Oleuropein, the Main Polyphenol of Olea europaea Leaf Extract, Has an Anti-Cancer Effect on Human BRAF Melanoma Cells and Potentiates the Cytotoxicity of Current Chemotherapies




 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. MTT Assay
2.3. Sample Preparation for Mass Spectrometry Analysis
2.4. Evaluation of Apoptosis
2.5. Cell Cycle Analysis
2.6. Invasion Assay
2.7. Plate Colony Forming Assay
2.8. Western Blotting Analysis
3. Olive Leaf Extract’s Preparation
3.1. Plant Material
3.2. Solvents and Reagents
3.3. Extraction and Lyophylisation
3.4. Sample Preparation for Mass Spectrometry Analysis
3.5. Statistical Analysis
4. Results
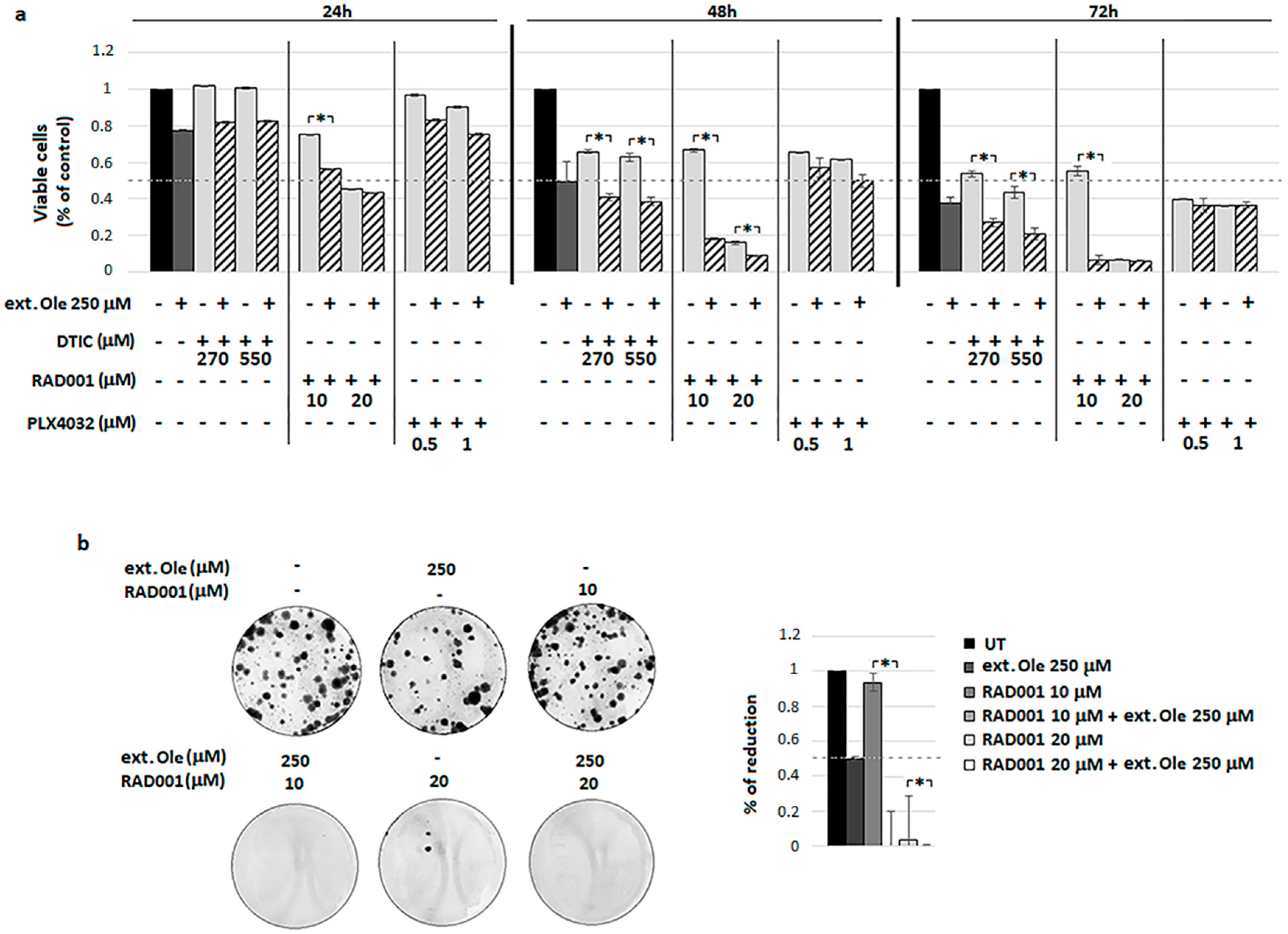
4.1. Ole Efficacy on BRAF Melanoma Cells
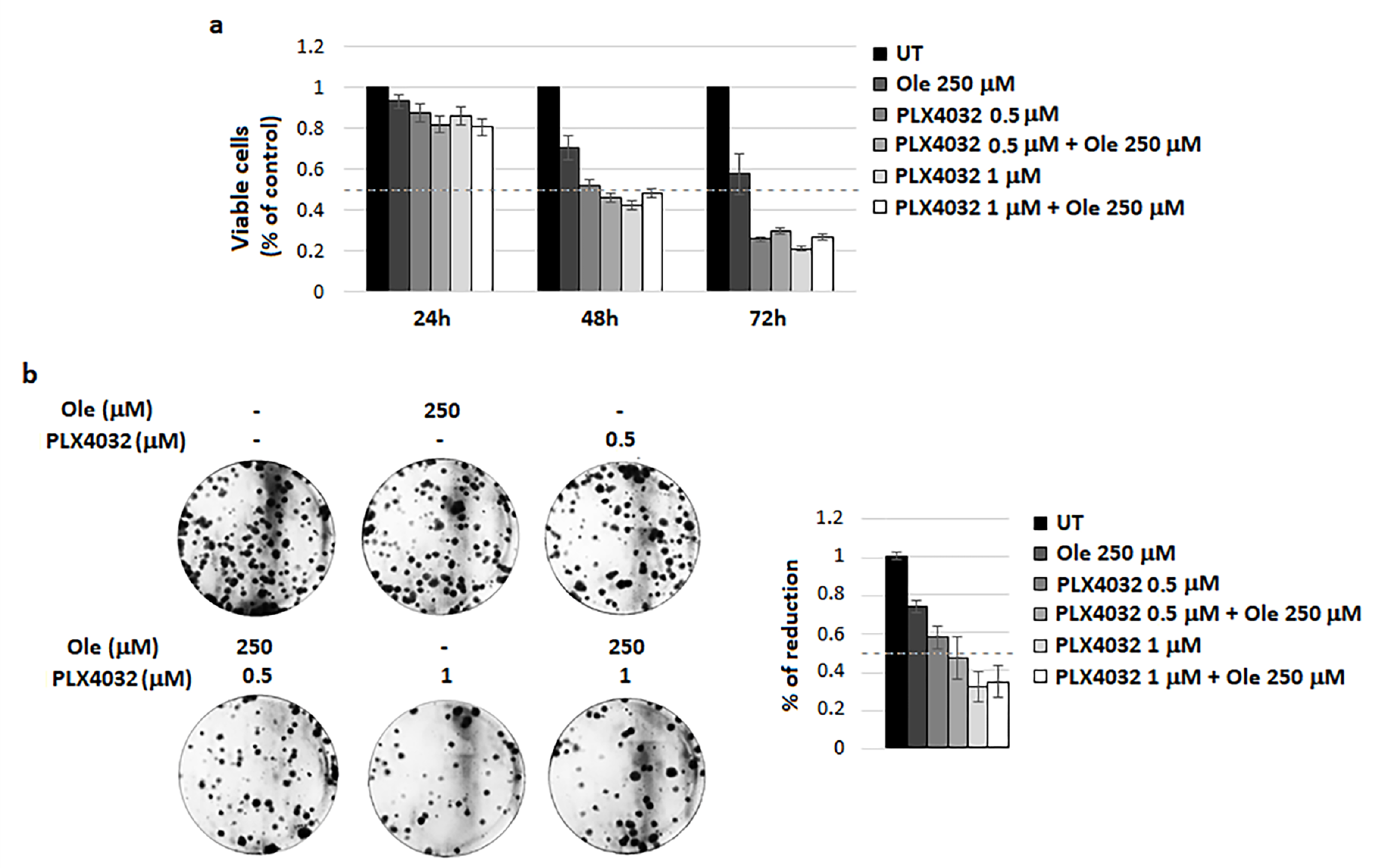
4.2. Combination of Ole with Vemurafenib (PLX4032)
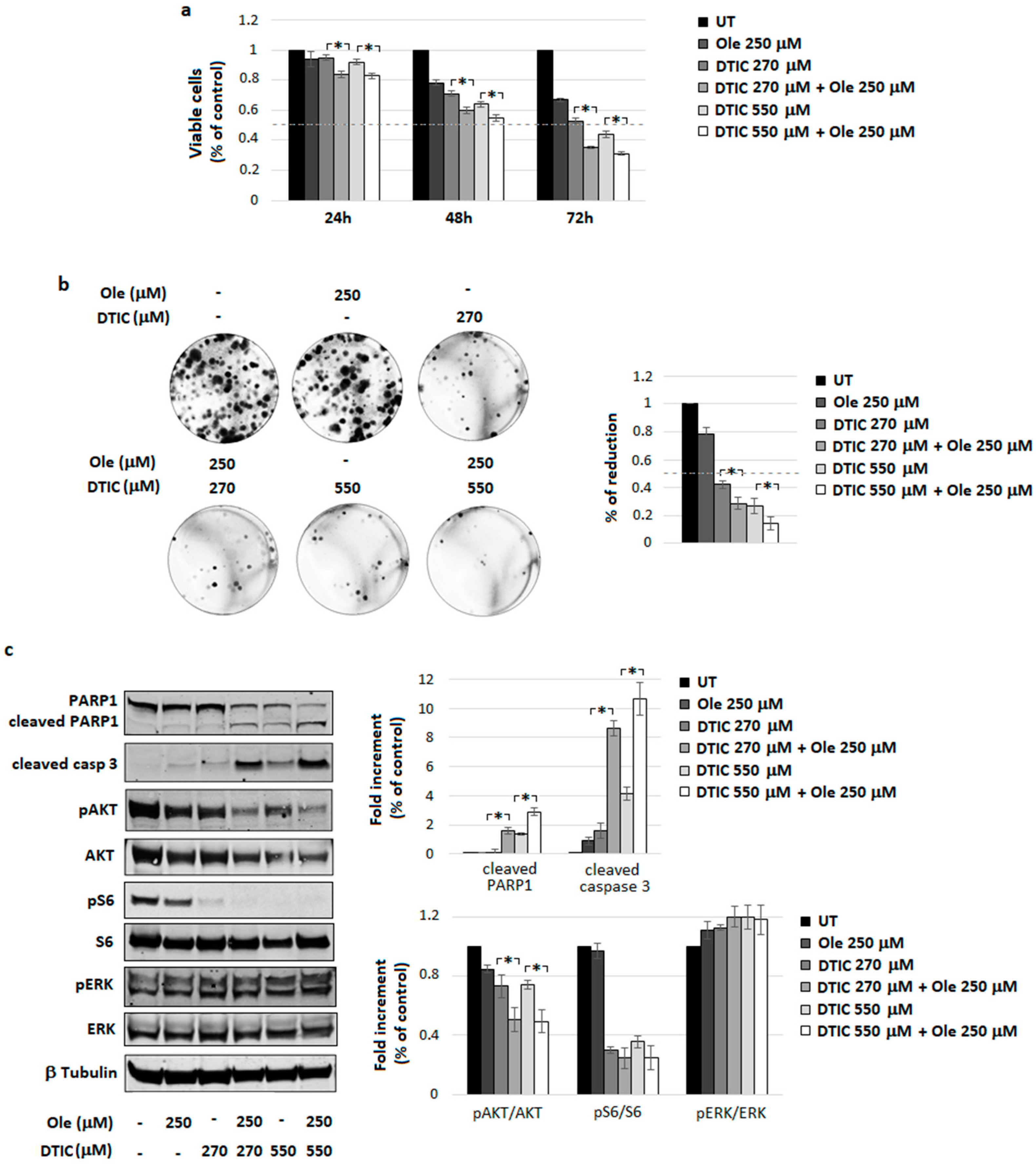
4.3. Combination of Ole with Dacarbazine (DTIC)
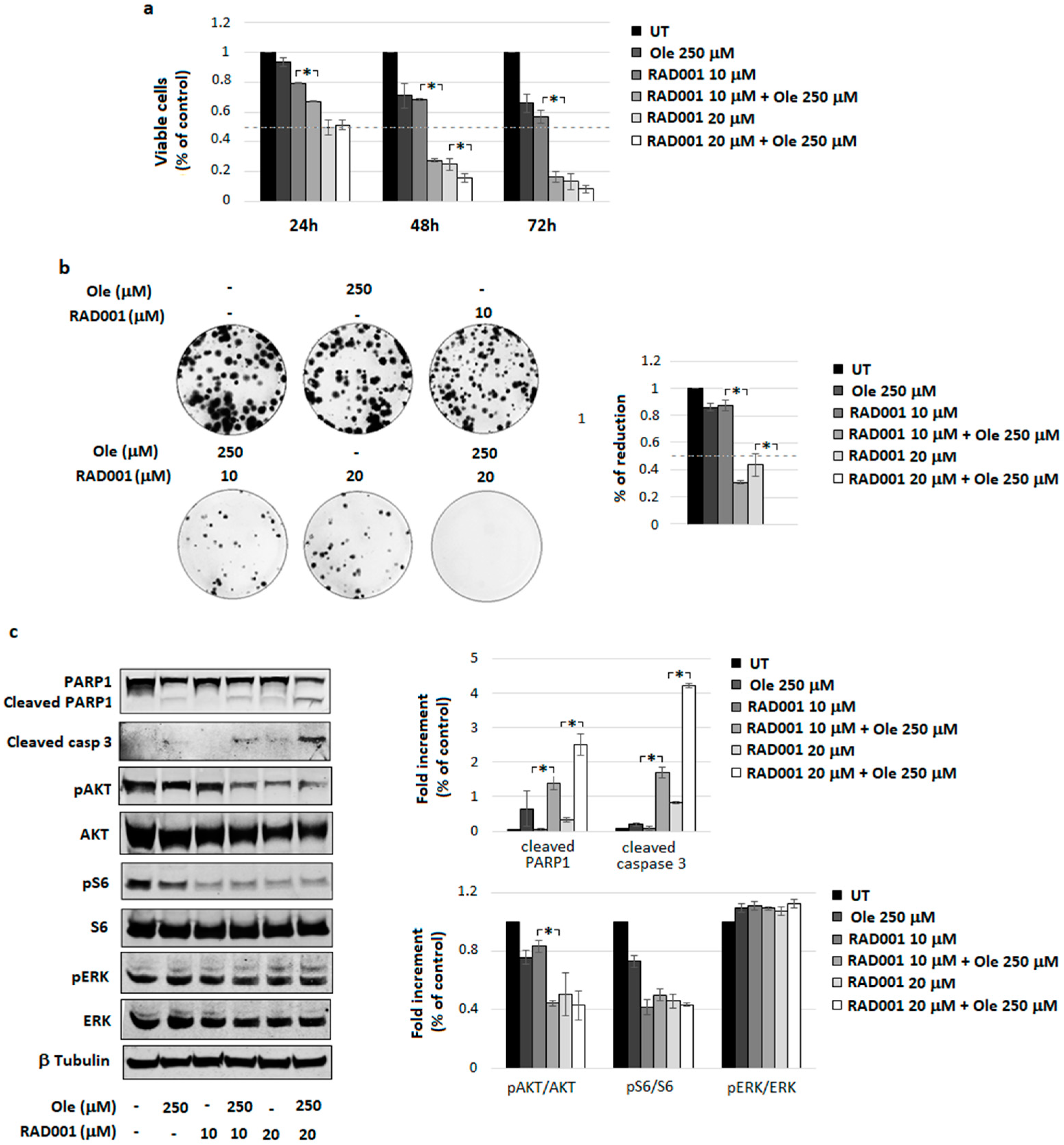
4.4. Combination of Ole with Everolimus (RAD001)
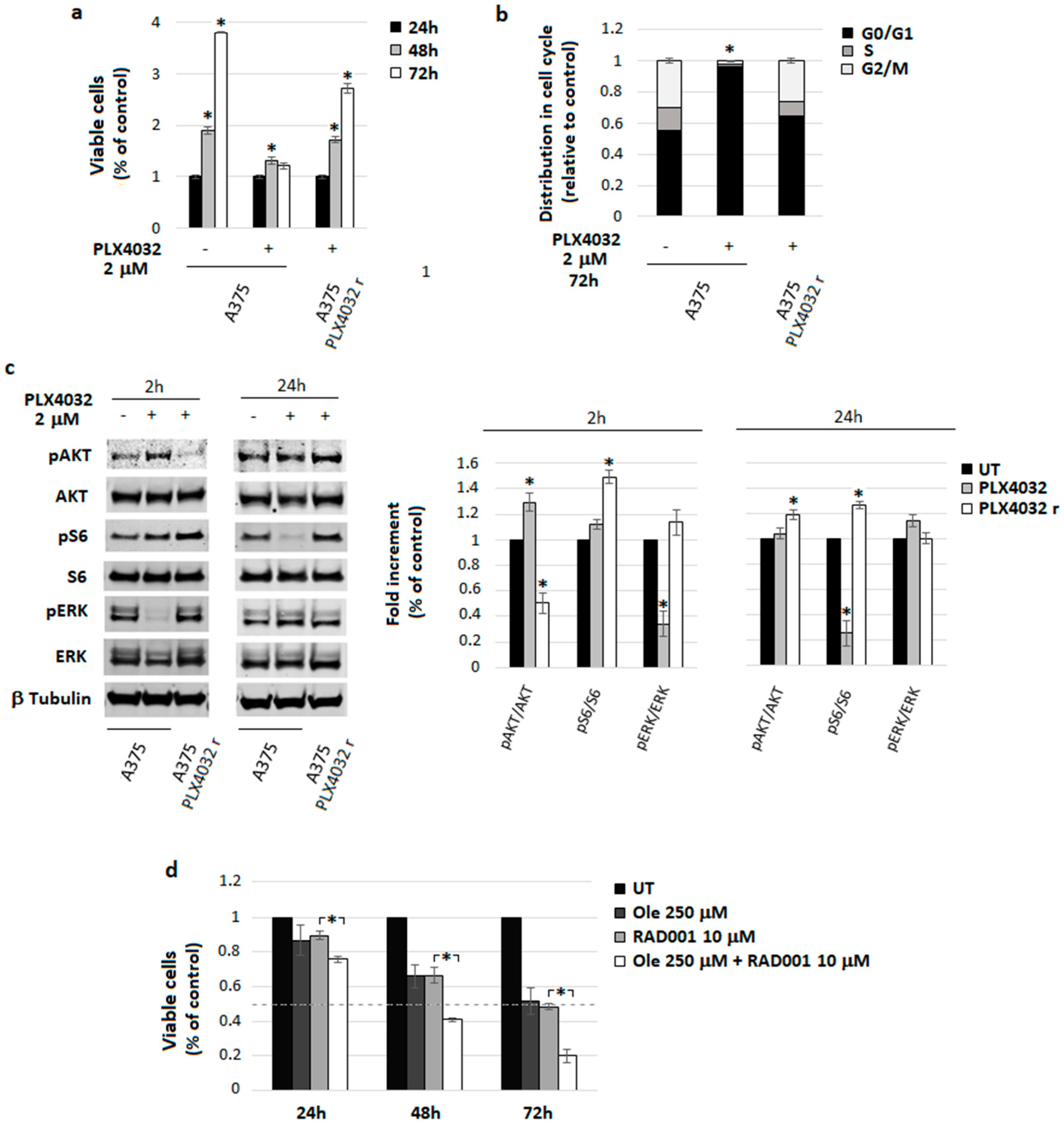
4.5. A Freshly Prepared Olive Leaf Extract Potentiates RAD001 Treatment on BRAF Melanoma Cells
5. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.; Becker, J.C.; Kappel, A.; Terheyden, P.; Bröcker, E.-B.; Goetz, R.; Rapp, U.R. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J. Carcinog. 2004, 3, 6. [Google Scholar] [CrossRef][Green Version]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Wellbrock, C.; Ogilvie, L.; Hedley, D.; Karasarides, M.; Martin, J.; Niculescu-Duvaz, D.; Springer, C.J.; Marais, R. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004, 64, 2338–2342. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant melanoma: Genetics and therapeutics in the genomic era. Genes Dev. 2006, 20, 2149–2182. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A. The role of the PI3K-AKT pathway in melanoma. Cancer J. 2012, 18, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Valls-Pedret, C.; Lamuela-Raventós, R.M.; Medina-Remón, A.; Quintana, M.; Corella, D.; Pintó, X.; Martínez-González, M.Á.; Estruch, R.; Ros, E. Polyphenol-rich foods in the Mediterranean diet are associated with better cognitive function in elderly subjects at high cardiovascular risk. J. Alzheimer’s Dis. 2012, 29, 773–782. [Google Scholar] [CrossRef]
- Santoro, A.; Pini, E.; Scurti, M.; Palmas, G.; Berendsen, A.; Brzozowska, A.; Pietruszka, B.; Szczecinska, A.; Cano, N.; Meunier, N.; et al. Combating inflammaging through a Mediterranean whole diet approach: The NU-AGE project’s conceptual framework and design. Mech. Ageing Dev. 2014, 136–137, 3–13. [Google Scholar] [CrossRef] [PubMed]
- van den Brandt, P.A.; Schulpen, M. Mediterranean diet adherence and risk of postmenopausal breast cancer: Results of a cohort study and meta-analysis. Int. J. Cancer 2017, 140, 2220–2231. [Google Scholar] [CrossRef] [PubMed]
- Sofi, F.; Abbate, R.; Gensini, G.F.; Casini, A. Accruing evidence on benefits of adherence to the Mediterranean diet on health: An updated systematic review and meta-analysis. Am. J. Clin. Nutr. 2010, 92, 1189–1196. [Google Scholar] [CrossRef]
- Ruiz-Canela, M.; Martínez-González, M.A. Olive oil in the primary prevention of cardiovascular disease. Maturitas 2011, 68, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Franco, M.; Toledo, E.; Luchsinger, J.; Willett, W.C.; Hu, F.B.; Martinez-Gonzalez, M.A. Olive oil and prevention of chronic diseases: Summary of an International conference. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-López, M.-J.; Molina, J.J.M.; Mir, M.V.; Rey, E.F.; Martín, F.; de la Serrana, H.L.-G. Extra virgin olive oil (EVOO) consumption and antioxidant status in healthy institutionalized elderly humans. Arch. Gerontol. Geriatr. 2013, 57, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Cicerale, S.; Lucas, L.J.; Keast, R.S.J. Antimicrobial, antioxidant and anti-inflammatory phenolic activities in extra virgin olive oil. Curr. Opin. Biotechnol. 2012, 23, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, B.; Toietta, G.; Maggio, R.; Arciello, M.; Tarocchi, M.; Galli, A.; Balsano, C. Effects of the olive-derived polyphenol oleuropein on human health. Int. J. Mol. Sci. 2014, 15, 18508–18524. [Google Scholar] [CrossRef] [PubMed]
- Crespo, M.C.; Tomé-Carneiro, J.; Dávalos, A.; Visioli, F. Pharma-Nutritional Properties of Olive Oil Phenols. Transfer of New Findings to Human Nutrition. Foods 2018, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Hassen, I.; Casabianca, H.; Hosni, K. Biological activities of the natural antioxidant oleuropein: Exceeding the expectation—A mini-review. J. Funct. Foods 2015, 18, 926–940. [Google Scholar] [CrossRef]
- Angeloni, C.; Malaguti, M.; Barbalace, M.C.; Hrelia, S. Bioactivity of Olive Oil Phenols in Neuroprotection. Int. J. Mol. Sci. 2017, 18, 2230. [Google Scholar] [CrossRef] [PubMed]
- Shamshoum, H.; Vlavcheski, F.; Tsiani, E. Anticancer effects of oleuropein. Biofactors 2017, 43, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Nadeem, M.; Gilani, S.A.; Khan, S.; Sajid, M.W.; Amir, R.M. Antitumor Perspectives of Oleuropein and Its Metabolite Hydroxytyrosol: Recent Updates. J. Food Sci. 2018, 83, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Papachristodoulou, A.; Tsoukala, M.; Benaki, D.; Kostidis, S.; Gioti, K.; Aligiannis, N.; Pratsinis, H.; Kletsas, D.; Skaltsounis, A.-L.; Mikros, E.; et al. Oleuropein is a Powerful Sensitizer of Doxorubicin-mediated Killing of Prostate Cancer Cells and Exerts Its Action via Induction of Autophagy. J. Cancer Res. Treat. 2018, 4, 61–68. [Google Scholar] [CrossRef]
- Choupani, J.; Alivand, M.R.; Derakhshan, S.M.; Zaeifizadeh, M.; Khaniani, M.S. Oleuropein inhibits migration ability through suppression of epithelial-mesenchymal transition and synergistically enhances doxorubicin-mediated apoptosis in MCF-7 cells. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Ruzzolini, J.; Peppicelli, S.; Andreucci, E.; Bianchini, F.; Margheri, F.; Laurenzana, A.; Fibbi, G.; Pimpinelli, N.; Calorini, L. Everolimus selectively targets vemurafenib resistant BRAFV600E melanoma cells adapted to low pH. Cancer Lett. 2017, 408, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, M.; Tuccoli, A.; D’Aurizio, R.; Sarti, S.; Giannecchini, L.; Lubrano, S.; Marranci, A.; Evangelista, M.; Peppicelli, S.; Ippolito, C.; et al. Context-dependent miR-204 and miR-211 affect the biological properties of amelanotic and melanotic melanoma cells. Oncotarget 2017, 8, 25395–25417. [Google Scholar] [CrossRef] [PubMed]
- Pagnini, I.; Simonini, G.; Cavalli, L.; la Marca, G.; Iuliano, A.; Brandi, M.; Bellisai, F.; Frediani, B.; Galeazzi, M.; Cantarini, L.; et al. Bone status of children born from mothers with autoimmune diseases treated during pregnancy with prednisone and/or low molecular weight heparin. Pediatr. Rheumatol. 2014, 12, 47. [Google Scholar] [CrossRef]
- Romani, A.; Mulas, S.; Heimler, D. Polyphenols and secoiridoids in raw material (Olea europaea L. leaves) and commercial food supplements. Eur. Food Res. Technol. 2017, 243, 429–435. [Google Scholar] [CrossRef]
- Romani, A.; Scardigli, A.; Pinelli, P. An environmentally friendly process for the production of extracts rich in phenolic antioxidants from Olea europaea L. and Cynara scolymus L. matrices. Eur. Food Res. Technol. 2017, 243, 1229–1238. [Google Scholar] [CrossRef]
- Robles-Almazan, M.; Pulido-Moran, M.; Moreno-Fernandez, J.; Ramirez-Tortosa, C.; Rodriguez-Garcia, C.; Quiles, J.L.; Ramirez-Tortosa, M. Hydroxytyrosol: Bioavailability, toxicity, and clinical applications. Food Res. Int. 2018, 105, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Carrera-González, M.P.; Ramírez-Expósito, M.J.; Mayas, M.D.; Martínez-Martos, J.M. Protective role of oleuropein and its metabolite hydroxytyrosol on cancer. Trends Food Sci. Technol. 2013, 31, 92–99. [Google Scholar] [CrossRef]
- Mouawad, R.; Sebert, M.; Michels, J.; Bloch, J.; Spano, J.-P.; Khayat, D. Treatment for metastatic malignant melanoma: Old drugs and new strategies. Crit. Rev. Oncol. Hematol. 2010, 74, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 2011, 29, 4688–4695. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Grammer, T.C.; Blenis, J. Evidence for MEK-independent pathways regulating the prolonged activation of the ERK-MAP kinases. Oncogene 1997, 14, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.C.; Lai, F.; Thorne, R.F.; Yang, F.; Liu, H.; Hersey, P.; Zhang, X.D. MEK-independent survival of B-RAFV600E melanoma cells selected for resistance to apoptosis induced by the RAF inhibitor PLX4720. Clin. Cancer Res. 2011, 17, 721–730. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Perna, D.; Karreth, F.A.; Rust, A.G.; Perez-Mancera, P.A.; Rashid, M.; Iorio, F.; Alifrangis, C.; Arends, M.J.; Bosenberg, M.W.; Bollag, G.; et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc. Natl. Acad. Sci. USA 2015, 112, E536–E545. [Google Scholar] [CrossRef]
- Chi, M.; Ye, Y.; Zhang, X.D.; Chen, J. Insulin induces drug resistance in melanoma through activation of the PI3K/Akt pathway. Drug Des. Dev. Ther. 2014, 8, 255–262. [Google Scholar] [CrossRef]
- Grossi, C.; Rigacci, S.; Ambrosini, S.; Ed Dami, T.; Luccarini, I.; Traini, C.; Failli, P.; Berti, A.; Casamenti, F.; Stefani, M. The polyphenol oleuropein aglycone protects TgCRND8 mice against Aß plaque pathology. PLoS ONE 2013, 8, e71702. [Google Scholar] [CrossRef] [PubMed]
- Miceli, C.; Santin, Y.; Manzella, N.; Coppini, R.; Berti, A.; Stefani, M.; Parini, A.; Mialet-Perez, J.; Nediani, C. Oleuropein Aglycone Protects against MAO-A-Induced Autophagy Impairment and Cardiomyocyte Death through Activation of TFEB. Oxidative Med. Cell. Longev. 2018, 2018, 8067592. [Google Scholar] [CrossRef] [PubMed]
- Rigacci, S.; Miceli, C.; Nediani, C.; Berti, A.; Cascella, R.; Pantano, D.; Nardiello, P.; Luccarini, I.; Casamenti, F.; Stefani, M. Oleuropein aglycone induces autophagy via the AMPK/mTOR signalling pathway: A mechanistic insight. Oncotarget 2015, 6, 35344–35357. [Google Scholar] [CrossRef] [PubMed]
- Luccarini, I.; Pantano, D.; Nardiello, P.; Cavone, L.; Lapucci, A.; Miceli, C.; Nediani, C.; Berti, A.; Stefani, M.; Casamenti, F. The Polyphenol Oleuropein Aglycone Modulates the PARP1-SIRT1 Interplay: An In Vitro and In Vivo Study. J. Alzheimer’s Dis. 2016, 54, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Liman, R.; Çoban, F.; Ciğerci, I.; Bulduk, İ.; Bozkurt, S. Antiangiogenic and Apoptotic Effects of Oleuropein on Breast Cancer Cells. Br. J. Pharm. Res. 2017, 16, 1–10. [Google Scholar] [CrossRef]
- Elamin, M.H.; Elmahi, A.B.; Daghestani, M.H.; Al-Olayan, E.M.; Al-Ajmi, R.A.; Alkhuriji, A.F.; Hamed, S.S.; Elkhadragy, M.F. Synergistic Anti-Breast-Cancer Effects of Combined Treatment with Oleuropein and Doxorubicin In Vivo. Altern. Ther. Health Med. 2017. [Google Scholar] [CrossRef]
- Menendez, J.A.; Vazquez-Martin, A.; Colomer, R.; Brunet, J.; Carrasco-Pancorbo, A.; Garcia-Villalba, R.; Fernandez-Gutierrez, A.; Segura-Carretero, A. Olive oil’s bitter principle reverses acquired autoresistance to trastuzumab (HerceptinTM) in HER2-overexpressing breast cancer cells. BMC Cancer 2007, 7. [Google Scholar] [CrossRef]
- Notarnicola, M.; Pisanti, S.; Tutino, V.; Bocale, D.; Rotelli, M.T.; Gentile, A.; Memeo, V.; Bifulco, M.; Perri, E.; Caruso, M.G. Effects of olive oil polyphenols on fatty acid synthase gene expression and activity in human colorectal cancer cells. Genes Nutr. 2011, 6, 63–69. [Google Scholar] [CrossRef]
- Hamdi, H.K.; Castellon, R. Oleuropein, a non-toxic olive iridoid, is an anti-tumor agent and cytoskeleton disruptor. Biochem. Biophys. Res. Commun. 2005, 334, 769–778. [Google Scholar] [CrossRef]
- Goldsmith, C.; Bond, D.; Jankowski, H.; Weidenhofer, J.; Stathopoulos, C.; Roach, P.; Scarlett, C. The Olive Biophenols Oleuropein and Hydroxytyrosol Selectively Reduce Proliferation, Influence the Cell Cycle, and Induce Apoptosis in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2018, 19, 1937. [Google Scholar] [CrossRef]
- Haris Omar, S. Oleuropein in Olive and its Pharmacological Effects. Sci. Pharm. 2010, 78, 133–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, J.; Huang, B.; Chen, A.; Li, X. Oleuropein inhibits the proliferation and invasion of glioma cells via suppression of the AKT signaling pathway. Oncol. Rep. 2016, 36, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.-M.; Chai, E.-Q.; Cai, H.-Y.; Miao, G.-Y.; Ma, W. Oleuropein induces apoptosis via activation of caspases and suppression of phosphatidylinositol 3-kinase/protein kinase B pathway in HepG2 human hepatoma cell line. Mol. Med. Rep. 2015, 11, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Pazoki-Toroudi, H.; Amani, H.; Ajami, M.; Nabavi, S.F.; Braidy, N.; Kasi, P.D.; Nabavi, S.M. Targeting mTOR signaling by polyphenols: A new therapeutic target for ageing. Ageing Res. Rev. 2016, 31, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Lim, D.Y.; Jung, J.I.; Cho, H.J.; Park, S.Y.; Kwon, G.T.; Kang, Y.-H.; Lee, K.W.; Choi, M.-S.; Park, J.H.Y. Dietary oleuropein inhibits tumor angiogenesis and lymphangiogenesis in the B16F10 melanoma allograft model: A mechanism for the suppression of high-fat diet-induced solid tumor growth and lymph node metastasis. Oncotarget 2017, 8, 32027–32042. [Google Scholar] [CrossRef] [PubMed]
- Samara, P.; Christoforidou, N.; Lemus, C.; Argyropoulou, A.; Ioannou, K.; Vougogiannopoulou, K.; Aligiannis, N.; Paronis, E.; Gaboriaud-Kolar, N.; Tsitsilonis, O.; et al. New semi-synthetic analogs of oleuropein show improved anticancer activity in vitro and in vivo. Eur. J. Med. Chem. 2017, 137, 11–29. [Google Scholar] [CrossRef] [PubMed]
- Vanella, L. Antiproliferative effect of oleuropein in prostate cell lines. Int. J. Oncol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Talorete, T.P.N.; Yamada, P.; Isoda, H. Anti-proliferative and apoptotic effects of oleuropein and hydroxytyrosol on human breast cancer MCF-7 cells. Cytotechnology 2009, 59, 45–53. [Google Scholar] [CrossRef]
- Cárdeno, A.; Sánchez-Hidalgo, M.; Rosillo, M.A.; de la Lastra, C.A. Oleuropein, a Secoiridoid Derived from Olive Tree, Inhibits the Proliferation of Human Colorectal Cancer Cell Through Downregulation of HIF-1α. Nutr. Cancer 2013, 65, 147–156. [Google Scholar] [CrossRef]
- Yao, J.; Wu, J.; Yang, X.; Yang, J.; Zhang, Y.; Du, L. Oleuropein induced apoptosis in HeLa cells via a mitochondrial apoptotic cascade associated with activation of the c-Jun NH2-terminal kinase. J. Pharmacol. Sci. 2014, 125, 300–311. [Google Scholar] [CrossRef]
- de Bock, M.; Thorstensen, E.B.; Derraik, J.G.B.; Henderson, H.V.; Hofman, P.L.; Cutfield, W.S. Human absorption and metabolism of oleuropein and hydroxytyrosol ingested as olive (Olea europaea L.) leaf extract. Mol. Nutr. Food Res. 2013, 57, 2079–2085. [Google Scholar] [CrossRef] [PubMed]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef] [PubMed]
- Samet, I.; Han, J.; Jlaiel, L.; Sayadi, S.; Isoda, H. Olive (Olea europaea) Leaf Extract Induces Apoptosis and Monocyte/Macrophage Differentiation in Human Chronic Myelogenous Leukemia K562 Cells: Insight into the Underlying Mechanism. Oxidative Med. Cell. Longev. 2014, 2014, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, S.A.; Timotijevic, G.S.; Miljkovic, D.M.; Radovic, J.M.; Maksimovic-Ivanic, D.D.; Dekanski, D.P.; Stosic-Grujicic, S.D. Multiple antimelanoma potential of dry olive leaf extract. Int. J. Cancer 2011, 128, 1955–1965. [Google Scholar] [CrossRef] [PubMed]
- Aponte, M.; Ungaro, F.; d’Angelo, I.; De Caro, C.; Russo, R.; Blaiotta, G.; Dal Piaz, F.; Calignano, A.; Miro, A. Improving in vivo conversion of oleuropein into hydroxytyrosol by oral granules containing probiotic Lactobacillus plantarum 299v and an Olea europaea standardized extract. Int. J. Pharm. 2018, 543, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, R. Anti-cancer properties of olive oil secoiridoid phenols: A systematic review of in vivo studies. Food Funct. 2016, 7, 4145–4159. [Google Scholar] [CrossRef] [PubMed]
- Tresserra-Rimbau, A.; Lamuela-Raventos, R.M.; Moreno, J.J. Polyphenols, food and pharma. Current knowledge and directions for future research. Biochem. Pharmacol. 2018, 156, 186–195. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruzzolini, J.; Peppicelli, S.; Andreucci, E.; Bianchini, F.; Scardigli, A.; Romani, A.; La Marca, G.; Nediani, C.; Calorini, L. Oleuropein, the Main Polyphenol of Olea europaea Leaf Extract, Has an Anti-Cancer Effect on Human BRAF Melanoma Cells and Potentiates the Cytotoxicity of Current Chemotherapies. Nutrients 2018, 10, 1950. https://doi.org/10.3390/nu10121950
Ruzzolini J, Peppicelli S, Andreucci E, Bianchini F, Scardigli A, Romani A, La Marca G, Nediani C, Calorini L. Oleuropein, the Main Polyphenol of Olea europaea Leaf Extract, Has an Anti-Cancer Effect on Human BRAF Melanoma Cells and Potentiates the Cytotoxicity of Current Chemotherapies. Nutrients. 2018; 10(12):1950. https://doi.org/10.3390/nu10121950
Chicago/Turabian StyleRuzzolini, Jessica, Silvia Peppicelli, Elena Andreucci, Francesca Bianchini, Arianna Scardigli, Annalisa Romani, Giancarlo La Marca, Chiara Nediani, and Lido Calorini. 2018. "Oleuropein, the Main Polyphenol of Olea europaea Leaf Extract, Has an Anti-Cancer Effect on Human BRAF Melanoma Cells and Potentiates the Cytotoxicity of Current Chemotherapies" Nutrients 10, no. 12: 1950. https://doi.org/10.3390/nu10121950
APA StyleRuzzolini, J., Peppicelli, S., Andreucci, E., Bianchini, F., Scardigli, A., Romani, A., La Marca, G., Nediani, C., & Calorini, L. (2018). Oleuropein, the Main Polyphenol of Olea europaea Leaf Extract, Has an Anti-Cancer Effect on Human BRAF Melanoma Cells and Potentiates the Cytotoxicity of Current Chemotherapies. Nutrients, 10(12), 1950. https://doi.org/10.3390/nu10121950