
Biodiversity and Potential Activity of Microorganisms in Underground Gas Storage Horizons

 , , ,
, , ,  ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Site Description and Water Sample Collection
2.2. Media Composition and Cultivation Conditions
2.3. Enumeration of Microorganisms and Determination of Microbial Growth
2.4. Illumina MiSeq Sequencing and Analysis of 16S rRNA Genes of Prokaryotes from Water Samples and Sulfidogenic Enrichment
2.5. Functional Annotation
2.6. Scanning Electron Microscopy (SEM) and Elemental Analysis
2.7. Nucleotide Sequence Accession Number
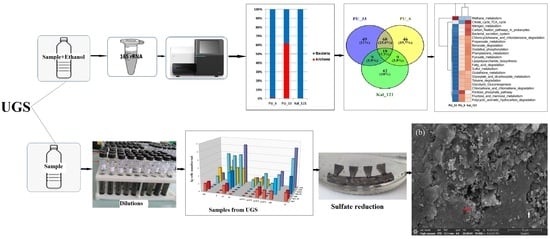
3. Results and Discussion
3.1. Physicochemical Properties of the Water Samples and Characterization of the Cultivable Prokaryotes
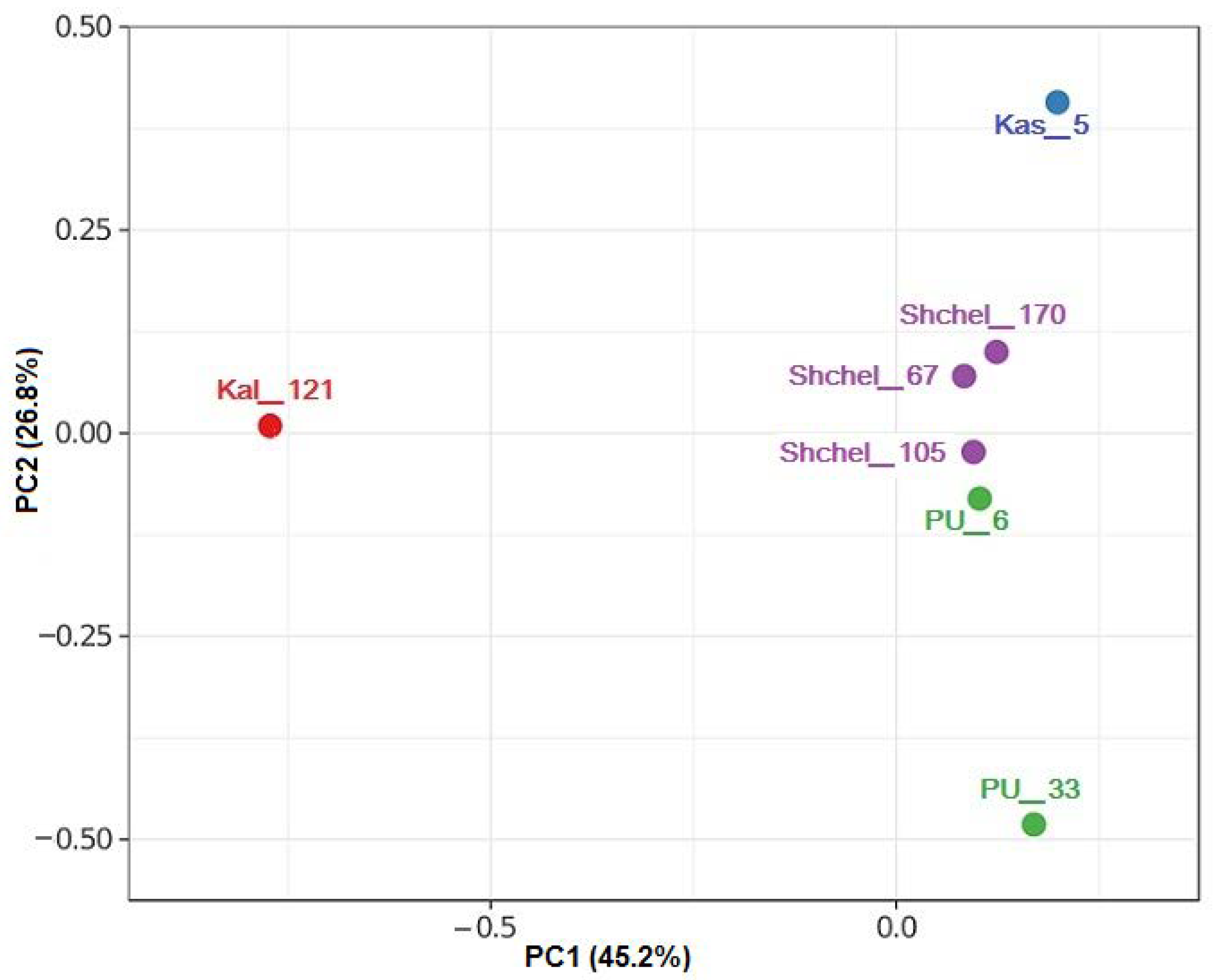
3.2. Composition of Microbial Communities in Water Samples from UGSs
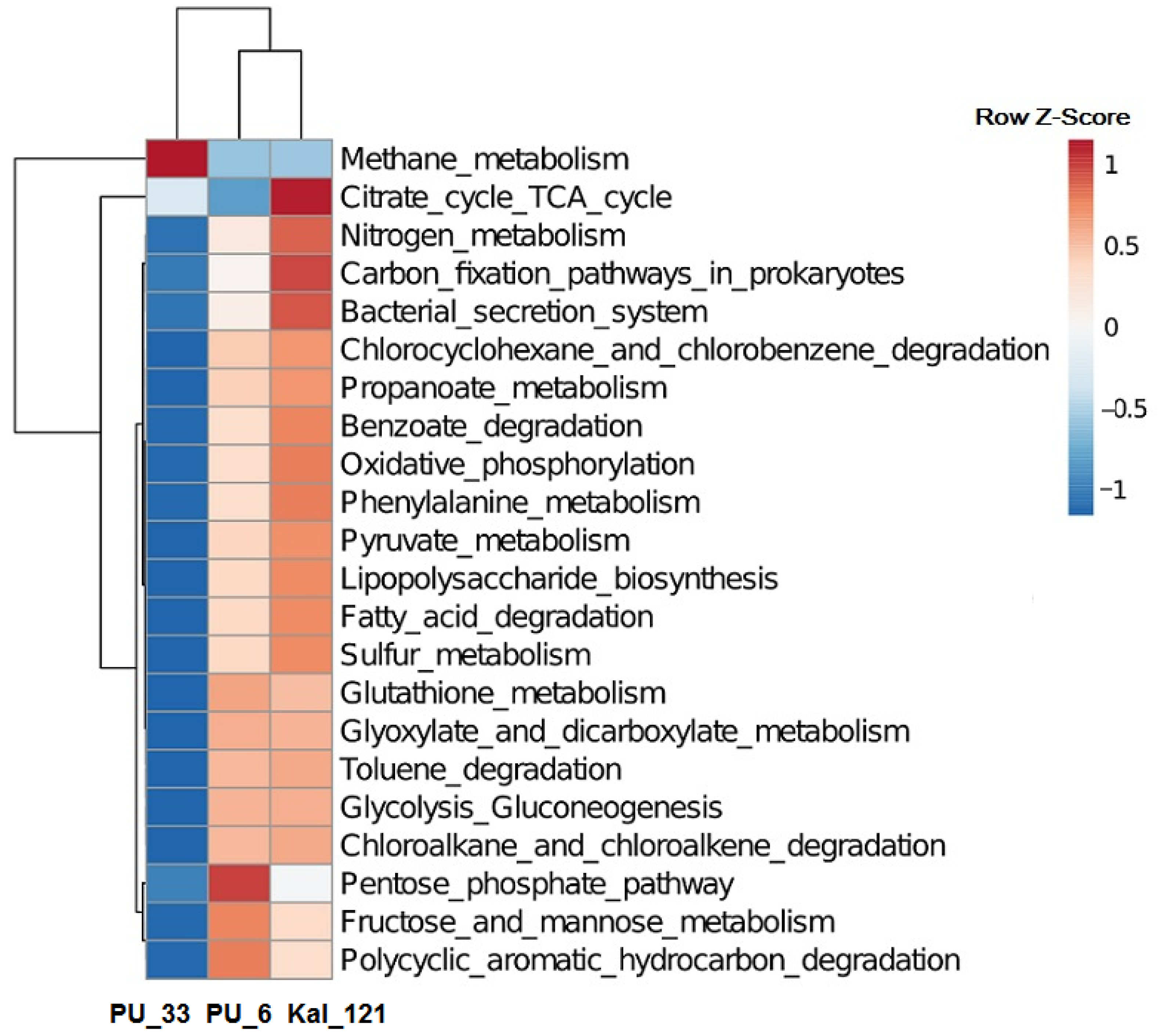
3.3. Functional Potential of Microbial Communities from UGSs
3.4. Sulfide Production and Steel Corrosion by Anaerobic Enrichments from UGS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molíková, A.; Vítězová, M.; Vítěz, T.; Buriánková, I.; Huber, H.; Dengler, L.; Hanišáková, N.; Onderka, V.; Urbanová, I. Underground gas storage as a promising natural methane bioreactor and reservoir? J. Energy Storage 2021, 47, 103631. [Google Scholar] [CrossRef]
- Zivar, D.; Kumar, S.; Foroozesh, J. Underground hydrogen storage: A comprehensive review. Int. J. Hydrog. Energy 2020, 46, 23436–23462. [Google Scholar] [CrossRef]
- Agarwal, R. Transition to a hydrogen-based economy: Possibilities and challenges. Sustainability 2022, 14, 15975. [Google Scholar] [CrossRef]
- Reitenbach, V.; Ganzer, L.; Albrecht, D.; Hagemann, B. Influence of added hydrogen on underground gas storage: A review of key issues. Environ. Earth Sci. 2015, 73, 6927–6937. [Google Scholar] [CrossRef]
- Gregory, S.P.; Barnett, M.J.; Field, L.P.; Milodowski, A.E. Subsurface microbial hydrogen cycling: Natural occurrence and implications for industry. Microorganisms 2019, 7, 53. [Google Scholar] [CrossRef]
- Dopffel, N.; Jansen, S.; Gerritse, J. Microbial side effects of underground hydrogen storage—Knowledge gaps, risks and opportunities for successful implementation. Int. J. Hydrog. Energy 2021, 46, 8594–8606. [Google Scholar] [CrossRef]
- Carden, P.; Paterson, L. Physical, chemical and energy aspects of underground hydrogen storage. Int. J. Hydrog. Energy 1979, 4, 559–569. [Google Scholar] [CrossRef]
- Barsuk, N.E.; Khaidina, M.P.; Khan, S.A. “Green gas” in the European gas transport system. Gazov. Promyshlennost 2018, 10, 104–109. (In Russian) [Google Scholar]
- Chapelle, F.H.; O’Neill, K.; Bradley, P.M.; Methé, B.A.; Ciufo, S.A.; Knobel, L.L.; Lovley, D.R. A hydrogen-based subsurface microbial community dominated by methanogens. Nature 2002, 415, 312–315. [Google Scholar] [CrossRef]
- Takai, K.; Mormille, M.R.; McKinley, J.P.; Brockman, F.J.; Holben, W.E.; Kovacik, J.K., Jr.; Fredrickson, J.K. Shifts in archaeal communities associated with lithological and geochemical variations in subsurface Cretaceous rock. Environ. Microbiol. 2003, 5, 309–320. [Google Scholar] [CrossRef]
- Harris, S.; Smith, R.; Suflita, J. In situ hydrogen consumption kinetics as an indicator of subsurface microbial activity. FEMS Microbiol. Ecol. 2007, 60, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Zhang, K.; Wang, L.Y.; Liu, J.F.; Yang, S.Z.; Gu, J.D.; Mu, B.Z. Different diversity and distribution of archaeal community in the aqueous and oil phases of production fluid from high-temperature petroleum reservoirs. Front. Microbiol. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Magot, M.; Ollivier, B.; Patel, B.K.C. Microbiology of petroleum reservoirs. Antonie Van Leeuwenhoek 2000, 77, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Gieg, L.M.; Davidova, I.A.; Duncan, K.E.; Suflita, J.M. Methanogenesis, sulfate reduction and crude oil biodegradation in hot Alaskan oilfields. Environ. Microbiol. 2010, 12, 3074–3086. [Google Scholar] [CrossRef] [PubMed]
- Belyaev, S.S.; Borzenkov, I.A. Microbiological transformation of low-molecular-weight carbon compounds in the deep subsurface. In Biogeochemistry of Global Change; Oremland, R.S., Ed.; Chapman & Hall: New York, NY, USA, 1993; pp. 825–838. [Google Scholar] [CrossRef]
- Nazina, T.N.; Shestakova, N.M.; Ivoilov, V.S.; Kostrukova, N.K.; Belyaev, S.S.; Ivanov, M.V. Radiotracer assay of microbial processes in petroleum reservoirs. Adv. Biotechnol. Microbiol. 2017, 2, 555591. [Google Scholar] [CrossRef]
- Bonch-Osmolovskaya, E.A.; Miroshnichenko, M.L.; Lebedinsky, A.V.; Chernyh, N.A.; Nazina, T.N.; Ivoilov, V.S.; Belyaev, S.S.; Boulygina, E.S.; Lysov, Y.P.; Perov, A.N.; et al. Radioisotopic, culture-based, and oligonucleotide microchip analyses of thermophilic microbial communities in a continental high-temperature petroleum reservoir. Appl. Environ. Microbiol. 2003, 69, 6143–6151. [Google Scholar] [CrossRef]
- Anantharaman, K.; Brown, C.T.; Hug, L.A.; Sharon, I.; Castelle, C.J.; Probst, A.J.; Thomas, B.C.; Singh, A.; Wilkins, M.J.; Karaoz, U.; et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 2016, 24, 13219. [Google Scholar] [CrossRef]
- Basso, O.; Lascourreges, J.F.; Le Borgne, F.; Le Goff, C.; Magot, M. Characterization by culture and molecular analysis of the microbial diversity of a deep subsurface gas storage aquifer. Res. Microbiol. 2009, 160, 107–116. [Google Scholar] [CrossRef]
- Ranchou-Peyruse, M.; Auguet, J.C.; Mazière, C.; Restrepo-Ortiz, C.X.; Guignard, M.; Dequidt, D.; Chiquet, P.; Cézac, P.; Ranchou-Peyruse, A. Geological gas-storage shapes deep life. Environ. Microbiol. 2019, 21, 3953–3964. [Google Scholar] [CrossRef]
- Turkiewicz, A.; Steliga, T.; Kluk, D.; Gminski, Z. Biomonitoring studies and preventing the formation of biogenic H2S in the Wierzchowice underground gas storage facility. Energies 2021, 14, 5463. [Google Scholar] [CrossRef]
- Ivanova, A.E.; Borzenkov, I.A.; Tarasov, A.L.; Milekhina, E.I.; Belyaev, S.S. A microbiological study of an underground gas storage in the process of gas injection. Microbiology 2007, 76, 453–460. [Google Scholar] [CrossRef]
- Ivanova, A.E.; Borzenkov, I.A.; Tarasov, A.L.; Milekhina, E.I.; Belyaev, S.S. A microbiological study of an underground gas storage in the process of gas extraction. Microbiology 2007, 76, 461–468. [Google Scholar] [CrossRef]
- Staniszewska, A.; Kunicka-Styczyńska, A.; Otlewska, A.; Gawor, J.; Gromadka, R.; Żuchniewicz, K.; Ziemiński, K. High-throughput sequencing approach in analysis of microbial communities colonizing natural gas pipelines. Microbiol. Open 2019, 8, e00806. [Google Scholar] [CrossRef]
- Šmigáň, P.; Greksák, M.; Kozánkova, J.; Buzek, F.; Onderka, V.; Wolf, I. Methanogenic bacteria as a key factor involved in changes of town gas stored in an underground reservoir. FEMS Microbiol. Lett. 1990, 73, 221–224. [Google Scholar] [CrossRef]
- Buzek, F.; Onderka, V.; Vančura, P.; Wolf, I. Carbon isotope study of methane production in a town gas storage reservoir. Fuel 1994, 73, 747–752. [Google Scholar] [CrossRef]
- Thaysen, E.M.; McMahon, S.; Strobel, G.J.; Butler, I.B.; Ngwenya, B.T.; Heinemann, N.; Wilkinson, M.; Hassanpouryouzband, A.; McDermott, C.I.; Edlmann, K. Estimating microbial growth and hydrogen consumption in hydrogen storage in porous media. Renew. Sustain. Energy Rev. 2021, 151, 111481. [Google Scholar] [CrossRef]
- Tarasov, A.L.; Borzenkov, I.A.; Chernykh, N.A.; Belyayev, S.S. Isolation and investigation of anaerobic microorganisms involved in methanol transformation in an underground gas storage facility. Microbiology 2011, 80, 172–179. [Google Scholar] [CrossRef]
- Tarasov, A.L.; Borzenkov, I.A.; Belyayev, S.S. Investigation of the trophic relations between anaerobic microorganisms from an underground gas repository during methanol utilization. Microbiology 2011, 80, 180–187. [Google Scholar] [CrossRef]
- Nazina, T.N.; Abukova, L.A.; Tourova, T.P.; Babich, T.L.; Bidzhieva, S.K.; Filippova, D.S.; Safarova, E.A. Diversity and possible activity of microorganisms in underground gas storage aquifers. Microbiology 2021, 90, 619–629. [Google Scholar] [CrossRef]
- Panfilov, M. Underground and pipeline hydrogen storage. Compendium of Hydrogen Energy, Volume 2: Hydrogen Storage. Transp. Infrastruct. 2016, 91–115. [Google Scholar] [CrossRef]
- Postgate, J.R. The Sulfate-Reducing Bacteria, 2nd ed.; Cambridge University Press: Cambridge, UK, 1984; p. 151. [Google Scholar]
- Pfennig, N.; Lippert, K.D. Über das vitamin B12—Bedürfnis phototropher Schweferelbakterien. Arch. Microbiol. 1966, 55, 245–256. [Google Scholar]
- Wolin, E.A.; Wolin, M.J.; Wolfe, R.S. Formation of methane by bacterial extracts. J. Biol. Chem. 1963, 238, 2882–2886. [Google Scholar] [CrossRef] [PubMed]
- Trüper, H.G.; Schlegel, H.G. Sulfur metabolism in Thiorhodaceae. I. Quantitative measurements on growing cells of Chromatium okenii. Antonie Van Leeuwenhoek 1964, 30, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Bidzhieva, S.K.; Sokolova, D.S.; Tourova, T.P.; Nazina, T.N. Bacteria of the genus Sphaerochaeta from low-temperature heavy oil reservoirs (Russia). Microbiology 2018, 87, 757–765. [Google Scholar] [CrossRef]
- Gohl, D.M.; MacLean, A.; Hauge, A.; Becker, A.; Walek, D.; Beckman, K.B. An optimized protocol for high-throughput amplicon-based microbiome profiling. Protoc. Exch. 2016. [Google Scholar] [CrossRef]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Hugerth, L.W.; Muller, E.E.L.; Hu, Y.O.O.; Lebrun, L.A.M.; Roume, H.; Lundin, D.; Wilmes, P.; Andersson, A.F. Systematic design of 18S rRNA gene primers for determining eukaryotic diversity in microbial consortia. PLoS ONE 2014, 9, e95567. [Google Scholar] [CrossRef]
- Merkel, A.Y.; Tarnovetskii, I.Y.; Podosokorskaya, O.A.; Toshchakov, S.V. Analysis of 16S rRNA primer systems for profiling of thermophilic microbial communities. Microbiology 2019, 88, 671–680. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- SILVAngs. Version: 1.9.10/1.4.9; SILVA: r138.1. Available online: https://www.arb-silva.de/ngs/ (accessed on 14 March 2023).
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Colwell, R.K. EstimateS: Statistical Estimation of Species Richness and Shared Species from Samples. Version 9. User’s Guide and Application. 2013. Available online: http://purl.oclc.org/estimates (accessed on 9 May 2019).
- Oliveros, J.C. (2007–2015) Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html.
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Nagpal, S.; Haque, M.M.; Singh, R.; Mande, S.S. iVikodak—A platform and standard workflow for inferring, analyzing, comparing, and visualizing the functional potential of microbial communities. Front. Microbiol. 2019, 9, 3336. [Google Scholar] [CrossRef]
- KEGG PATHWAY Database. Available online: https://www.genome.jp/kegg/pathway.html (accessed on 30 March 2023).
- Youssef, N.; Elshahed, M.S.; McInerney, M.J. Microbial processes in oil fields: Culprits, problems and opportunities. Adv. Appl. Microbiol. 2009, 66, 141–251. [Google Scholar] [CrossRef]
- Widdel, F.; Musat, F.; Knittel, K.; Galushko, A. Anaerobic degradation of hydrocarbons with sulphate as electron donor. In Sulphate-Reducing Bacteria. Environmental and Engineered Systems; Barton, L.L., Hamilton, W.A., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 265–303. [Google Scholar]
- Kieft, T.L. Microbiology of the deep continental biosphere. In Their World: A Diversity of Microbial Environments Advances in Environmental Microbiology; Hurst, C.J., Ed.; Springer: Cham, Switzerland, 2016; pp. 225–249. [Google Scholar] [CrossRef]
- Kaye, J.Z.; Sylvan, J.B.; Edwards, K.J.; Baross, J.A. Halomonas and Marinobacter ecotypes from hydrothermal vent, subseafloor and deep-sea environments. FEMS Microbiol. Ecol. 2010, 75, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Fathepure, B.Z. Recent studies in microbial degradation of petroleum hydrocarbons in hypersaline environments. Front. Microbiol. 2014, 5, 173. [Google Scholar] [CrossRef]
- Tourova, T.P.; Sokolova, D.S.; Semenova, E.M.; Ershov, A.P.; Grouzdev, D.S.; Nazina, T.N. Genomic and physiological characterization of halophilic bacteria of the genera Halomonas and Marinobacter from petroleum reservoirs. Microbiology 2022, 91, 235–248. [Google Scholar] [CrossRef]
- Van Hamme, J.D.; Ward, O.P. Physical and metabolic interactions of Pseudomonas sp. strain JA5-B45 and Rhodococcus sp. strain F9-D79 during growth on crude oil and effect of a chemical surfactant on them. Appl. Environ. Microbiol. 2001, 67, 4874–4879. [Google Scholar] [CrossRef]
- Kuyukina, M.S.; Ivshina, I.B. Application of Rhodococcus in bioremediation of contaminated environments. In Biology of Rhodococcus; Microbiology, Monographs; Alvarez, H., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 16, pp. 231–262. [Google Scholar] [CrossRef]
- Nazina, T.N.; Shumkova, E.S.; Sokolova, D.S.; Babich, T.L.; Zhurina, M.V.; Xue, Y.-F.; Osipov, G.A.; Poltaraus, A.B.; Tourova, T.P. Identification of hydrocarbon-oxidizing Dietzia bacteria from petroleum reservoirs based on phenotypic properties and analysis of the 16S rRNA and gyrB genes. Microbiology 2015, 84, 377–388. [Google Scholar] [CrossRef]
- Takii, S.; Hanada, S.; Tamaki, H.; Ueno, Y.; Sekiguchi, Y.; Ibe, A.; Matsuura, K. Dethiosulfatibacter aminovorans gen. nov., sp. nov., a novel thiosulfate-reducing bacterium isolated from a sulfate-reducing mixed culture enriched with Casamino acids from coastal marine sediment. Int. J. Syst. Evol. Microbiol. 2007, 57, 2320–2326. [Google Scholar] [CrossRef] [PubMed]
- Jabari, L.; Gannoun, H.; Cayol, J.-L.; Hamdi, M.; Fauque, G.; Ollivier, B.; Fardeau, M.-L. Characterization of Defluviitalea saccharophila gen. nov., sp. nov., a thermophilic bacterium isolated from an upflow anaerobic filter treating abattoir wastewaters, and proposal of Defluviitaleaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2012, 62, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, A.; Tindall, B.J.; Bardin, V.; Blanchet, D.; Jeanthon, C. Petrimonas sulfuriphila gen. nov., sp. nov., a mesophilic fermentative bacterium isolated from a biodegraded oil reservoir. Int. J. Syst. Evol. Microbiol. 2005, 55, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Schoberth, S.; Tanner, R.S.; Wolfe, R.S. Acetobacterium, a new genus of hydrogen-oxidizing, carbon dioxide-reducing, anaerobic bacteria. Int. J. Syst. Bacteriol. 1977, 27, 355–361. [Google Scholar] [CrossRef]
- Braun, M.; Gottschalk, G. Acetobacterium wieringae sp. nov., a new species producing acetic acid from molecular hydrogen and carbon dioxide. Zentralbl. Bakteriol. Parasitenkd. Infektionskr. Hyg. Abt. 1 Orig. 1982, 3, 368–376. [Google Scholar] [CrossRef]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W.; et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2022, 51, D678–D689. [Google Scholar] [CrossRef]
- Schomburg, I.; Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G.; Schomburg, D. Brenda, the enzyme database: Updates and major new developments. Nucleic Acids Res. 2004, 32, 431–433. [Google Scholar] [CrossRef]
- Gieg, L.M.; Jack, T.R.; Foght, J.M. Biological souring and mitigation in oil reservoirs. Appl. Microbiol. Biotechnol. 2011, 92, 263–282. [Google Scholar] [CrossRef]
- Vigneron, A.; Alsop, E.B.; Chambers, B.; Lomans, B.P.; Head, I.M.; Tsesmetzis, N. Complementary microorganisms in highly corrosive biofilms from an offshore oil production facility. Appl. Environ. Microbiol. 2016, 82, 2545–2554. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Sproer, C.; Rainey, F.A.; Burghardt, J.; Pauker, O.; Hippe, H. Phylogenetic analysis of the genus Desulfotomaculum: Evidence for the misclassification of Desulfotomaculum guttoideum and description of Desulfotomaculum orientis as Desulfosporosinus orientis gen. nov., comb. nov. Int. J. Syst. Bacteriol. 1997, 47, 1134–1139. [Google Scholar] [CrossRef]
- Panova, I.A.; Ikkert, O.; Avakyan, M.R.; Kopitsyn, D.S.; Mardanov, A.V.; Pimenov, N.V.; Shcherbakova, V.A.; Ravin, N.V.; Karnachuk, O.V. Desulfosporosinus metallidurans sp. nov., an acidophilic, metal-resistant sulfate-reducing bacterium from acid mine drainage. Int. J. Syst. Evol. Microbiol. 2021, 71, 004876. [Google Scholar] [CrossRef]
- Vandieken, V.; Niemann, H.; Engelen, B.; Cypionka, H. Marinisporobacter balticus gen. nov., sp. nov., Desulfosporosinus nitroreducens sp. nov. and Desulfosporosinus fructosivorans sp. nov., new spore-forming bacteria isolated from subsurface sediments of the Baltic Sea. Int. J. Syst. Evol. Microbiol. 2017, 67, 1887–1893. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Cheng, C.L.; Huang, P.J.; Lin, S.Y. Application of scanning electron microscopy and X-ray microanalysis: FE-SEM, ESEM-EDS, and EDS mapping for studying the characteristics of topographical microstructure and elemental mapping of human cardiac calcified deposition. Anal. Bioanal. Chem. 2014, 406, 359–366. [Google Scholar] [CrossRef]
- Costa-Bauzá, A.; Grases, F.; Julià, F. The power of desktop scanning electron microscopy with elemental analysis for analyzing urinary stones. Urolithiasis 2023, 51, 50. [Google Scholar] [CrossRef]
- Safarova, E.A.; Filippova, D.S.; Stolyarov, V.E. Features of operation of underground gas storage facilities in joint storage of methane and hydrogen. Sci. J. Russian Gas Soc. 2021, 3, 58–62. (In Russian) [Google Scholar]
- Colman, D.R.; Poudel, S.; Stamps, B.W.; Boyd, E.S.; Spear, J.R. The deep, hot biosphere: Twenty-five years of retrospection. Proc. Natl. Acad. Sci. USA 2017, 114, 6895–6903. [Google Scholar] [CrossRef] [PubMed]
- Berta, M.; Dethlefsen, F.; Ebert, M.; Schäfer, D.; Dahmke, A. Geochemical effects of millimolar hydrogen concentrations in groundwater: An experimental study in the context of subsurface hydrogen storage. Environ. Sci. Technol. 2018, 52, 4937–4949. [Google Scholar] [CrossRef]
- Schieche, D.; Murty, M.V.S.; Kermode, R.I.; Bhattacharyya, D. Biohydrogenation of fumarate using Desulfovibrio desulfuricans: Experimental results and kinetic rate modelling. J. Chem. Technol. Biotechnol. 1997, 70, 316–322. [Google Scholar] [CrossRef]
- Miller, J.F.; Shah, N.N.; Nelson, C.M.; Ludlow, J.M.; Clark, D.S. Pressure and temperature effects on growth and methane production of the extreme thermophile Methanococcus jannaschi. Appl. Environ. Microbiol. 1988, 54, 3039–3042. [Google Scholar] [CrossRef]
- Laban, M. Hydrogen storage in salt caverns: Chemical modelling and analysis of large-scale hydrogen storage in underground salt caverns. Master’s Thesis, Delft University of Technology, Delft, The Netherlands, 2020. [Google Scholar]
- Bo, Z.; Zeng, L.; Chen, Y.; Xie, Q. Geochemical reactions-induced hydrogen loss during underground hydrogen storage in sandstone reservoirs. Int. J. Hydrog. Energy 2021, 46, 19998–20009. [Google Scholar] [CrossRef]
- Zeng, L.; Keshavarz, A.; Xie, Q.; Iglauer, S. Hydrogen storage in Majiagou carbonate reservoir in China: Geochemical modelling on carbonate dissolution and hydrogen loss. Int. J. Hydrog. Energy 2022, 47, 24861–24870. [Google Scholar] [CrossRef]
- Saeed, M.; Jadhawar, P.; Bagala, S. Geochemical effects on storage gases and reservoir rock during underground hydrogen storage: A depleted North Sea oil reservoir case study. Hydrogen 2023, 4, 323–337. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazina, T.N.; Abukova, L.A.; Tourova, T.P.; Babich, T.L.; Bidzhieva, S.K.; Loiko, N.G.; Filippova, D.S.; Safarova, E.A. Biodiversity and Potential Activity of Microorganisms in Underground Gas Storage Horizons. Sustainability 2023, 15, 9945. https://doi.org/10.3390/su15139945
Nazina TN, Abukova LA, Tourova TP, Babich TL, Bidzhieva SK, Loiko NG, Filippova DS, Safarova EA. Biodiversity and Potential Activity of Microorganisms in Underground Gas Storage Horizons. Sustainability. 2023; 15(13):9945. https://doi.org/10.3390/su15139945
Chicago/Turabian StyleNazina, Tamara N., Leyla A. Abukova, Tatiana P. Tourova, Tamara L. Babich, Salimat K. Bidzhieva, Nataliya G. Loiko, Dina S. Filippova, and Elisaveta A. Safarova. 2023. "Biodiversity and Potential Activity of Microorganisms in Underground Gas Storage Horizons" Sustainability 15, no. 13: 9945. https://doi.org/10.3390/su15139945
APA StyleNazina, T. N., Abukova, L. A., Tourova, T. P., Babich, T. L., Bidzhieva, S. K., Loiko, N. G., Filippova, D. S., & Safarova, E. A. (2023). Biodiversity and Potential Activity of Microorganisms in Underground Gas Storage Horizons. Sustainability, 15(13), 9945. https://doi.org/10.3390/su15139945