Hydrocortisone, Ascorbic Acid and Thiamine (HAT Therapy) for the Treatment of Sepsis. Focus on Ascorbic Acid

Abstract
1. Introduction
2. Vitamin C
3. Hydrocortisone
4. Thiamine
5. Hydrocortisone, Ascorbic Acid, and Thiamine (HAT) in Combination
6. Conclusions
Funding
Conflicts of Interest
References
- Fleischmann, C.; Scherag, A.; Adhikari, N.K.; Hartog, C.S.; Tsaganos, T.; Angus, D.C.; Reinhart, K. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Gaieski, D.F.; Edwards, J.M.; Kallan, M.J.; Carr, B.G. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit. Care Med. 2013, 41, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, E.K.; Rubenstein, A.R.; Radin, G.T.; Wiener, R.S.; Walkey, A.J. Two decadesof mortality trends among patients with severe sepsis: A comparative meta-analysis. Crit. Care Med. 2014, 42, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Kaukonen, K.M.; Bailey, M.; Suzuki, S.; Pilcher, D.; Bellomo, R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA 2014, 311, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Torio, C.M.; Moore, B.J. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2013: Statistical Brief #240; Healthcare Costs and Utilization Project (HCUP) Statistical Briefs; Agency for Healthcare Research and Quality: Rockville, MD, USA, 2016.
- Karlsson, S.; Ruokonen, E.; Varpula, T.; Ala-Kokko, T.I.; Pettila, V. Long-term outcome and quality-adjusted life years after severe sepsis. Crit. Care Med. 2009, 37, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Yende, S.; Austin, S.; Rhodes, A.; Finfer, S.; Opal, S.; Thompson, T.; Bozza, F.; Angus, D.C. Long-term qualityof life among survivors of severe sepsis: Analyses of two international trials. Crit. Care Med. 2016, 44, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.M.; Chu, H.; Chao, P.W.; Lee, Y.J.; Kuo, S.C.; Chen, T.J.; Tseng, C.M. Long-term mortality and major adverse cardiovascular events in sepsis survivors. A Nationwide population-based study. Am. J. Respir. Crit. Care Med. 2016, 194, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Prescott, H.C.; Langa, K.M.; Liu, V.; Escobar, G.J. Increased 1-year healthcare use in survivors of severe sepsis. Am. J. Respir. Crit. Care Med. 2014, 190, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.; Lei, X.; Prescott, H.C.; Kipnis, P.; Iwashyna, T.J. Hospital readmission and healthcare utilization following sepsis in community settings. J. Hosp. Med. 2014, 9, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.A.; Pike, F.; Alvarez, K.; Angus, D.C.; Newman, A.B. Bidirectional relationship between cognitive function and pneumonia. Am. J. Respir. Crit. Care Med. 2013, 188, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.C.; Angus, D.C. Enhancing recovery from sepsis. JAMA 2018, 319, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Osler, W. The Evolution of Modern Medicine; Yale University Press: New Haven, CT, USA, 1921. [Google Scholar]
- Calvano, S.E.; Xiao, W.; Richards, D.R.; Felciano, R.M.; Baker, H.V.; Cho, R.J.; Moldawer, L.L.; Lowry, S.F. A network-based analysis of systemic inflammation in humans. Nature 2005, 437, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.M.; Huang, S.J.; McLean, A.S. Genome-wide transcription profiling of human sepsis: A systematic review. Crit Care 2010, 14, R237. [Google Scholar] [CrossRef] [PubMed]
- Landry, D.W.; Oliver, J.A. Pathogenesis of vasodilatory shock. N. Engl. J. Med. 2001, 345, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Slutsky, A.S. Sepsis and endothelial permeability. N. Engl. J. Med. 2010, 363, 689–691. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Harrison, F.E. Role of vitamin C in the function of the vascular endothelium. Antioxid. Redox Signal. 2013, 19, 2068–2083. [Google Scholar] [CrossRef] [PubMed]
- Spoelstra-de Man, A.M.; Elbers, P.W.; Oudermans-van Straaten, H.M. Making sense of early high-dose intravenous vitamin C in ischemia/reperfusion injury. Crit Care 2018, 22, 70. [Google Scholar] [CrossRef] [PubMed]
- Prauchner, C.A. Oxidative stress in sepsis: Pathophysiological implications justifying antioxidant co-therapy. Burns 2017, 43, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Fessel, J.P.; May, A.K.; Roberts, L.J. Plasma biomarkers of oxidant stress and development of organ failure in severe sepsis. Shock 2011, 36, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Gupta, E.; Kaushik, S.; Kumar Srivastava, V.; Mehta, S.K. Evaluation of oxidative stress and antioxidant status: Correlation with the severity of sepsis. Scand. J. Immunol. 2018, 87, e12653. [Google Scholar] [CrossRef] [PubMed]
- Quoilin, C.; Mouithys-Mickalad, A.; Lecart, S.; Fontaine-Aupart, M.P.; Hoebeke, M. Evidence of oxidative stress and mitochondrial respiratory chain dysfunction in an in vitro model of sepsis-induced kidney injury. Biochim. Biophys. Acta 2014, 1837, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.J.; Tsalik, E.L.; van Velkinburgh, J.C.; Glickman, S.W. An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci. Transl. Med. 2013, 5, 195ra195. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.J.; Tipper, J.L.; Bruse, S.; Baron, R.M.; Tsalik, E.L.; Huntley, J.; Choi, A.M. Integrative “omic” analysis of experimental bacteremia identifies a metabolic signature that distinguishes human sepsis from systemic inflammatory response syndromes. Am. J. Respir. Crit. Care Med. 2014, 190, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Beesley, S.J.; Weber, G.; Sarge, T.; Nikravan, S. Septic cardiomyopathy. Crit. Care Med. 2018, 46, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Haileselassie, B.; Su, E.; Pozios, I.; Nino, D.F.; Liu, H.; Lu, D.Y.; Ventoulis, I. Myocardial oxidative stress correlates with left ventricular dysfunction on strain echocardiography in a rodent model of sepsis. Intensive Care Med. Exp. 2017, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F. Oxidative stress and mitochondrial dysfunction in sepsis. Br. J. Anaesth. 2011, 107, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Alamdari, N.; Constantin-Teodosiu, D.; Murton, A.J.; Gardiner, S.M.; Bennett, T.; Layfield, R.; Greenhaff, P.L. Temporal changes in the involvement of pyruvate dehydrogenase complex in muscle lactate accumulation during lipopolysaccharide infusion in rats. J. Physiol. 2008, 586, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.W. Potential dysregulation of the pyruvate dehydrogenase complex by bacterial toxins and insulin. J. Trauma 2009, 67, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Artenstein, A.W.; Higgins, T.L.; Opal, S.M. Sepsis and scientific revolutions. Crit. Care Med. 2013, 41, 2770–2772. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003, 101, 3765–3777. [Google Scholar] [CrossRef] [PubMed]
- Blythe, R.; Cook, D.; Graves, N. Septicemia: The impact on the health system and patients of dealying new treatments with uncertain evidence: A case study of the sepsis bundle. F1000Research 2018, 7, 500. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; Davies, M.J.; Webster, N.R. Ascorbyl radical formation in patients with sepsis: Effect of ascorbate loading. Free Radic. Biol Med. 1996, 20, 139–143. [Google Scholar] [CrossRef]
- Borrelli, E.; Roux-Lombard, P.; Grau, G.E.; Giradin, E.; Ricou, B.; Dayer, J.; Suter, P.M. Plasma concentrations of cytokines, their soluble receptors, and antioxidant vitamins can predict the development of multiple organ failure in patients at risk. Crit. Care Med. 1996, 24, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Evans-Olders, R.; Eintracht, S.; Hoffer, L.J. Metabolic origin of hypovitaminosis C in acutely hospitalized patients. Nutrition 2010, 26, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.; Cadenas, S.; Herrero, A.; Mendez, J.; Batja, G. Endotoxin depletes ascorbate in the guinea pig heart. Protective effects of vitamins C and E against oxidative stress. Life Sci. 1996, 59, 649–657. [Google Scholar] [CrossRef]
- Armour, J.; Tyml, K.; Lidington, D.; Wilson, J.X. Ascorbate prevents microvascular dysfunction in the skeletal muscle of the septic rat. J. Appl. Physiol. 2001, 90, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Victor, V.M.; Guayerbas, N.; Puerto, M.; De la Fuente, M. Changes in the ascorbic acid levels of peritoneal lymphocytes and macrophages of mice with endotoxin-induced oxidative stress. Free Radic. Res. 2001, 35, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wilson, J.X.; Tyml, K. Ascorbate inhibits iNOS expression and preserves vasoconstrictor responsiveness in skeletal muscle of septic mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R50–R56. [Google Scholar] [CrossRef] [PubMed]
- Tyml, K.; Li, F.; Wilson, J.X. Delayed ascorbate bolus protects against maldistribution of microvascular blood flow in septic rat skeletal muscle. Crit. Care Med. 2005, 33, 1823–1828. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.A.; Syed, A.A.; Knowlson, S.; Sculthorpe, R.; Farthing, D.; DeWilde, C.; Farthing, C.A.; Laruss, T.L.; Martin, E.; Brophy, D.F. Phase 1 safety trial of intravenous ascorbic acid in patients with severe sepsis. J. Transl. Med. 2014, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Marik, P.E.; Khangoora, V.; Rivera, R.; Hooper, M.H.; Catravas, J. Hydrocortisone, Vitamin C and Thiamine for the treatment of severe sepsis and septic shock: A retrospective before-after study. Chest 2017, 151, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- De Grooth, H.M.; Spoeistra-de Man, A.M.; Oudermans-van Straaten, H.M. Early plasma vitamin C concentration, organ dysfunction and ICU mortality [Abstract]. Intensive Care Med. 2014, 40, S199. [Google Scholar]
- Carr, A.C.; Rosengrave, P.C.; Bayer, S.; Chambers, S.; Mehrtens, J.; Shaw, G.M. Hypovitaminosis C and vitamin C deficiency in critically ill patients despite recommended enteral and parenteral intakes. Crit Care 2017, 21, 300. [Google Scholar] [CrossRef] [PubMed]
- De Grooth, H.J.; Manubulu-Choo, W.P.; Zandvliet, A.; Spoelstra-de Man, A.M.; Swart, E. Oudermans-van Straaten HM. Vitamin C pharmacokinetics in critically ill patients: A randomized trial of four intravenous regimens. Chest 2018, 153, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.X. Evaluation of vitamin C for adjuvant sepsis therapy. Antioxid. Redox Signal. 2013, 19, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Oudemans-van Straaten, H.M.; Spoelstra-de Man, A.M.; de Waard, M.C. Vitamin C revisited. Crit Care 2014, 18, 460. [Google Scholar] [CrossRef] [PubMed]
- Sagun, K.C.; Carcamo, J.M.; Golde, D.N. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J. 2005, 19, 1657–1667. [Google Scholar]
- Lowes, D.A.; Webster, N.R.; Galley, H.F. Dehydroascorbic acid as pre-conditioner: Protection from lipopolysaccharide induced mitochondrial damage. Free Radic. Res. 2010, 44, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Puskarich, M.A.; Kline, J.A.; Krabill, V.; Claremont, H.; Jones, A.E. Preliminary safety and efficacy of L-carnitine infusion for the treatment of vasopressor-dependent septic shock: A randomized control trial. JPEN 2014, 38, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Dhar-Mascareno, M.; Carcamo, J.M.; Golde, D.W. Hypoxia-reoxygenation-induced mitochondrial damage and apotosis in human endothelial cells inhibited by vitamin C. Free Radic. Biol. Med. 2005, 38, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, S.M. Effect of ascorbic acid on hepatic vasoregulatory gene expression during polymicrobial sepsis. Life Sci. 2004, 75, 2015–2026. [Google Scholar] [CrossRef] [PubMed]
- Szakmany, T. N-acetylcysteine for sepsis and systemic inflammatory response in adults. Cochrane Database Syst. Rev. 2012, 9, CD006616. [Google Scholar] [CrossRef] [PubMed]
- Molnar, Z. N-acetylcysteine as the magic bullet: Too good to be true. Crit. Care Med. 2008, 36, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Carcamo, J.M.; Pedraza, A.; Borquez-Ojeda, O.; Golde, D.W. Vitamin C suppresses TNF-alpha induced NFkB activation by inhibiting IkB-alpha phosphorylation. Biochemistry 2002, 41, 12995–13002. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Kim, Y.M.; Park, E.J.; Kim, J.W.; Park, S.W.; Kim, H.J. Ascorbic acid reduces HMGB1 secretion in lipopolysaccharide-activated RAW 264.7 cells and improves survival rate in septic mice by activation of Nrf2/HO-1 signals. Biochem. Pharmacol. 2015, 95, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Hagel, A.F.; Layritz, C.M.; Hagel, W.H.; Hagel, H.J.; Hagel, E.; Dauth, W.; Kressel, J.; Regnet, T. Intravenous infusion of ascorbic acid decreases serum histamine concentrations in patients with allergic and non-allergic diseases. Naunyn-Schmiedebergs Arch. Pharmacol. 2013, 386, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Yamazaki, M.; Ohashi, W.; Tanaka, S.; Hattori, K.; Todoroki, K. Critical role of endogenous histamine in promoting end-organ tissue injury in sepsis. Intensive Care Med. Exp. 2016, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Shaw, G.; Fowler, A.A.; Natarajan, R. Ascorbate-dependent vasopressor synthesis- a rationale for vitamin C administration in severe sepsis and septic shock? Crit Care 2015, 19, 418. [Google Scholar] [CrossRef] [PubMed]
- Klenner, F.R. The treatment of poliomyelitis and other virus diseases with vitamin C. South. Med. Surg. 1949, 111, 209–214. [Google Scholar] [PubMed]
- Pauling, L. Ascorbic acid and the common cold. Am. J. Clin. Nutr. 1971, 24, 1294. [Google Scholar] [CrossRef] [PubMed]
- Hemila, H.; Chalker, E. Vitamin C for preventing and treating the common cold. Cochrane Database Syst. Rev. 2013, 1, CD000980. [Google Scholar] [CrossRef] [PubMed]
- Hemila, H. Does vitamin C alleviate the symptoms of the common cold?—A review of current evidence. Scand. J. Infect. Dis. 1994, 26, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Furuya, A.; Uozaki, M.; Yamasaki, H.; Arakawa, T.; Arita, M.; Koyama, A.H. Antiviral effects of ascorbic and dehydroascorbic acids in vitro. Int. J. Mol. Med. 2008, 22, 541–545. [Google Scholar] [PubMed]
- Madhusudana, S.N. In vitro inactivation of the rabies virus by ascorbic acid. Int. J. Infect. Dis. 2004, 8, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R. Vitamin C and cellular immune functions. Protection against hypochlorous acid-mediated inactivation of glyceraldehyde-3-phosphate dehydrogenase and ATP generation in human leukocytes as a possible mechanism of ascorbate-mediated immunostimulation. Ann. N. Y. Acad. Sci. 1990, 587, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Manzella, J.P.; Roberts, N.J. Human macrophage and lymphocyte responses to mitogen stimulation after exposure to influenza virus, ascorbic acid, and hyperthermia. J. Immunol. 1979, 123, 1940–1944. [Google Scholar] [PubMed]
- Siegel, B.V. Enhancement of interferon production by poly(rI)-poly(rC) in mouse cell cultures by ascorbic acid. Nature 1975, 254, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.J.; Kim, J.H.; Hong, J.M.; Kang, J.S.; Kim, H.R. Vitamin C treatment of mouse bone marrow-derived dendritic cells enhanced CD8(+) memory T cell production capacity of these cells in vivo. Immunobiology 2014, 219, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999, 13, 1007–1024. [Google Scholar] [CrossRef] [PubMed]
- Paolini, M.; Pozzetti, L.; Pedulli, G.F.; Marchesi, E.; Canteli-Forti, G. The nature of prooxidant activity of vitamin C. Life Sci. 1999, 64, L-8. [Google Scholar] [CrossRef]
- Prat, A.G.; Turrens, J.F. Ascorbate- and hemoglobin-dependent brain chemiliminescence. Free Radic. Biol. Med. 1990, 8, 319–325. [Google Scholar] [CrossRef]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassu, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef] [PubMed]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.Y.; Lee, S.M. Protective effect of low dose of ascorbic acid on hepatobiliary function in hepatic ischemia/reperfusion in rats. J. Hepatol. 2002, 36, 72–77. [Google Scholar] [CrossRef]
- Park, S.W.; Lee, S.M. Antioxidant and prooxidant properties of ascorbic acid on hepatic dysfunction induced by cold ischemia/reperfusion. Eur. J. Pharmacol. 2008, 580, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Motl, J.; Radhakrishnan, J.; Ayoub, I.M.; Grmec, S.; Gazmuri, R.J. Vitamin C compromises cardiac resuscitability in a rat model of ventricular fibrillation. Am. J. Ther. 2014, 21, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Kuck, J.L.; Bastarache, J.A.; Shaver, C.M.; Fessel, J.P.; Dikalov, S.I.; May, J.M.; Ware, L.B. Ascorbic acid attenuates endothelial permeability triggered by cell-free hemoglobin. Biochem. Biophys. Res. Commun. 2018, 495, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Levine, M. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Padayatty, S.J.; Espey, M.G. Vitamin C: A concentration-function approach yields pharmacology and therapeutic discoveries. Adv. Nutr. 2011, 2, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Muhlhofer, A.; Mrosek, S.; Schlegel, B.; Trommer, W.; Rozario, F.; Bohles, H.; Schremmer, D.; Zoller, W.G.; Biesalski, H.K. High-dose intravenous vitamin C is not associated with an increase of pro-oxidative biomarkers. Eur. J. Clin. Nutr. 2004, 58, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Matsuda, T.; Miyagantani, Y.; Yukioka, T.; Matsuda, H.; Shimazaki, S. Reduction of resuscitation fluid volumes in severely burned patients using ascorbic acid administration: A randomized, prospective study. Arch. Surg. 2000, 135, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Basili, S.; Tanzilli, G.; Mangieri, E.; Raparelli, V.; Di Santo, S. Intravenous ascorbic acid infusion improves myocardial perfusion grade during elective percutaneous coronary intervention: Relationship with oxidative stress markers. JACC Cardiovasc. Interv. 2010, 3, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J. The effect of intravenous vitamin C infusion on periprocedural myocardial injury for patients undergoing elective percutaneous coronary intervention. Can. J. Cardiol. 2014, 30, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Hu, X. Efficacy and safety of vitamin C for atrial fibrillation after cardiac surgery: A meta-analysis with trial sequential analysis of randomized controlled trials. Int. J. Surg. 2017, 37, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zheng, X.; Liang, B.; Gao, J.; Gu, Z. Vitamins for prevention of contrast-induced acute kidney injury: A systematic review and trial sequential analysis. Am. J. Cardiovasc. Drugs 2018, 18, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Topete, D.; Cidlowski, J.A. One hormone, two actions: Anti-and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Busillo, J.M.; Cidlowski, J.A. The five Rs of glucocorticoid action during inflammation: Ready, reinforce, repress, resolve and restore. Trends Endocrinol. Metab. 2013, 24, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Glucocorticosteroids: Current and future directions. Br. J. Pharmacol. 2011, 163, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Franchimont, D.; Hiroi, N.; Frey, G.; Boettner, A.; Ehrhart-Bornstein, M.; O’Shea, J.J.; Chrousos, G.P.; Bornstein, S.R. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 2002, 16, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Keh, D.; Boehnke, T.; Weber-Cartens, S.; Schulz, C.; Ahlers, O.; Bercker, S.; Volk, H.D.; Doecke, W.D.; Falke, K.J.; Gerlach, H. Immunologic and hemodynamic effects of “low-dose” hydrocortisone in septic shock: A double-blind, randomized, placebo-controlled, crossover study. Am. J. Respir. Crit. Care Med. 2003, 167, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Rygard, S.L.; Butler, E.; Granholm, A.; Moller, M.H.; Cohen, J.; Finfer, S.; Perner, A.; Myburgh, J.; Venkatesh, B.; Delaney, A. Low-dose corticosteroids for adults with septic shock: A systematic review with meta-analysis and trial sequential analysis. Intensive Care Med. 2018, 44, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Minneci, P.C.; Deans, K.J.; Eichacker, P.Q.; Natanson, C. The effects of steroids during sepsis depend on dose and severity of illness: An updated meta-analysis. Clin. Microbiol. Infect. 2009, 15, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, M.; Wetterslev, J.; Gluud, C.; Zijlstra, J.G.; van der Horst, I.C.; Keus, F. Glucocorticosteroids for sepsis: Systematic review with meta-analysis and trial sequential analysis. Intensive Care Med. 2015, 41, 1220–1234. [Google Scholar] [CrossRef] [PubMed]
- Kalil, A.C.; Sun, J. Low-dose steroids for septic shock and severe sepsis; the use of Bayesian statistics to resolve clinical trial controversies. Intensive Care Med. 2011, 37, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, B.; Finfer, S.; Cohen, J.; Rajbhandari, D.; Arabi, Y.; Billot, L.; Glass, P.; Perner, A.; Bellomo, R.; Myburgh, J. Adjunctive glucocorticoid therapy in patients with septic shock. N. Engl. J. Med. 2018, 378, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Renault, A.; Brub-Buisson, C.; Megarbane, Z.B.; Quenot, J.P.; Siami, S. Hydrocortisone plus fludrocortisone for adults with septic shock. N. Engl. J. Med. 2018, 378, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Collie, J.T.; Greaves, R.F.; Jones, O.A.; Lam, Q.; Eastwood, G.M.; Bellomo, R. Vitamin B1 in critically ill patents: Needs and challenges. Clin. Chem. Lab. Med. 2017, 55, 1652–1668. [Google Scholar] [CrossRef] [PubMed]
- Manzetti, S.; Zhang, J.; van der Spoel, D. Thiamin function, metabolism, uptake, and transport. Biochemistry 2014, 53, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, A.M.; Telfer, A.B.; Shenkin, A. Thiamine deficiency in the critically ill. Intensive Care Med. 1988, 14, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Donnino, M.W.; Carney, E.; Cocchi, M.N.; Barbash, I.; Chase, M. Thiamine deficiency in critically ill patients with sepsis. J. Crit. Care 2010, 25, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Donnino, M.W.; Andersen, L.W.; Chase, M.; Berg, K.M.; Tidswell, M.; Giberson, T.; Wolfe, R.; Moskowitz, A. Randomized, double-blind, placebo-controlled trial of thiamine as a metabolic resuscitator in septic shock: A pilot study. Crit. Care Med. 2016, 44, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Hazell, A.S.; Faim, S.; Wertheimer, G.; Silva, V.R.; Marques, C.S. The impact of oxidative stress in thiamine deficiency: A multifactorial targeting issue. Neurochem. Int. 2013, 62, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Zhang, H. Interactions of oxidative stress with thiamine homeostasis promote neurodegeneration. Neurochem. Int. 2002, 40, 493–504. [Google Scholar] [CrossRef]
- Gioda, C.R.; de Oliveira Barreto, T.; Primola-Gomes, T.N.; de Lima, D.C.; Campos, P.P.; Cruz, J.S. Cardiac oxidative stress is involved in hart fialure induced by thiamine deprivation in rats. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H2039–H2045. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, A.; Anderson, L.W.; Cocchi, M.N.; Karlsson, M.; Patel, P.V.; Donnino, M.W. Thiamine as a renal protective agent in septic shock: A secondary analysis of a randomized, double-blind, placebo-controlled trial. Ann. Am. Thorac. Soc. 2017, 14, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Woolum, J.A.; Abner, E.L.; Kelly, A.; Thompson Bastin, M.L.; Morris, P.E.; Flannery, A.H. Effect of thiamine administration on lactate clearance and mortality in patients with septic shock. Crit. Care Med. 2018, 46, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N.; Khangoora, V.; Marik, P.E.; Catravas, J.D. Hydrocortisone and Ascorbic Acid synergistically protect and repair lipopolysaccharide-induced pulmonary endothelial barrier dysfunction. Chest 2017, 152, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Azari, O.; Kheirandish, R.; Azizi, S.; Farajli Abassi, M.; Bidi, M. Protective effects of hydrocortisone, Vitamin C and E alone or in combination against renal-ischemia-reperfusion injury rat. Iran. J. Pathol. 2015, 10, 272–280. [Google Scholar] [PubMed]
- Tavasoli, M.; Azari, O.; Kheirandish, R. Evaluation of combination therapy with hydrocortisone, vitamin C and vitamin E in a rat model of intestine ischemia-reperfusion injury. Comp. Clin. Pathol. 2018, 27, 433–439. [Google Scholar] [CrossRef]
- Kim, W.Y.; Jo, E.J.; Eom, J.S.; Mok, J.; Kim, M.H.; Kim, K.U.; Park, H.K.; Lee, M.K.; Lee, K. Combined vitamin, C.; hydrocortisone, and thiamine therapy for patients with severe pneumonia who were admitted to the intensive care unit: Propensity score-based analysis of a before-after cohort study. J. Crit. Care 2018, 47, 211–218. [Google Scholar] [CrossRef] [PubMed]



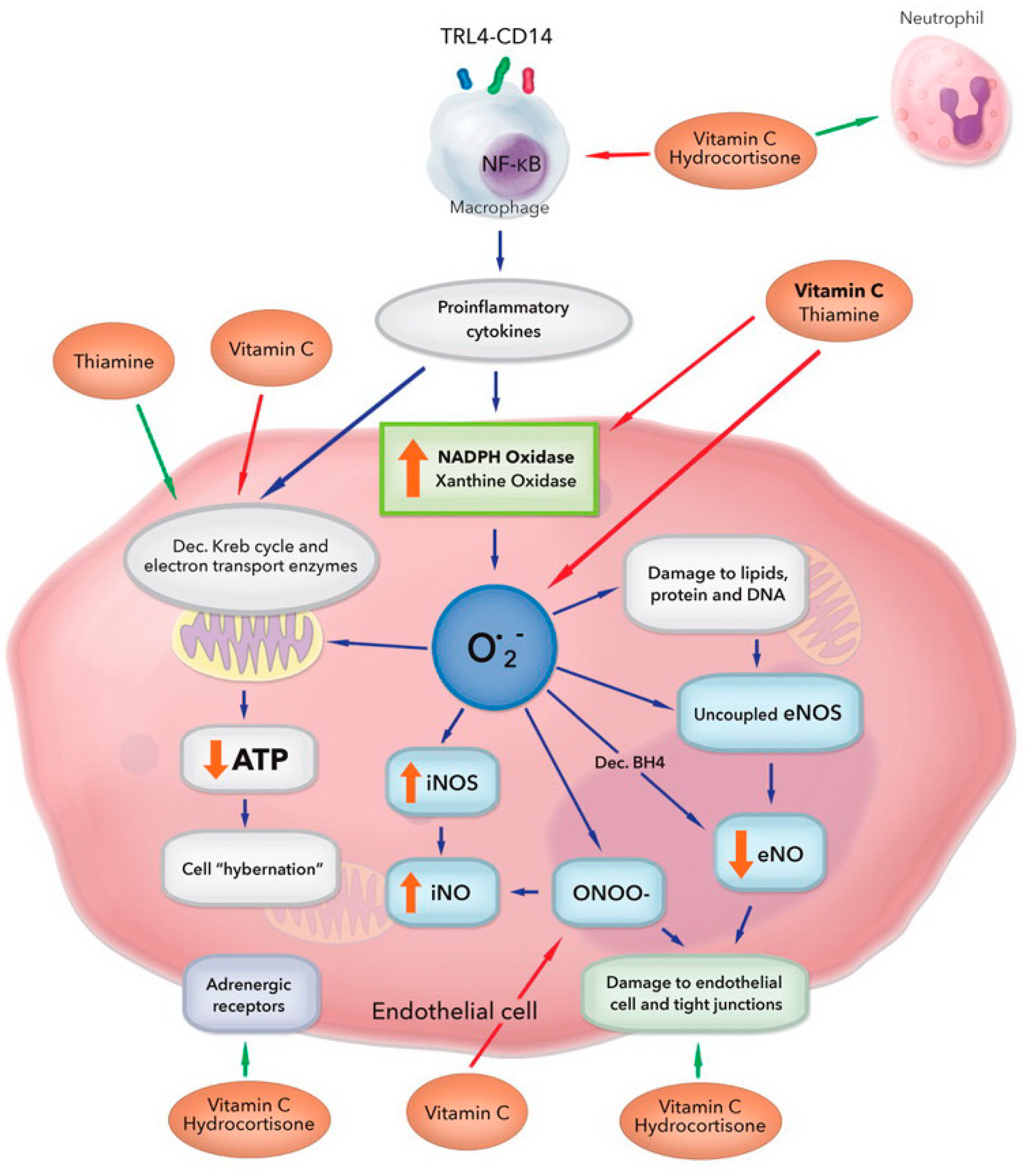{kind=link}
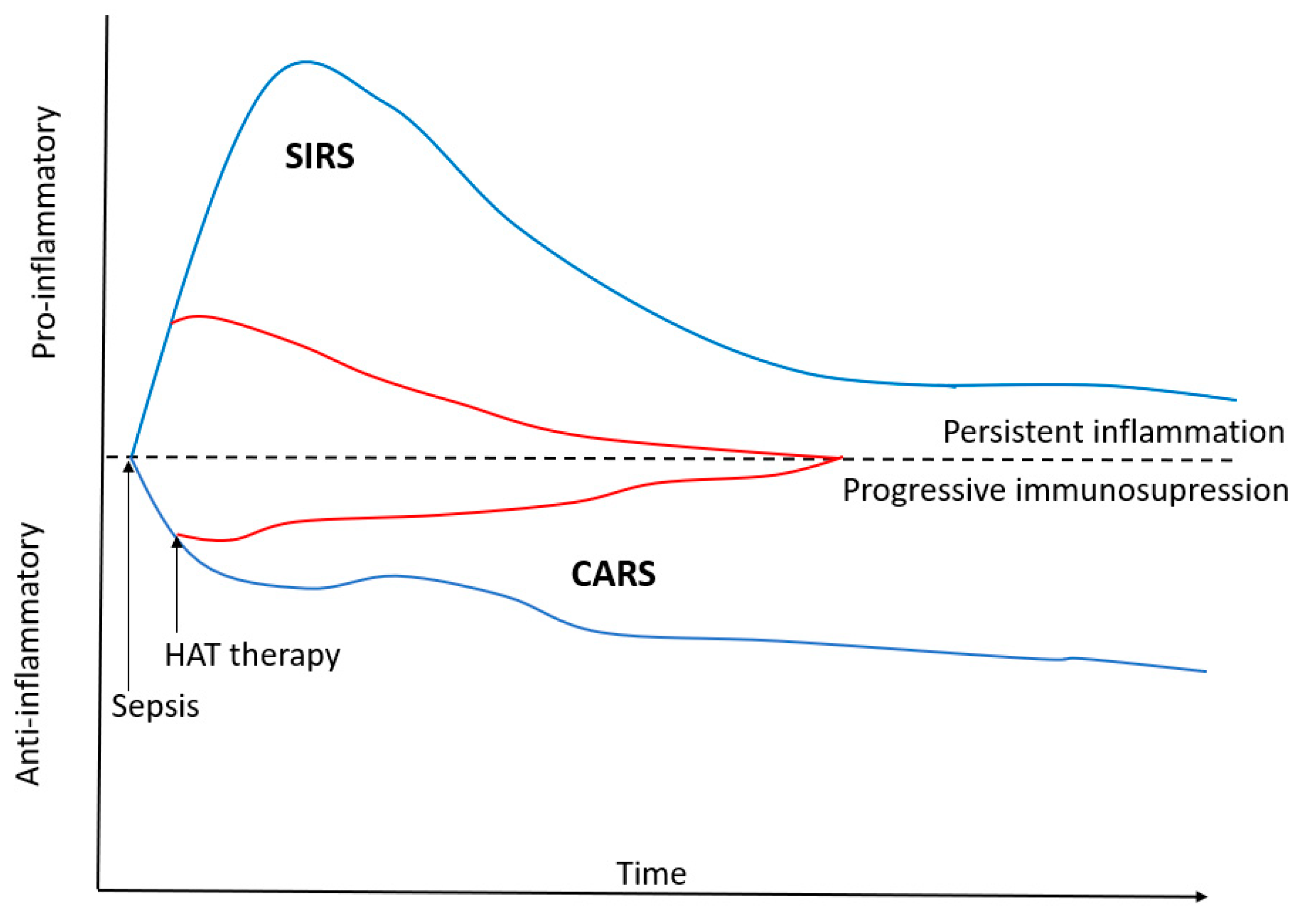{kind=link}
| Key Role | Mechanism |
|---|---|
| Antioxidant | Scavenges extracellular, intracellular and mitochondrial ROS; limits oxidation of mitochondrial proteins, enzymes, lipoproteins, cell membrane, etc. |
| Anti-inflammatory | Inhibits activation of NFκB, decreases HMGB1, inhibits histamine, prevents NETosis, inactivates HIF-1α |
| Microcirculation | Increases eNOS, decreases iNOS, preserves tight junctions |
| Immune function | Supports lymphocyte proliferation, increases neutrophil bacteriocidal action, improves chemotaxis, stimulates interferon production, decreases T regulatory cells (Tregs) |
| Anti-thrombotic | Decreases platelet activation and tissue factor expression, increases thrombomodulin |
| Synthesis of catecholamines | Acts as a cofactor in synthesis of epinephrine, dopamine and vasopressin.Increases adrenergic sensitivity |
| Wound Healing | Hydroxylation of procollagen, increased expression of collagen mRNA |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marik, P.E. Hydrocortisone, Ascorbic Acid and Thiamine (HAT Therapy) for the Treatment of Sepsis. Focus on Ascorbic Acid. Nutrients 2018, 10, 1762. https://doi.org/10.3390/nu10111762
Marik PE. Hydrocortisone, Ascorbic Acid and Thiamine (HAT Therapy) for the Treatment of Sepsis. Focus on Ascorbic Acid. Nutrients. 2018; 10(11):1762. https://doi.org/10.3390/nu10111762
Chicago/Turabian StyleMarik, Paul E. 2018. "Hydrocortisone, Ascorbic Acid and Thiamine (HAT Therapy) for the Treatment of Sepsis. Focus on Ascorbic Acid" Nutrients 10, no. 11: 1762. https://doi.org/10.3390/nu10111762
APA StyleMarik, P. E. (2018). Hydrocortisone, Ascorbic Acid and Thiamine (HAT Therapy) for the Treatment of Sepsis. Focus on Ascorbic Acid. Nutrients, 10(11), 1762. https://doi.org/10.3390/nu10111762