 2018. "Saccharin Increases Fasting Blood Glucose but Not Liver Insulin Resistance in Comparison to a High Fructose-Fed Rat Model" Nutrients 10, no. 3: 341. https://doi.org/10.3390/nu10030341
APA StyleLeibowitz, A., Bier, A., Gilboa, M., Peleg, E., Barshack, I., & Grossman, E. (2018). Saccharin Increases Fasting Blood Glucose but Not Liver Insulin Resistance in Comparison to a High Fructose-Fed Rat Model. Nutrients, 10(3), 341. https://doi.org/10.3390/nu10030341