
Effects of Different Nitrogen Sources on the Formation of Biogenic Jarosite



Abstract
:1. Introduction
2. Materials and Methods
2.1. Concentrated Solution of A. ferrooxidans LX5 and Nitrogen-Free 9K Solution
2.2. Effects of Nitrogen Sources on the Mineralization of A. ferrooxidans LX5
2.3. Analytical Methods
3. Results and Discussion
3.1. Change in the pH of a Solution during the Reaction with Different Nitrogen Sources
- ①
- Oxidation of Fe2+ to Fe3+ involving consumption of H+.
- ②
- Hydrolysis of Fe3+ to schwertmannite or jarosites and release of H+.
3.2. Effects of Nitrogen Source on Fe2+ Oxidization
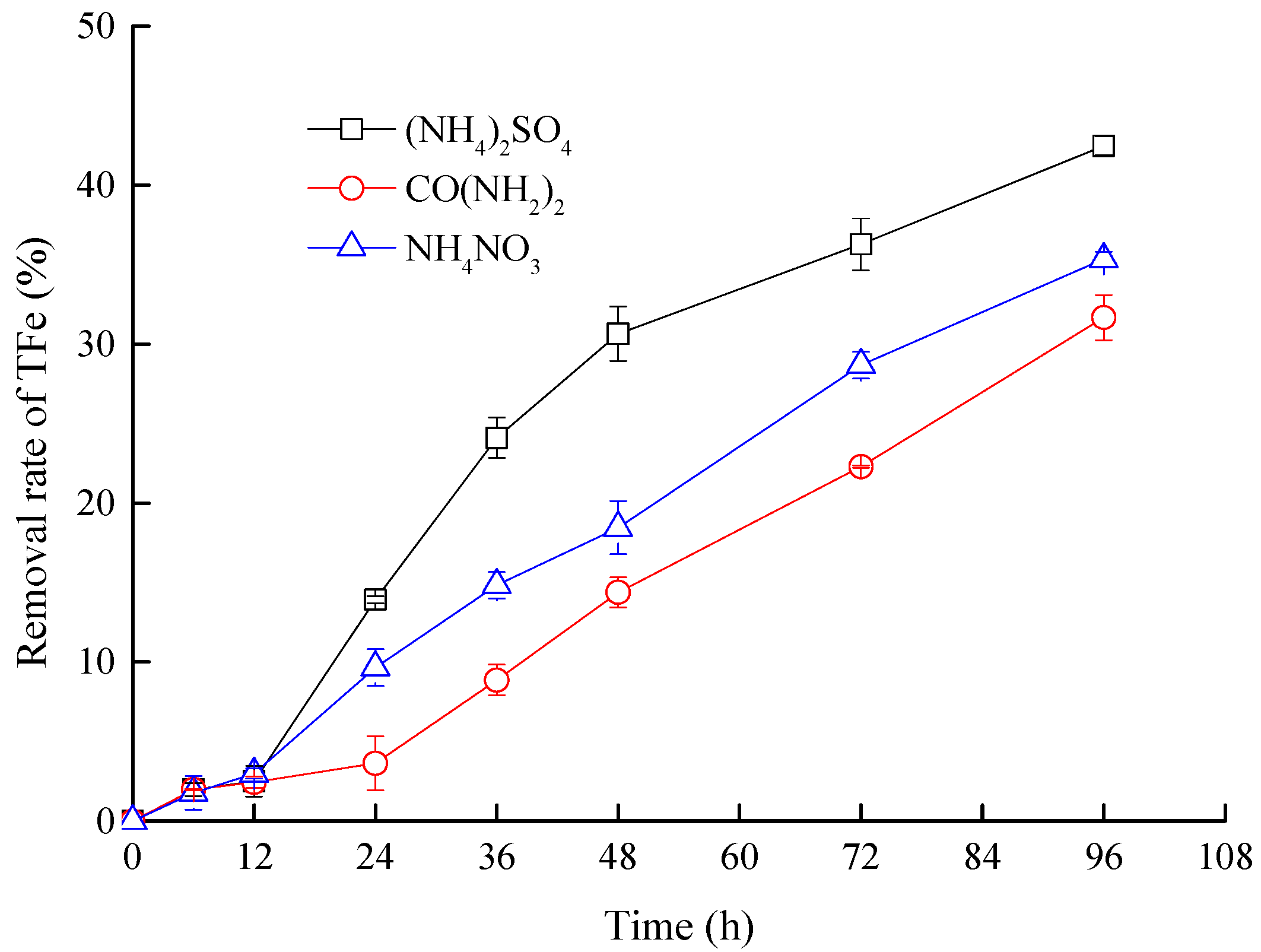
3.3. Effects of Nitrogen Sources on Mineralization Efficiency
3.4. Identification and Analysis of Sediments
3.4.1. XRD and Chemical Element Analysis
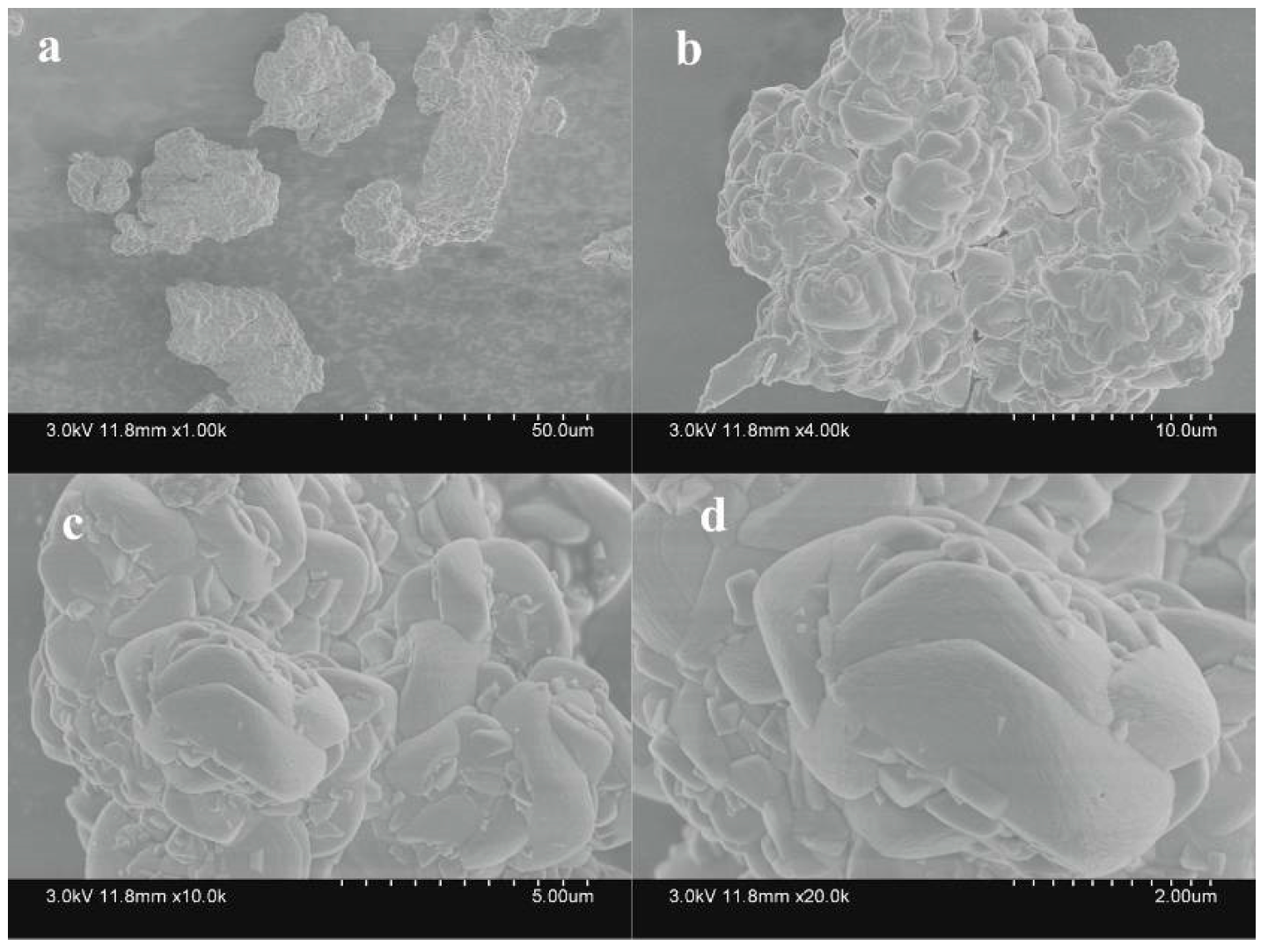
3.4.2. SEM Analysis
4. Conclusions
- (1)
- The pH of the solution was decreased by A. ferrooxidans. Carbamide supplementation yielded the highest pH value, followed by treatment with NH4NO3 and (NH4)2SO4.
- (2)
- The utilization efficiency of (NH4)2SO4 was the highest, followed by NH4NO3. The microbial activity of A. ferrooxidans LX5 was the lowest in the CO(NH2)2-dosing reaction system. The rate of TFe precipitation in the (NH4)2SO4-containing system was substantially higher than in the carbamide- and NH4NO4-containing systems. Compared with the other two nitrogen sources, (NH4)2SO4 was strongly conducive to the growth of A. ferrooxidans LX5.
- (3)
- The morphologies and chemical compositions of minerals varied slightly when different nitrogen sources were used. However, the peak positions on the XRD spectra were basically consistent. The resultant secondary mineral was a mixture of jarosite, ammonioiarosite, and carphosiderite.
- (4)
- Using (NH4)2SO4 as a nitrogen source, A. ferrooxidans LX5 oxidized Fe2+ to Fe3+ within 36 h at a low pH of 2.5. At the end of the 96 h experiment, approximately 42.48% of soluble Fe yielded secondary iron-containing minerals, which were effectively removed. The enhanced biological oxidation of Fe2+ compared with traditional neutralization technique has significant practical implications for lime neutralization. It minimizes the usage of neutralizers, and decreases sludge generation.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rambabu, K.; Banat, F.; Pham, Q.M.; Ho, S.-H.; Ren, N.-Q.; Show, P.L. Biological Remediation of Acid Mine Drainage: Review of Past Trends and Current Outlook. Environ. Sci. Ecotechnol. 2020, 2, 100024. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zhang, C.; Su, P.; Tang, Y.; Huang, Z.; Ma, T. A review of acid mine drainage: Formation mechanism, treatment technology, typical engineering cases and resource utilization. Process Saf. Environ. Prot. 2023, 170, 1240–1260. [Google Scholar] [CrossRef]
- Yang, J.; Wang, R.; Wang, H.; Song, Y. The Important Role of Dissolved Oxygen Supply Regulated by the Hydraulic Shear Force during the Biosynthesis of Iron Hydroxysulfate Minerals. Minerals 2020, 10, 518. [Google Scholar] [CrossRef]
- Sobron, P.; Rull, F.; Sobron, F.; Sanz, A.; Medina, J.; Nielsen, C. Raman spectroscopy of the system iron(III)–sulfuric acid–water: An approach to Tinto River’s (Spain) hydrogeochemistry. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2007, 68, 1138–1142. [Google Scholar] [CrossRef]
- Skousen, J.G.; Ziemkiewicz, P.F.; McDonald, L.M. Acid mine drainage formation, control and treatment: Approaches and strategies. Extr. Ind. Soc. 2019, 6, 241–249. [Google Scholar] [CrossRef]
- Yang, Y.; Li, B.; Li, T.; Liu, P.; Zhang, B.; Che, L. A review of treatment technologies for acid mine drainage and sustainability assessment. J. Water Process Eng. 2023, 55, 104213. [Google Scholar] [CrossRef]
- Raúl, M.G.; Carlos, R.C.; Manuel, O.; Macías, F. Seasonal variability of extremely metal rich acid mine drainages from the Tharsis mines (SW Spain). Environ. Pollut. 2020, 259, 113829. [Google Scholar]
- Tong, L.; Fan, R.; Yang, S.; Li, C. Development and Status of the Treatment Technology for Acid Mine Drainage. Min. Metall. Explor. 2020, 38, 315–327. [Google Scholar] [CrossRef]
- Akinpelu, E.A.; Ntwampe, S.K.O.; Fosso-Kankeu, E.; Nchu, F.; Angadam, J.O. Performance of microbial community dominated by Bacillus spp. in acid mine drainage remediation systems: A focus on the high removal efficiency of SO42−, Al3+, Cd2+, Cu2+, Mn2+, Pb2+, and Sr2+. Heliyon 2021, 7, e072412021. [Google Scholar] [CrossRef]
- Srivastava, S.; Agrawal, S.B.; Mondal, M.K. A review on progress of heavy metal removal using adsorbents of microbial and plant origin. Environ. Sci. Pollut. Res. 2015, 22, 15386–15415. [Google Scholar] [CrossRef]
- Magowo, W.E.; Sheridan, C.; Rumbold, K. Global Co-occurrence of Acid Mine Drainage and Organic Rich Industrial and Domestic Effluent: Biological sulfate reduction as a co-treatment-option. J. Water Process Eng. 2020, 38, 101650. [Google Scholar] [CrossRef]
- Naidu, G.; Ryu, S.; Thiruvenkatachari, R.; Choi, Y.; Jeong, S.; Vigneswaran, S. A critical review on remediation, reuse, and resource recovery from acid mine drainage. Environ. Pollut. 2019, 247, 1110–1124. [Google Scholar] [CrossRef] [PubMed]
- Daraz, U.; Li, Y.; Ahmad, I.; Iqbal, R.; Ditta, A. Remediation technologies for acid mine drainage: Recent trends and future perspectives. Chemosphere 2022, 311, 137089. [Google Scholar] [CrossRef] [PubMed]
- Gersberg, R.; Elkins, B.; Lyon, S.; Goldman, C. Role of aquatic plants in wastewater treatment by artificial wetlands. Pergamon 1986, 20, 363–368. [Google Scholar] [CrossRef]
- Singh, S.; Envelope, S.C.P. Impact of seasonal variation on the treatment response of constructed wetlands receiving acid mine drainage in a subtropical region. J. Water Process Eng. 2022, 49, 103182. [Google Scholar] [CrossRef]
- López, G.O.; Cortina, J.L. Integration of membrane technologies to enhance the sustainability in the treatment of metal-containing acidic liquid wastes. An overview. Sep. Purif. Technol. 2021, 265, 118485. [Google Scholar] [CrossRef]
- Kaur, G.; Couperthwaite, S.J.; Millar, G.J. Alternative neutralisation materials for acid mine drainage treatment. J. Water Process Eng. 2018, 22, 46–58. [Google Scholar] [CrossRef]
- Lauren, B.; Bethany, K. Calcium carbonate in waste flooring for neutralization of acid rock drainage. Mine Water Environ. 2023, 42, 70–77. [Google Scholar]
- Akeil, A.; Koldas, S. Acid mine drainage (AMD): Causes, treatment and case studies. J. Clean. Prod. 2006, 14, 1139–1145. [Google Scholar]
- Meruane, G.; Vargas, T. Bacterial oxidation of ferrous iron by Acidithiobacillus ferrooxidans in the pH range 2.5–7.0. Hydrometallurgy 2003, 71, 149–158. [Google Scholar] [CrossRef]
- Asta, M.P.; Cama, J.; Martínez, M.; Giménez, J. Arsenic removal by goethite and jarosite in acidic conditions and its environmental implications. J. Hazard. Mater. 2009, 171, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Drouet, C.; Baron, D.; Navrotsky, A. On the thermochemistry of the solid solution between jarosite and its chromate analog. Am. Mineral. 2015, 88, 1949–1954. [Google Scholar] [CrossRef]
- Chen, M.; Lu, G.; Guo, C.; Yang, C.; Wu, J.; Huang, W.; Yee, N.; Dang, Z. Sulfate migration in a river affected by acid mine drainage from the Dabaoshan mining area, South China. Chemosphere 2015, 119, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Mastrotheodoros, G.P.; Beltsios, K.G. Pigments-Iron-based red, yellow, and brown ochres. Archaeol. Anthropol. Sci. 2022, 14, 35. [Google Scholar] [CrossRef]
- Song, Y.; Wang, H.; Yang, J.; Cao, Y. Influence of Monovalent Cations on the Efficiency of Ferrous Ion Oxidation, Total Iron Precipitation, and Adsorptive Removal of Cr(VI) and As(III) in Simulated Acid Mine Drainage with Inoculation of Acidithiobacillus ferrooxidans. Metals 2018, 8, 596. [Google Scholar] [CrossRef]
- Ulatowska, J.; Stala, Ł.; Polowczyk, I. Comparison of Cr(VI) Adsorption Using Synthetic Schwertmannite Obtained by Fe3+ Hydrolysis and Fe2+ Oxidation: Kinetics, Isotherms and Adsorption Mechanism. Int. J. Mol. Sci. 2021, 22, 8175. [Google Scholar] [CrossRef]
- Gan, M.; Li, M.-M.; Zeng, J.; Liu, X.-X.; Zhu, J.-Y.; Hu, Y.-H.; Qiu, G.-Z. Acidithiobacillus ferrooxidans enhanced heavy metals immobilization efficiency in acidic aqueous system through bio-mediated coprecipitation. Trans. Nonferrous Met. Soc. China 2017, 27, 1156–1164. [Google Scholar] [CrossRef]
- Li, T.; Wang, Z.; Zhang, Z.; Feng, K.; Liang, J.; Wang, D.; Zhou, L. Organic carbon modified Fe3O4/schwertmannite for heterogeneous Fenton reaction featuring synergistic in-situ H2O2 generation and activation. Sep. Purif. Technol. 2021, 276, 119344. [Google Scholar] [CrossRef]
- Wang, M.; Liang, J.; Zhou, L. The formation of biogenic jarosite by Acidithiobacillus ferrooxidans in the presence of crystal seed and potassium. J. Nanjing Agric. Univ. 2013, 36, 97–102. [Google Scholar]
- Eftekhari, N.; Kargar, M.; Zamin, F.; Rastakhiz, N.; Manafi, Z. A Review on Various Aspects of Jarosite and Its Utilization Potentials. Ann. De Chim. Sci. Des Matériaux 2020, 44, 43–52. [Google Scholar] [CrossRef]
- Qin, S.; Liu, X.; Lu, M.; Li, D.; Feng, X.; Zhao, L. Acidithiobacillus ferrooxidans and mixed Acidophilic microbiota oxidation to remove sulphur impurity from iron concentrate. Biochem. Eng. J. 2022, 187, 108647. [Google Scholar] [CrossRef]
- Appia-Ayme, C.; Quatrini, R.; Denis, Y.; Denizot, F.; Silver, S.; Roberto, F.; Veloso, F.; Valdés, J.; Cárdenas, J.P.; Esparza, M.; et al. Microarray and bioinformatic analyses suggest models for carbon metabolism in the autotroph Acidithiobacillus ferrooxidans. Hydrometallurgy 2006, 83, 273–280. [Google Scholar] [CrossRef]
- Jiang, V.; Khare, S.D.; Banta, S. Computational Structure Prediction Provides a Plausible Mechanism for Electron Transfer by the Outer Membrane Protein Cyc2 from Acidithiobacillus ferrooxidans. Protein Sci. 2021, 30, 1640–1652. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Liu, Z.; Jiang, Y.; Dong, Y.; Shi, L. In vivo interactions between Cyc2 and Rus as well as Rus and Cyc1 of Acidithiobacillus ferrooxidans during extracellular oxidization of ferrous iron. Int. Biodeterior. Biodegrad. 2022, 173, 105453. [Google Scholar] [CrossRef]
- Zhu, M.L.; Pan, Y.G.; Dai, X.F. (p)ppGpp: The magic governor of bacterial growth economy. Curr. Genet. 2019, 65, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.L.; Yang, H.; Ding, A.Q.; Li, C.; Lin, Q. Metabolic regulation of bacteria with limited carbon and nitrogen sources. Acta Microbiol. Sin. 2023, 63, 946–962. [Google Scholar]
- Tsuyoshi, S.; Shinji, T.; Kyoko, F.; Yamaryo, K.; Inagaki, K.; Tano, T. Utilization of Amino Acids as a Sole Source of Nitrogen by Obligate Chemolithoautotroph Thiobacillus ferrooxidans. Agric. Biol. Chem. 1987, 51, 2229–2236. [Google Scholar]
- Briceo, P.G.D.; Gerardo, A.C.P.; Marco, A.M.G. Early reprecipitation of sulfate salts in coal biodesulfurization processes using acidophilic chemolithotrophic bacteria. World J. Microbiol. Biotechnol. 2020, 36, 81. [Google Scholar]
- Tuovinen, O.H.; Kelly, D.P. Biology of Thiobacillus ferrooxidans in relation to the microbiological leaching of sulphide ores. Z. Allg. Mikrobiol. 1972, 12, 311–346. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.C.; Gou, J.X.; Li, Y.; Yang, L. Study of the effects of culture condition on the growth of Thiobacillus ferrooxidans and the jarosite precipitates. J. Saf. Environ. 2012, 12, 28–31. [Google Scholar]
- Song, Y.W.; Wang, H.R.; Liang, J.R.; Zhou, L.X. Effects of temperature and pH on the formation of biogenic Fe(III) hydroxysulfate precipitates. Acta Sci. Circumstantiae 2016, 36, 3683–3690. [Google Scholar]
- Wang, H.R.; Yang, L.L.; Wang, R.; Yang, J.; Cao, X.Y.; Song, Y.W. Fe2+ oxidation and mineralization properties of A. ferrooxidans biofilm immobilized on three fillers. Acta Sci. Circumstantiae 2022, 42, 160–168. [Google Scholar]
- Song, Y.; Yang, L.; Wang, H.; Sun, X.; Bai, S.; Wang, N.; Liang, J.; Zhou, L. The coupling reaction of Fe2+ bio-oxidation and resulting Fe3+ hydrolysis drastically improve the formation of iron hydroxysulfate minerals in AMD. Environ. Technol. 2019, 42, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, S.L.; Ding, Y.; Tao, X.X. Influence factors on growth and metabolism of pyrite sulfur-removing bacteria. Ind. Miner. Process. 2005, 10, 9–12. [Google Scholar]
- Wang, H.; Guo, Q.; Guo, Z.; Luo, H.; Li, H.; Yang, J.; Song, Y. Assessment of the induced effect of selected iron hydroxysulfates biosynthesized using Acidithiobacillus ferrooxidans for biomineralization of acid mine drainage. Water Sci. Technol. 2023, 87, 1879–1892. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, F.; Xie, X.Y. Effect of simulated inorganic anion leaching solution of electroplating sludge on the bioactivity of Acidithiobacillus ferrooxidans. J. Environ. Sci. 2014, 35, 1377–1383. [Google Scholar]
- Liu, F.W.; Qiao, X.X.; Xing, K.; Shi, J.; Zhou, L.X.; Dong, Y.; Bi, W.L.; Zhang, J. Effect of Nitrate Ions on Acidithiobacillus ferrooxidans-Mediated Bio-oxidation of Ferrous Ions and Pyrite. Curr. Microbiol. 2020, 77, 1070–1080. [Google Scholar] [CrossRef]
- Fryi, V.; Lazarof, F.N.; Packer, L. Sulfate-dependent iron oxidation by Thiobacillus ferrooxidans: Characterization of a new EPR detectable electron transport component on the reducing side of rusticyanin. Arch. Biochem. Bio 1986, 246, 650–654. [Google Scholar] [CrossRef]
- Gramp, J.P.; Jones, F.S.; Bigham, J.M.; Tuovinen, O.H. Monovalent cation concentrations determine the types of Fe(III) hydroxysulfate precipitates formed in bioleach solutions. Hydrometallurgy 2008, 94, 29–33. [Google Scholar] [CrossRef]
- Huang, H.; Geng, K.; Wang, C.; Wu, X.; Wei, C. Impact of Fulvic Acid and Acidithiobacillus ferrooxidan Inoculum Amount on the Formation of Secondary Iron Minerals. Int. J. Environ. Res. Public Health 2023, 20, 4736. [Google Scholar] [CrossRef]
- Karamanov, A.; Pelino, M. Evaluation of the degree of crystallisation in glass-ceramics by density measurements. J. Eur. Ceram. Soc. 1999, 19, 649–654. [Google Scholar] [CrossRef]
- Sun, Z.P.; Ni, Y.; Yue, W.Q.; Yan, F.; Liu, F.W.; Li, X.W. Preparation of silver nanoparticles loaded Schwertmannite and catalytic degradation performance on methyl orange. Acta Sci. Circumstantiae 2022, 42, 61–70. [Google Scholar]
- Zhang, Z.; Wang, L.; Zhou, B.; Wang, S.; Fan, L.; Hu, S.; Wu, Y. Adsorption Performance and Mechanism of Synthetic Schwertmannite to Remove Low-Concentration Fluorine in Water. Bull. Environ. Contam. Toxicol. 2021, 107, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Dutrizac, J.E. Factors affecting alkali jarosite precipitation. Metall. Trans. B 1983, 14, 531–539. [Google Scholar] [CrossRef]
- Regenspurg, S.; Brand, A.; Peiffer, S. Formation and stability of schwertmannite in acid mining lakes. Geochim. Cosmochim Acta 2004, 68, 1185–1197. [Google Scholar] [CrossRef]
- Gaowa, N.; Hironori, O.; Shuqin, B.; Kotaro, Y.; Takushi, Y. Uptake mechanism of silicic acid by schwertmannite and its stabilization. J. Environ. Chem. Eng. 2023, 11, 111136. [Google Scholar]
- Bigham, J.; Schwertmann, U.; Traina, S.; Winland, R.; Wolf, M. Schwertmannite and the chemical modeling of iron in acid sulfate waters. Geochim. Cosmochim Acta 1996, 60, 2111–2121. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (h) | Nitrogen | ||
|---|---|---|---|
| (NH4)2SO4 | CO(NH2)2 | NH4NO3 | |
| 0–12 | 124 | 69 | 90 |
| 12–24 | 601 | 100 | 330 |
| 24–36 | 42 | 118 | 24 |
| 36–72 | - | 135 | 78 |
| 72–96 | - | 39 | 40 |
| Nitrogen | K (wt.%) | N (wt.%) | Fe (wt.%) | SO42− (wt.%) |
|---|---|---|---|---|
| (NH4)2SO4 | 1.10 | 0.22 | 32.8 | 21.6 |
| CO(NH2)2 | 1.79 | 0.35 | 20.9 | 22.3 |
| NH4NO3 | 1.43 | 0.18 | 28.6 | 21.0 |
| KFe3(SO4)2(OH)6 | 7.80 | 33.5 | 38.3 | |
| NH4Fe3(SO4)2(OH)6 | 2.92 | 35.0 | 40.0 | |
| H3OFe3(SO4)2(OH)6 | 34.8 | 40.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.; Hu, W.; Zi, X.; Wang, X.; Liang, J.; Zhou, L. Effects of Different Nitrogen Sources on the Formation of Biogenic Jarosite. Sustainability 2023, 15, 15765. https://doi.org/10.3390/su152215765
Huang H, Hu W, Zi X, Wang X, Liang J, Zhou L. Effects of Different Nitrogen Sources on the Formation of Biogenic Jarosite. Sustainability. 2023; 15(22):15765. https://doi.org/10.3390/su152215765
Chicago/Turabian StyleHuang, Haitao, Weitong Hu, Xiang Zi, Xiaomeng Wang, Jianru Liang, and Lixiang Zhou. 2023. "Effects of Different Nitrogen Sources on the Formation of Biogenic Jarosite" Sustainability 15, no. 22: 15765. https://doi.org/10.3390/su152215765
APA StyleHuang, H., Hu, W., Zi, X., Wang, X., Liang, J., & Zhou, L. (2023). Effects of Different Nitrogen Sources on the Formation of Biogenic Jarosite. Sustainability, 15(22), 15765. https://doi.org/10.3390/su152215765