
Sequential Formation of CO2 Hydrates in a Confined Environment: Description of Phase Equilibrium Boundary, Gas Consumption, Formation Rate and Memory Effect


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Apparatus
2.2. Materials
2.3. Experimental Procedure
3. Results and Discussion
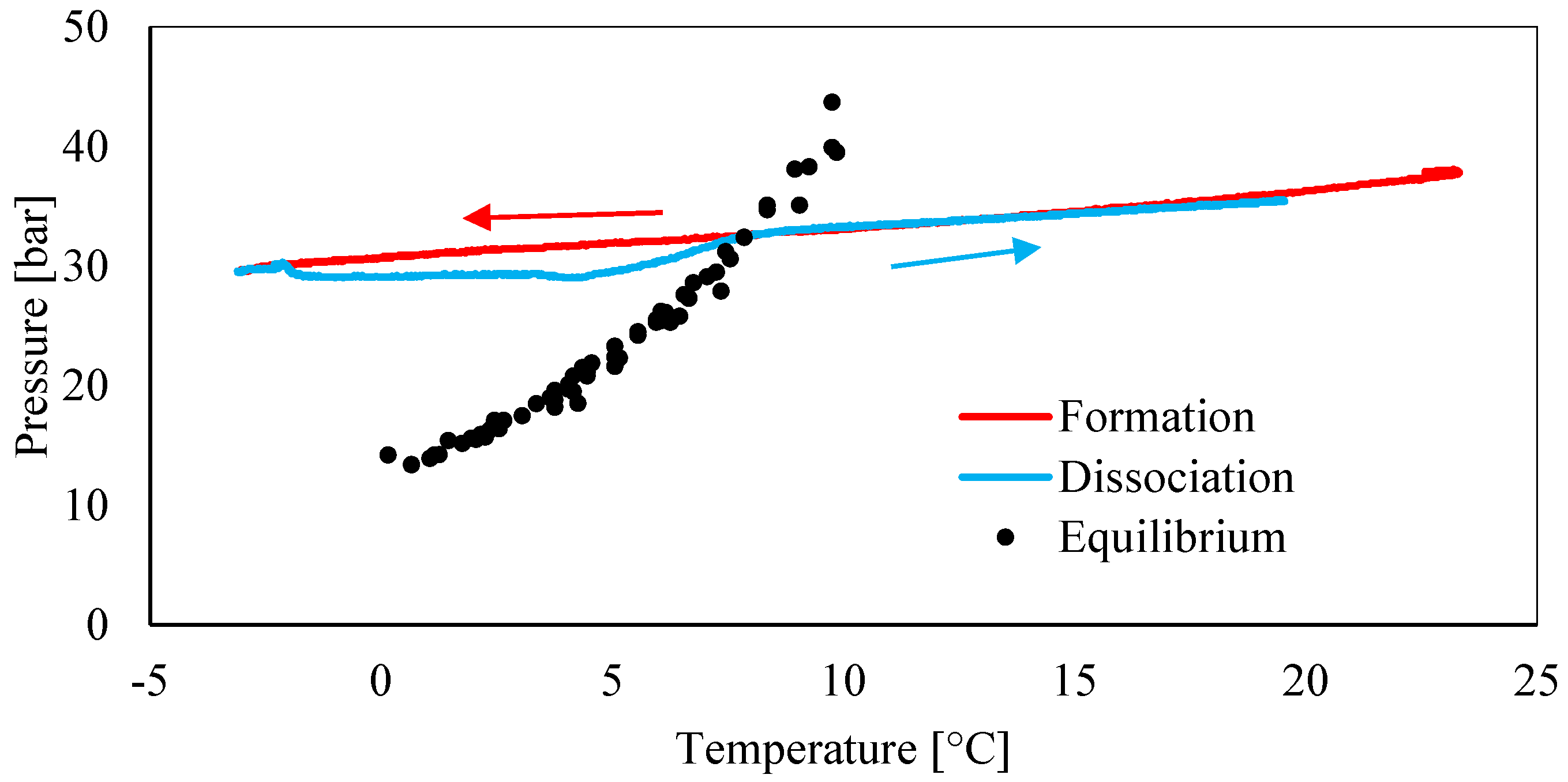
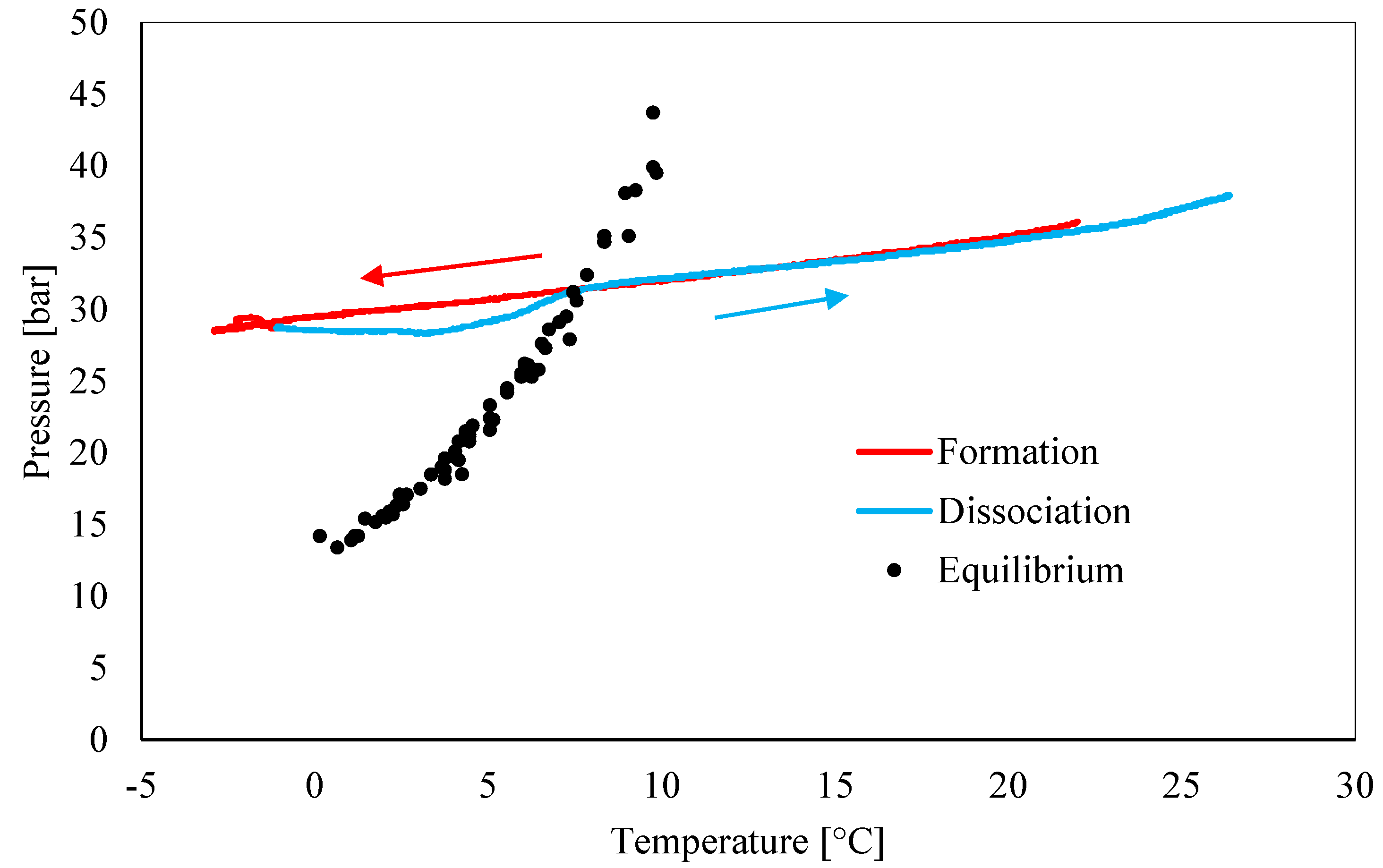
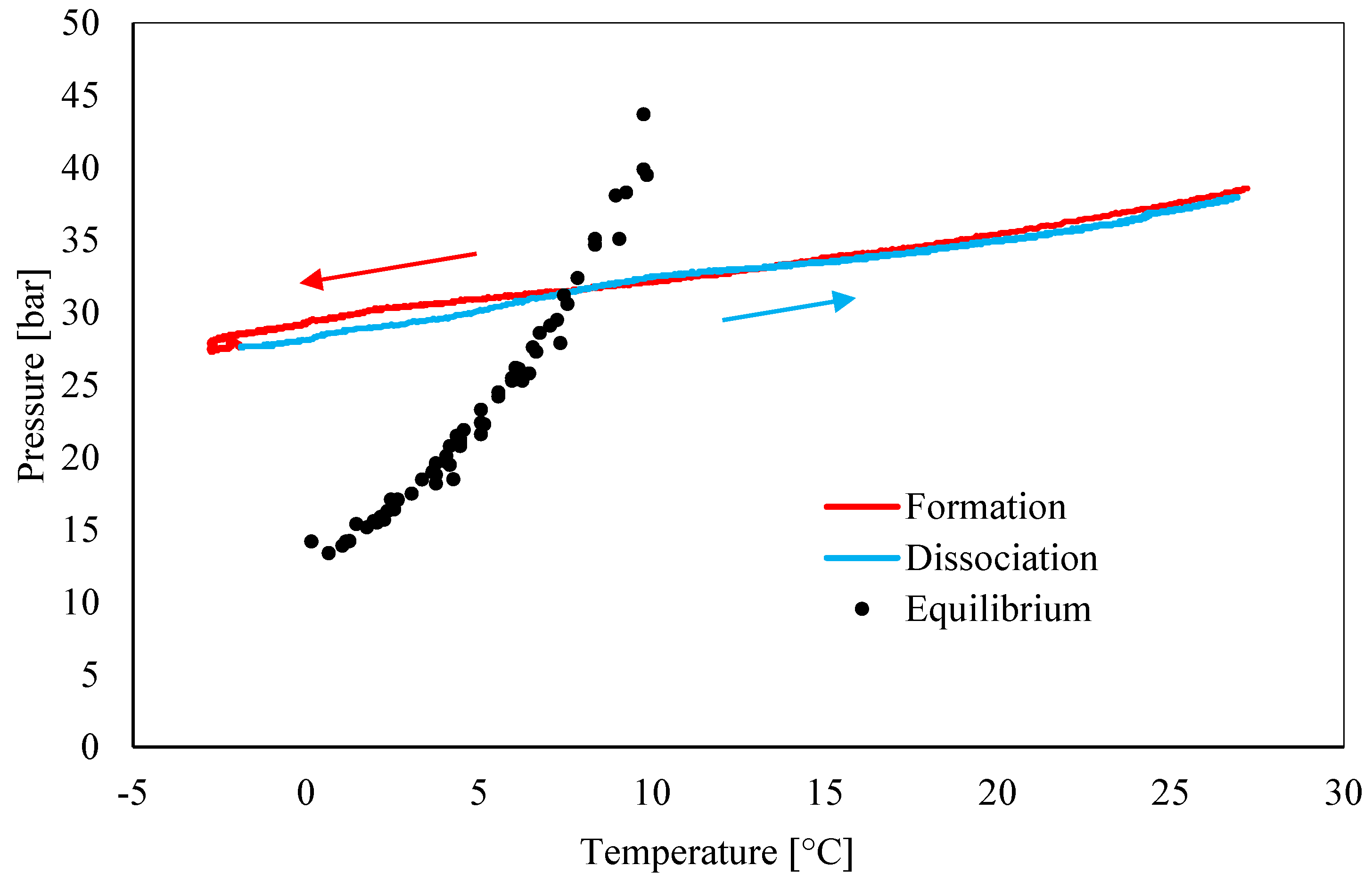
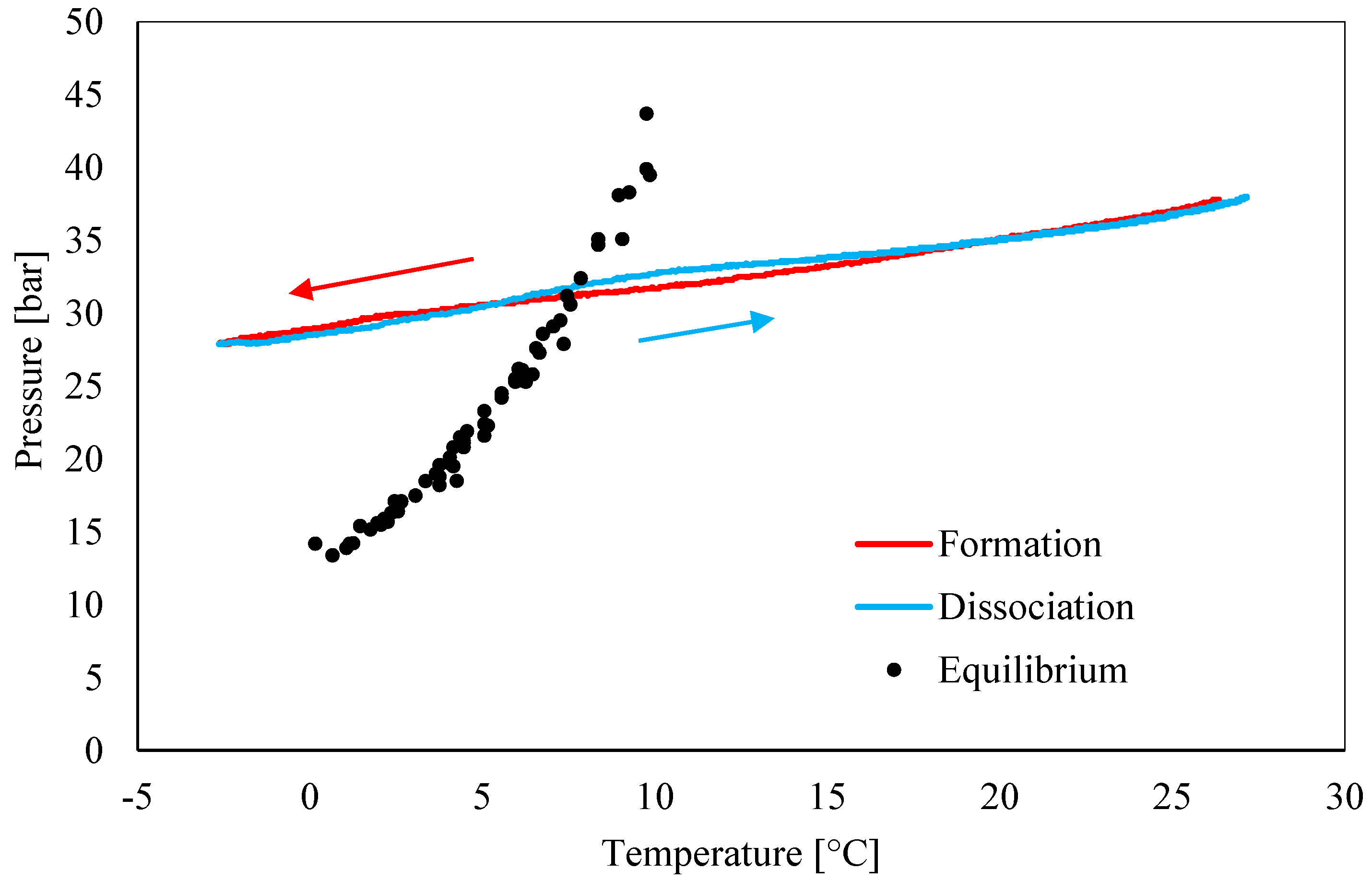
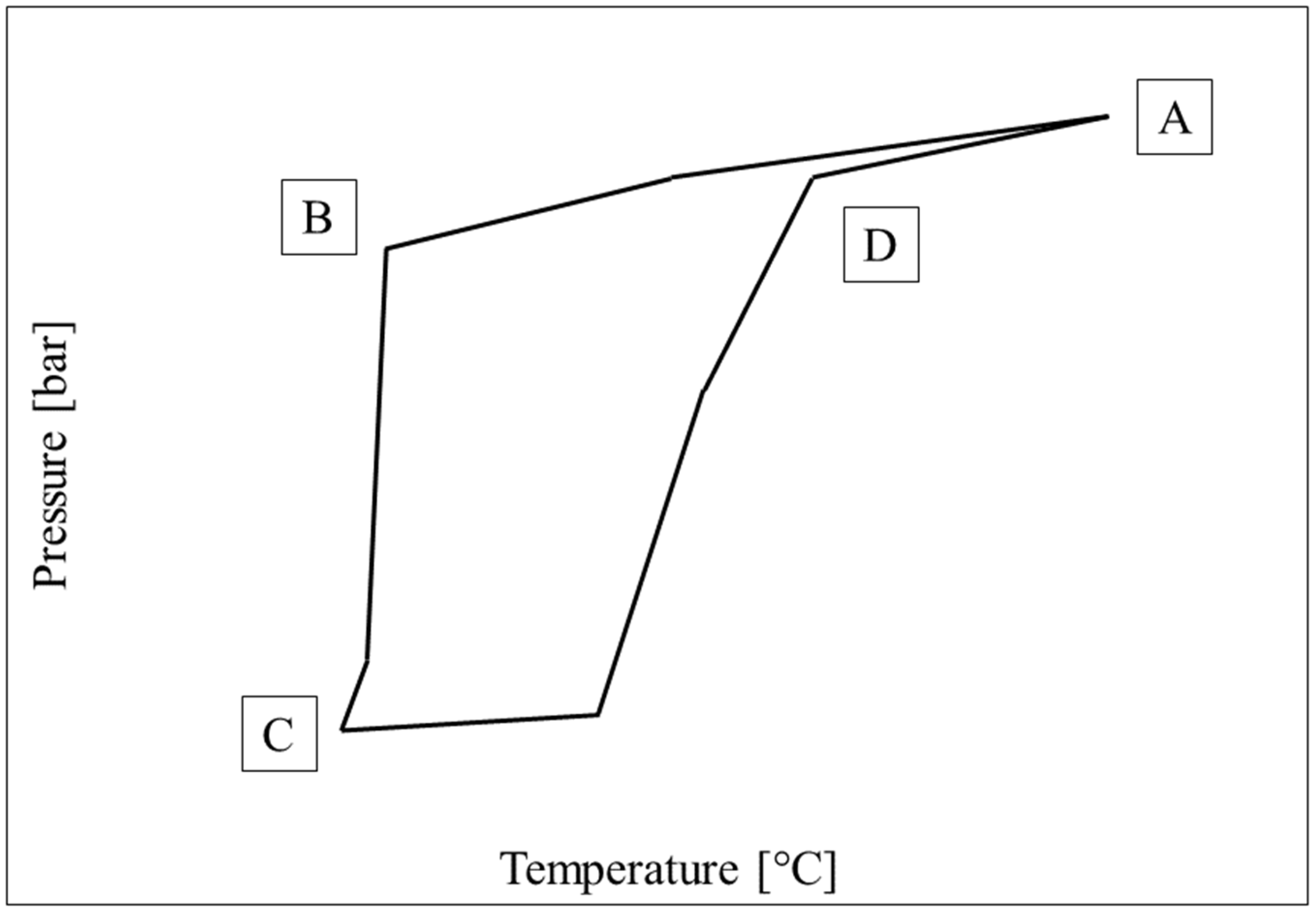
3.1. Pressure–Temperature Diagrams and Comparison with Equilibrium Phase Boundaries for CO2 Hydrates
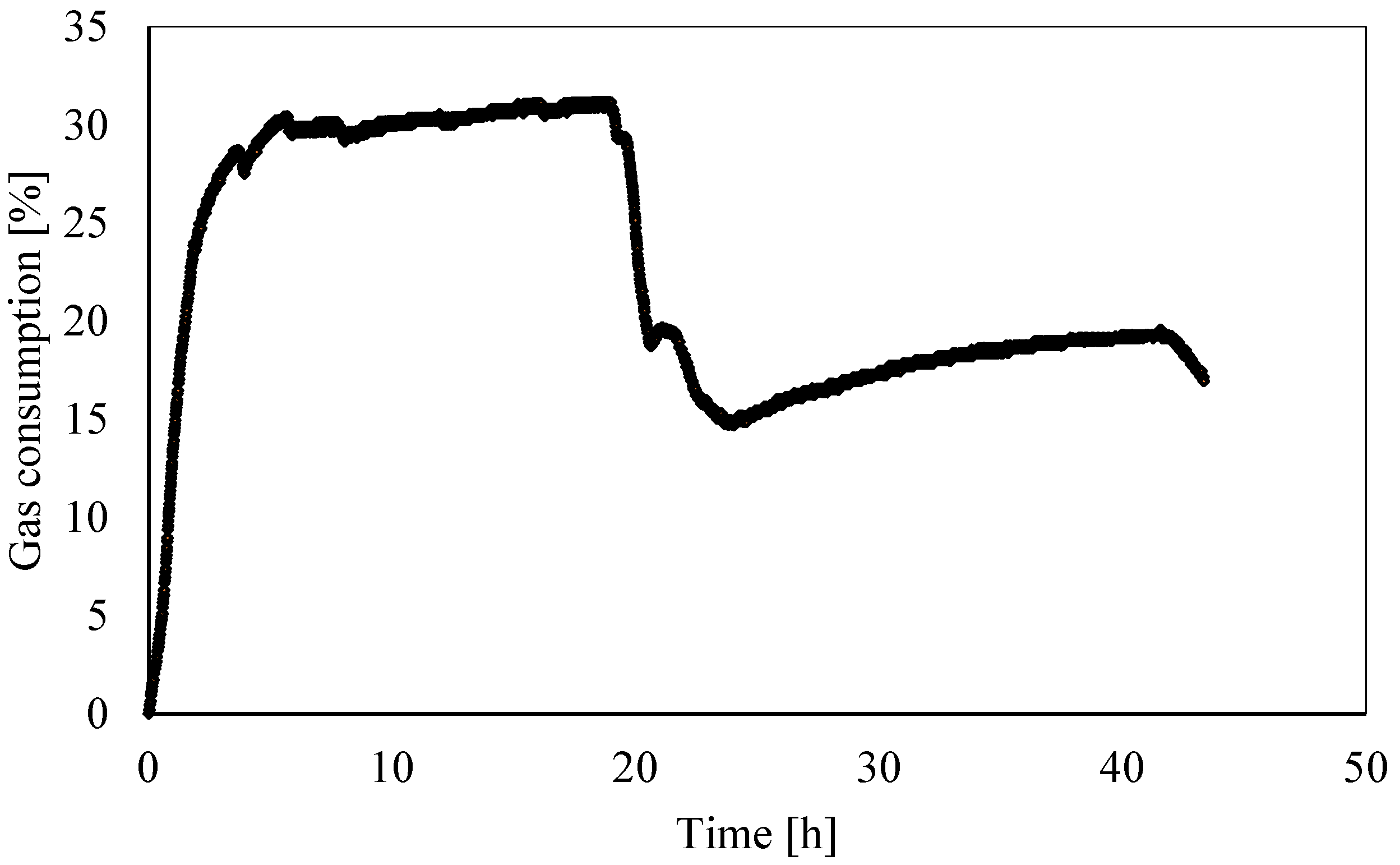
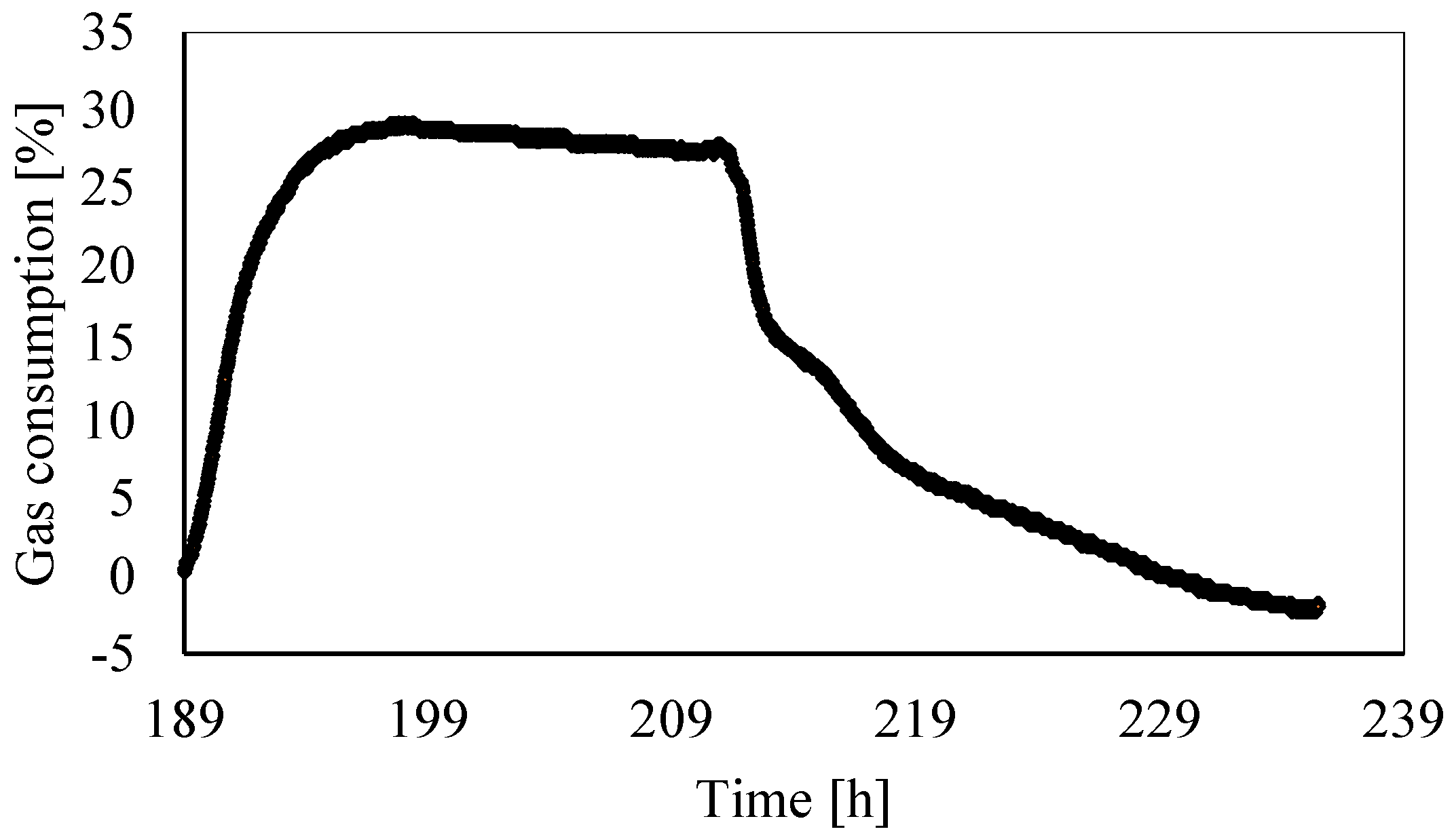
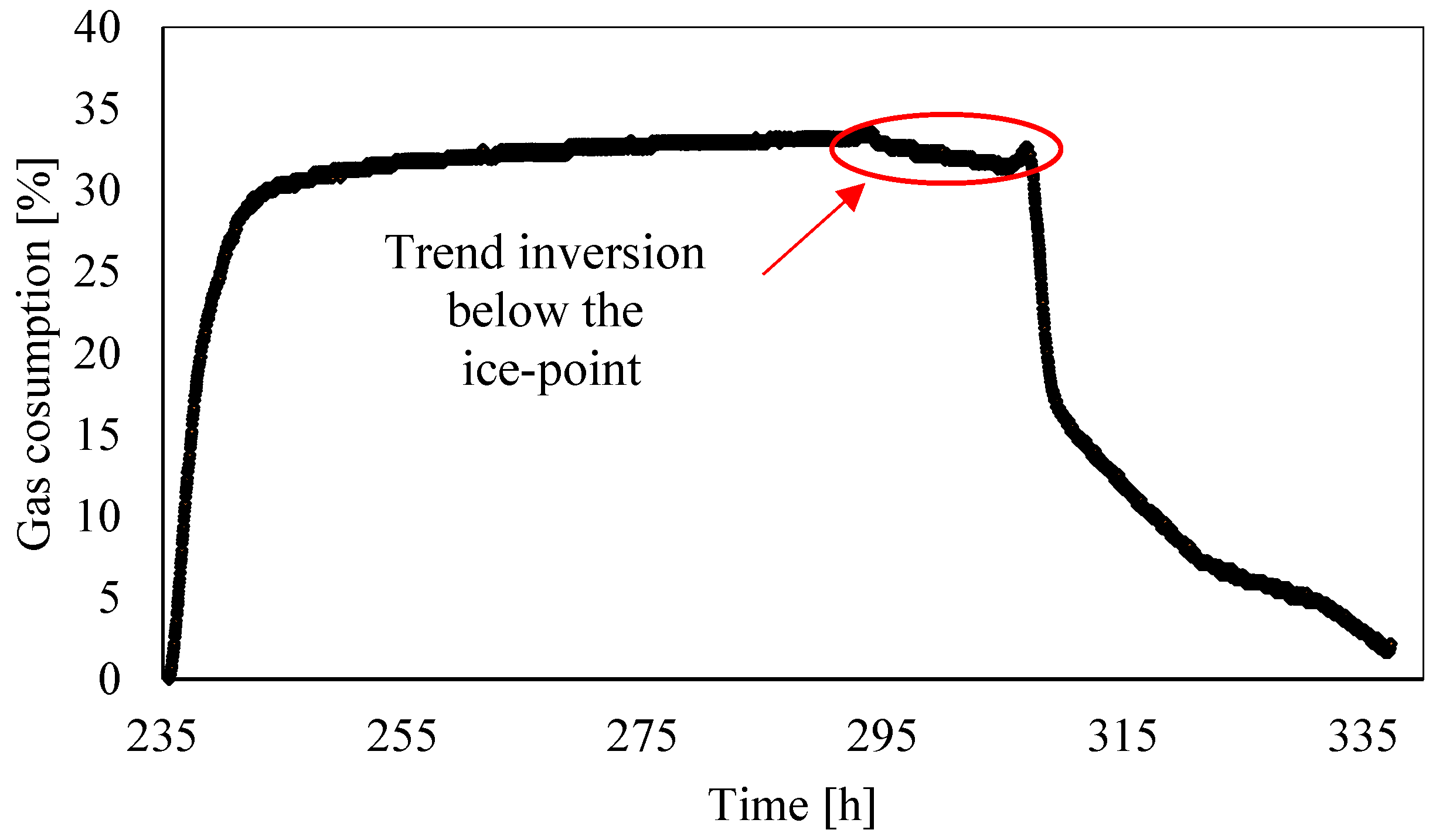
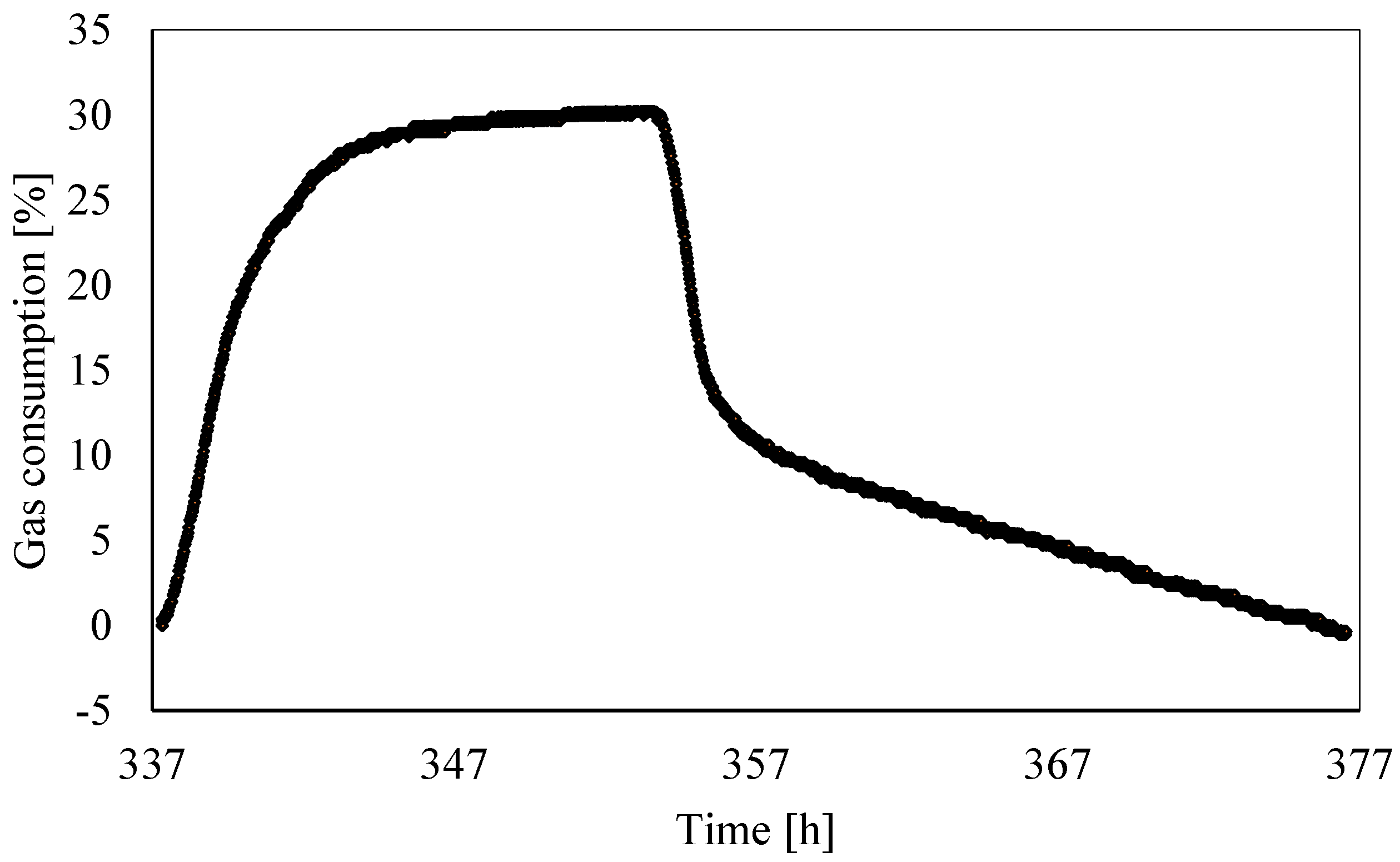
3.2. Analysis of Gas Consumption over Time
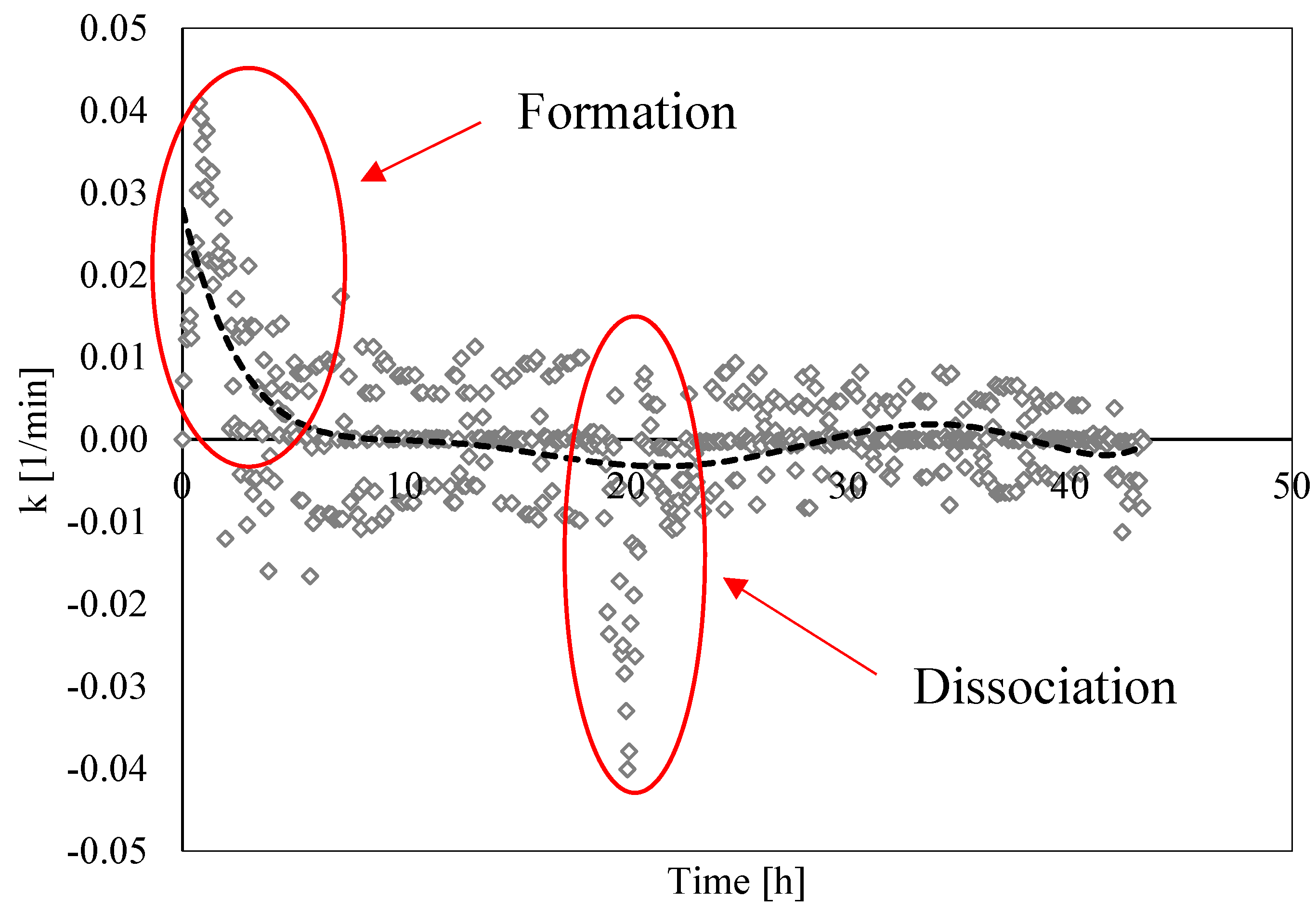
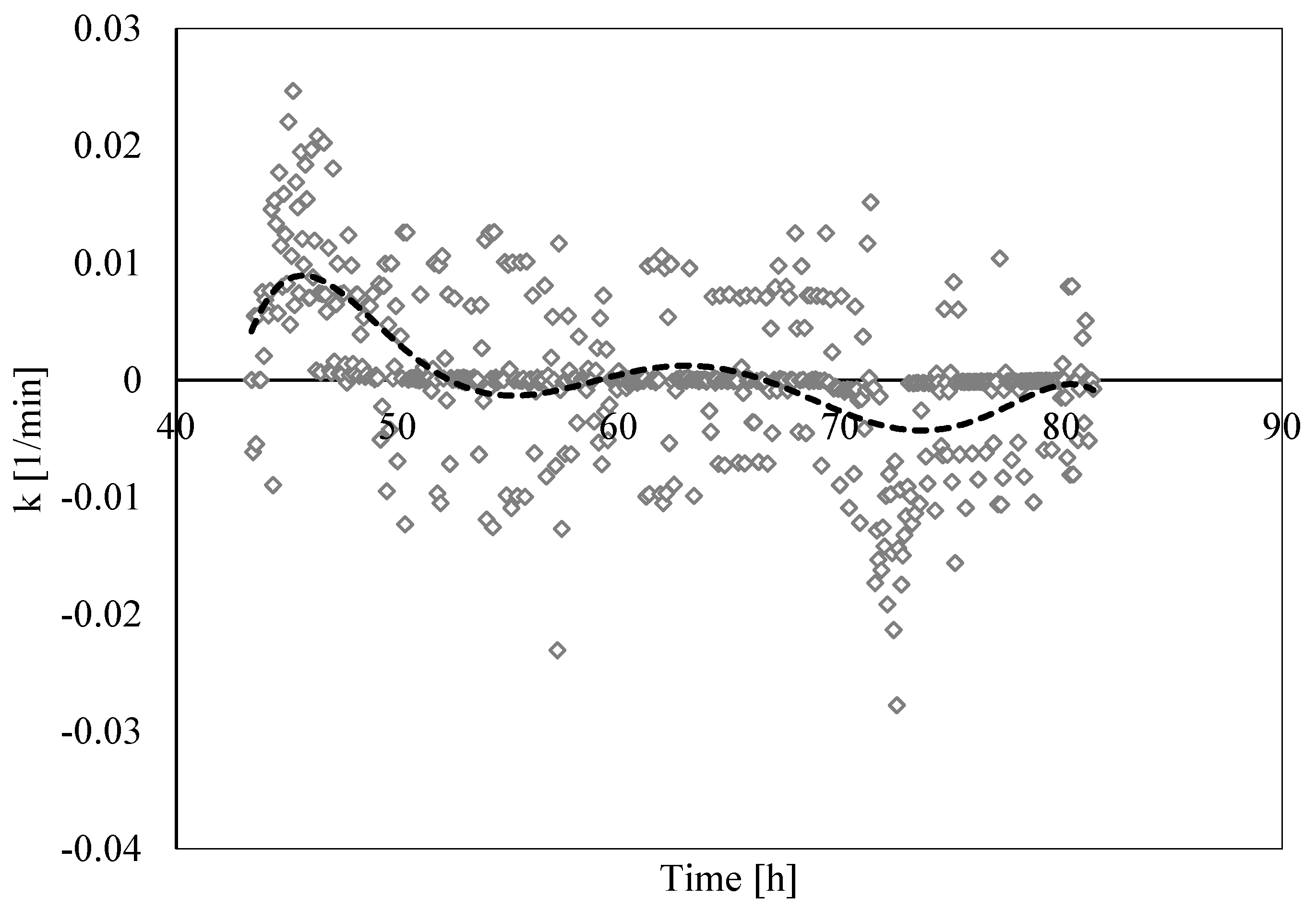
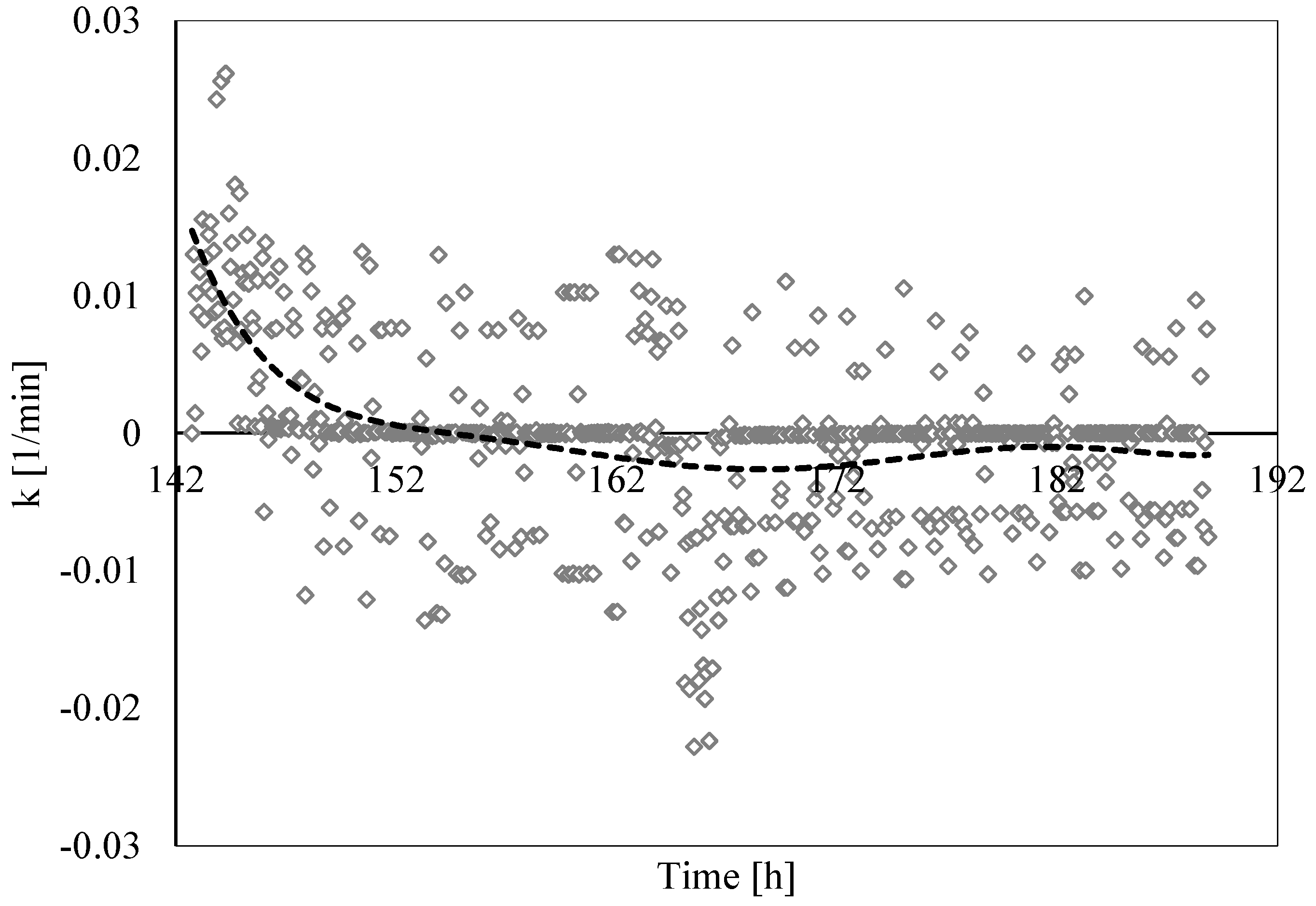
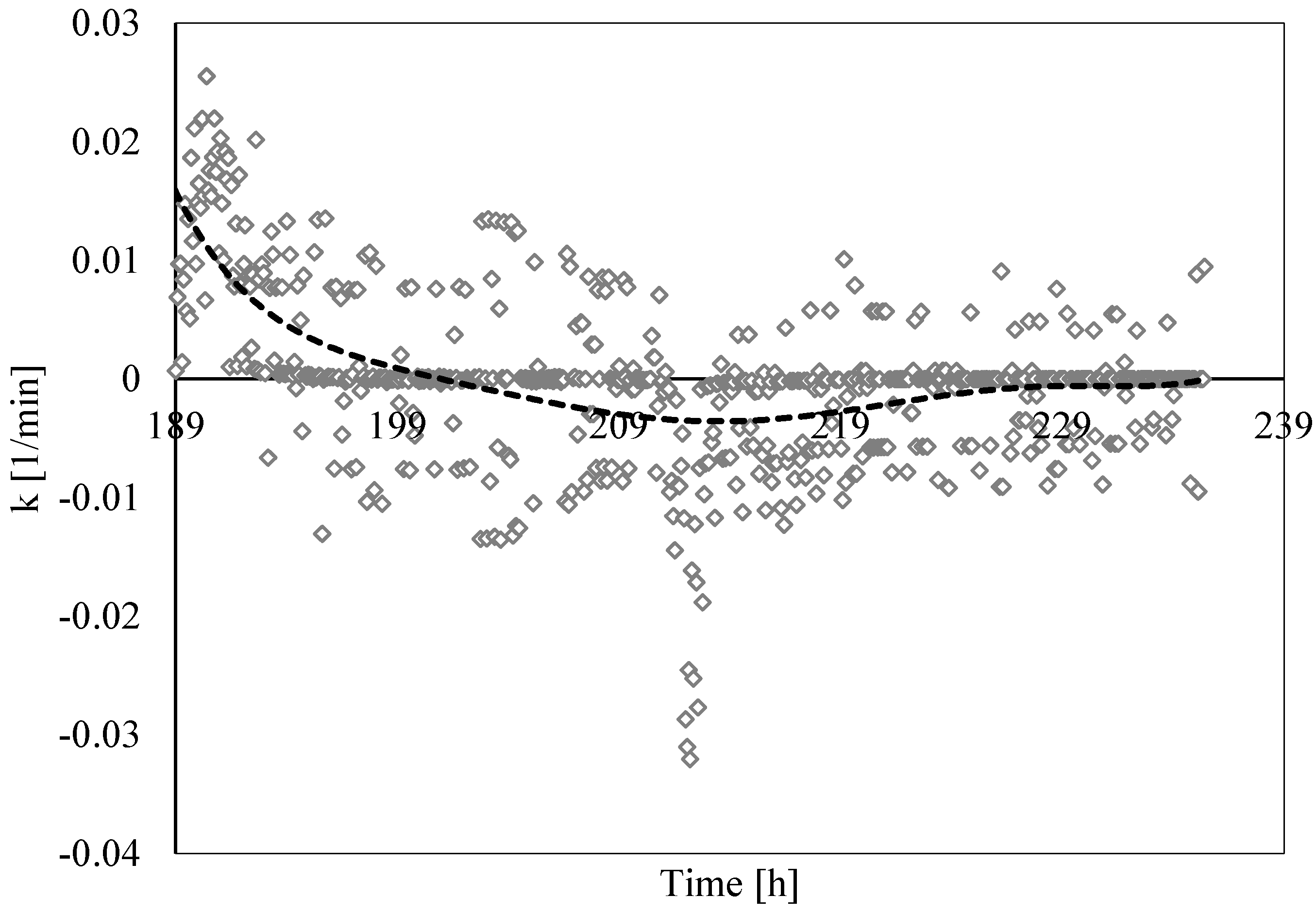
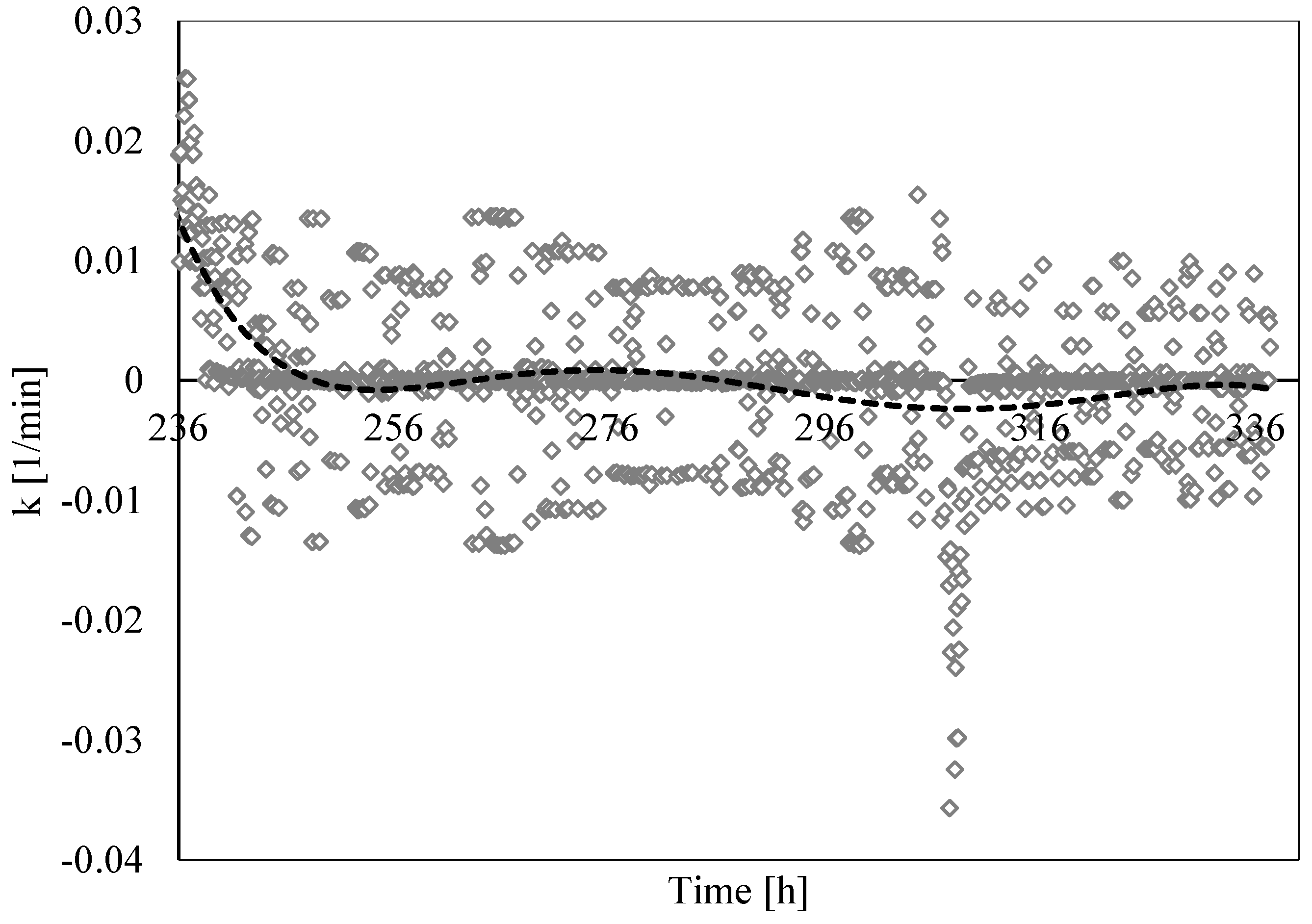
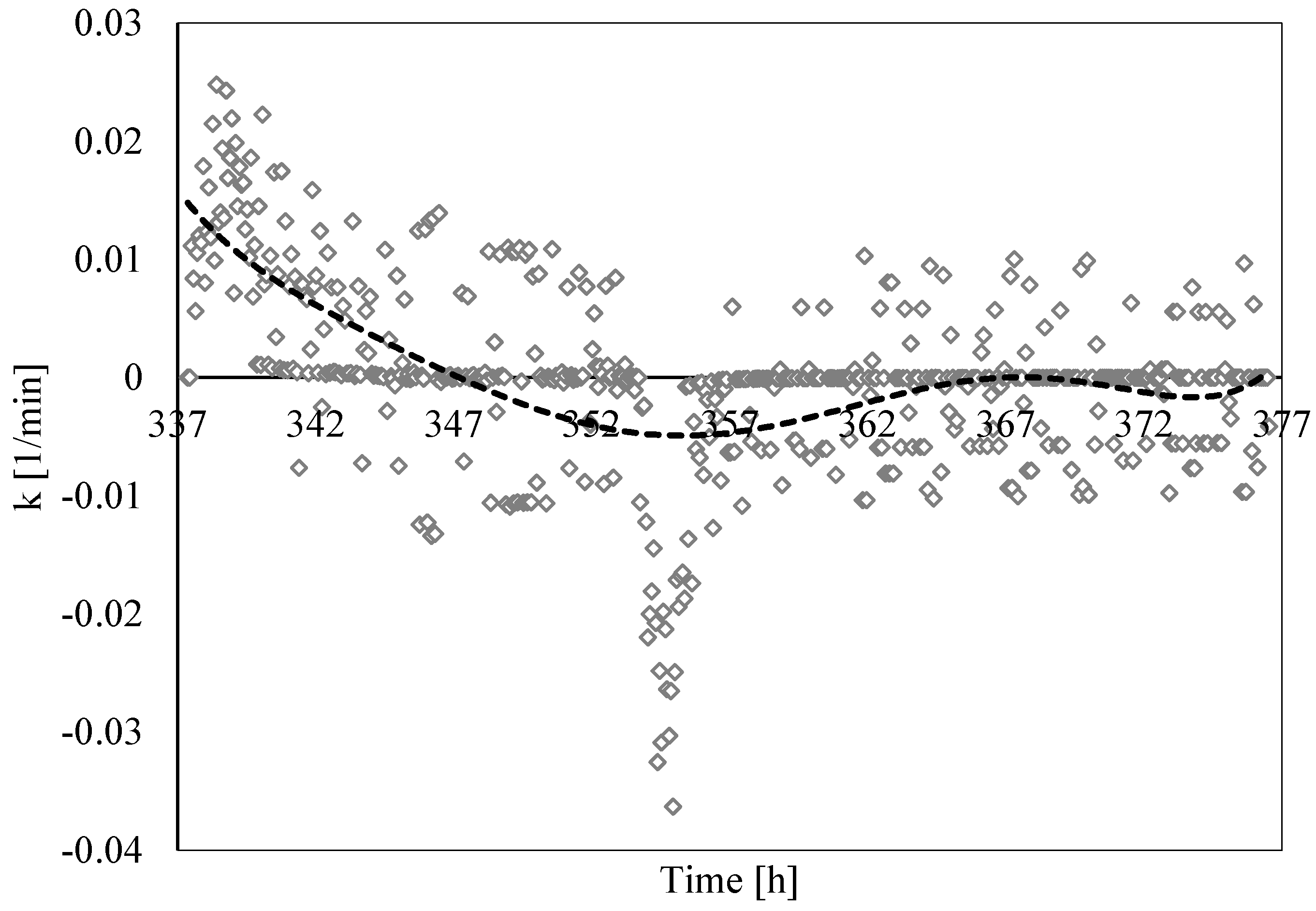
3.3. Analysis of Formation and Dissociation Rate Constant
4. Conclusions
- (1)
- Data about pressure and temperature conditions and the comparison between experimental and theoretical equilibrium proved the persistence of the so-called “memory effect” during tests. The difference between formation and dissociation diagrams constantly decreased from Test 1 to Test 3, while in the remaining three tests, the two curves were perfectly overlapped. In particular, in Test 1, the difference between the formation and the dissociation curves reached values up to 4.5 bar, while in Test 6, such a difference was completely negligible (absent or lower than 0.1 bar).
- (2)
- Conversely to the evidence presented in the literature, this work led us to assert that the “memory effect” also partially persists at temperatures equal or slightly above 25 °C. In the literature, there is a substantial lack of information about the properties of such a phenomenon in the presence of carbon dioxide and the present work proved the need for further insights.
- (3)
- The gas consumption allowed us to quantify the percentage of carbon dioxide dissolved in water and its variation over time as a function of the local thermodynamic conditions. Moreover, this parameter assumed a dual behavior in the presence of temperatures, respectively, above or below the freezing point. In the latter case, it showed a partial increase. Its trend proved the expected competition between ice and hydrates formation; in addition, it proved that ice formation also caused a partial release of gas molecules. Similar to point 2, here, further research will focus on defining if the released quantity was related to the portion of CO2 trapped into hydrates, dissolved in water, or both.
- (4)
- The analysis of the formation/dissociation rate constant confirmed what was asserted in the Labile Cluster Theory and extended its conclusion to the whole process (both formation and dissociation). During experiments, the thermodynamic conditions varied and, especially during hydrates dissociation, constantly remained close to the phase boundary equilibrium; consequently, labile clusters and hydrates nuclei formed and dissolved along the whole process.
- (5)
- Finally, such a parameter allowed us to characterize and explain the differences which often occur between experimental and theoretical equilibrium values. Hydrates formation constantly occurred during the dissociation phase, even if clearly less pronounced than hydrates dissociation. This secondary phenomenon affected the thermodynamic conditions and moved them away from the equilibrium. The analysis of parameter “k” allowed us to well identify and quantify it in order to give an idea of the uncertainty associated with the measurements.
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koh, C.A.; Sloan, E.D. Natural gas hydrates: Recent advances and challenges in energy and environmental applications. AIChE J. 2007, 53, 1636–1643. [Google Scholar] [CrossRef]
- Rossi, F.; Gambelli, A.M. Thermodynamic phase equilibrium of single-guest hydrate and formation data of hydrate in presence of chemical additives: A review. Fluid Phase Equilibr. 2021, 536, 112958. [Google Scholar] [CrossRef]
- Brewer, K.C.H.P.G. Clathrate hydrates in nature. Ann. Rev. Mar. Sci. 2009, 1, 303–327. [Google Scholar]
- Sloan, E.D.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Gambelli, A.M.; Filipponi, M.; Rossi, F. How methane release may affect carbon dioxide storage during replacement processes in natural gas hydrate reservoirs. J. Pet. Sci. Eng. 2021, 205, 108895. [Google Scholar] [CrossRef]
- Li, X.Y.; Li, X.S.; Wang, Y.; Liu, J.W.; Hu, H.Q. The optimization mechanism for gas hydrate dissociation by depressurization in the sediment with different water saturations and different particle sizes. Energy 2021, 215, 119129. [Google Scholar] [CrossRef]
- Song, Y.; Tian, M.; Zheng, J.N.; Yang, M. Thermodynamic analysis and ice behavior during the depressurization process of methane hydrate reservoir. Energy 2022, 250, 123801. [Google Scholar] [CrossRef]
- Dong, S.; Yang, M.; Chen, M.; Zheng, J.N.; Song, Y. Thermodynamics analysis and temperature response mechanism during methane hydrate production by depressurization. Energy 2022, 241, 122902. [Google Scholar] [CrossRef]
- Lv, J.; Cheng, Z.; Duan, J.; Wang, S.; Xue, K.; Liu, Y.; Mu, H. Enhanced CH4 recovery from hydrate-bearing sand packs via CO2 replacement assisted thermal stimulation method. J. Nat. Gas Sci. Eng. 2021, 96, 104326. [Google Scholar] [CrossRef]
- Yang, Z.; Si, H.; Zhong, D. AI-based composition model for energy utilization efficiency optimization of gas hydrate recovery by combined method of depressurization and thermal stimulation. J. Nat. Gas Sci. Eng. 2021, 92, 104001. [Google Scholar] [CrossRef]
- Tupsakhare, S.S.; Castaldi, M.J. Efficiency enhancements in methane recovery from natural gas hydrates using injection of CO2/N2 gas mixture simulating in-situ combustion. Appl. Energy 2019, 236, 825–836. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Castellani, B.; Nicolini, A.; Rossi, F. Water Salinity as Potential Aid for Improving the Carbon Dioxide Replacement Process’ Effectiveness in Natural Gas Hydrate Reservoirs. Processes 2020, 8, 1298. [Google Scholar] [CrossRef]
- Hassanpouryouzband, A.; Yang, J.; Tohidi, B.; Chuvilin, E.; Istomin, V.; Bukhanov, B.; Cheremisin, A. CO2 Capture by Injection of Flue Gas or CO2-N2 Mixtures into Hydrate Reservoirs: Dependence of CO2 Capture Efficiency on Gas Hydrate Reservoir Conditions. Environ. Sci. Technol. 2018, 52, 4324–4330. [Google Scholar] [CrossRef]
- Ota, M.; Morohashi, K.; Abe, Y.; Watanabe, M.; Lee Smith, R., Jr.; Inomata, H. Replacement of CH4 in the hydrate by use of liquid CO2. Energy Convers. Manag. 2005, 46, 1680–1691. [Google Scholar] [CrossRef]
- Ota, M.; Abe, Y.; Watanabe, M.; Lee Smith, R., Jr.; Inomata, H. Methane recovery from methane hydrate using pressurized CO2. Fluid Phase Equilibr. 2005, 228–229, 553–559. [Google Scholar] [CrossRef]
- Lee, S.; Lee, Y.; Lee, J.; Lee, H.; Seo, Y. Experimental verification of methane-carbon dioxide replacement in natural gas hydrate using a differential scanning calorimeter. Environ. Sci. Technol. 2013, 47, 13184–13190. [Google Scholar] [CrossRef]
- Ersland, G.; Husebo, J.; Graue, A.; Baldwin, B.A.; Howard, J.; Stevens, J. Measuring gas hydrate formation and exchange with CO2 in Bentheim sandstone using MRI tomography. Chem. Eng. J. 2010, 158, 25–31. [Google Scholar] [CrossRef]
- Feron, P.H.M.; Hendriks, C.A. CO2 Capture Process Principles and Costs. Oil Gas Sci. Technol. 2005, 60, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Kamath, V.A.; Holder, G.D.; Angert, P.F. 3 phase interfacial heat-transfer during the dissociation of propane hydrates. Chem. Eng. Sci. 1984, 39, 1435–1442. [Google Scholar] [CrossRef]
- Selim, M.S.; Sloan, E.D. Heat and mass transfer during the dissociation of hydrates in porous media. AIChE J. 1989, 35, 1049–1052. [Google Scholar] [CrossRef]
- Kvamme, B.; Coffin, R.B.; Zhao, J.; Wei, N.; Zhou, S.; Li, P.; Saeidi, N.; Chien, Y.C.; Dunn-Rankin, D.; Sun, W.; et al. Stages in the dynamics of hydrate formation and consequences for design of experiments for hydrate formation in sediments. Energies 2019, 12, 3399. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Tong, S.; Lu, K.; Chen, C.M.; Su, F.Y.; Zhou, J.; Lu, Z.H.; Wang, X.; Feng, G.; Zhang, R. Reduced graphene oxide supported Ni-Ce catalysts for CO2 methanation: The support and ceria promotion effects. J. CO2 Util. 2019, 34, 676–687. [Google Scholar] [CrossRef]
- Kamath, V.A.; Holder, G.D. Dissociation heat-transfer characteristics of methane hydrates. AIChE J. 1987, 33, 347–350. [Google Scholar] [CrossRef]
- Katsuki, D.; Ohmura, R.; Ebinuma, T.; Narita, H. Visual observation of dissociation of methane hydrate crystals in a glass micro model: Production and transfer of methane. Int. J. App. Phys. 2008, 104, 083514. [Google Scholar] [CrossRef]
- Almenningen, S.; Flatlandsmo, J.; Ferno, M.A.; Ersiand, G. Multiscale laboratory verification of depressurization for production of sedimentary methane hydrates. SPE J. 2017, 22, 138–147. [Google Scholar] [CrossRef]
- Hachikubo, A.; Takeya, S.; Chuvilin, E.; Istomin, V. Preservation phenomena of methane hydrate in pore spaces. Phys. Chem. Chem. Phys. 2011, 13, 17449–17452. [Google Scholar] [CrossRef]
- Kou, X.; Feng, J.C.; Li, X.S.; Wang, Y.; Chen, Z.Y. Memory effect of gas hydrate: Influencing factors of hydrate reformation and dissociation behaviors. Appl. Energy 2022, 306, 118015. [Google Scholar] [CrossRef]
- Li, Y.; Wu, N.; Ning, F.; Gao, D.; Hao, X.; Chen, Q.; Liu, C.; Sun, J. Hydrate-induced clogging of sand-control screen and its implication on hydrate production operation. Energy 2020, 206, 118030. [Google Scholar] [CrossRef]
- Li, Y.; Chen, J.; Gambelli, A.M.; Zhao, X.; Gao, Y.; Rossi, F. In situ experimental study on the effect of mixed inhibitors on the phase equilibrium of carbon dioxide hydrate. Chem. Eng. Sci. 2022, 248, 117230. [Google Scholar] [CrossRef]
- Roozeboom, H.W.B. Sur l’hydrate de l’acide sulfureux. Recl. Trav. Chim. Pays Bas 1884, 3, 29–58. [Google Scholar] [CrossRef]
- Wilson, P.W.; Haymet, A.D.J. Hydrate formation and re-formation in nucleating THF/water mixtures show no evidence to support a “memory” effect. Chem. Eng. J. 2010, 161, 146–150. [Google Scholar] [CrossRef]
- Li, Y.; Wu, N.; He, C.; Sun, Z.; Zhang, Z.; Hao, X.; Chen, Q.; Bu, Q.; Liu, C.; Sun, J. Nucleation probability and memory effect of methane-propane mixed gas hydrate. Fuel 2021, 291, 120103. [Google Scholar] [CrossRef]
- Takeya, S.; Hori, A.; Hondoh, T.; Uchida, T. Freezing-Memory Effect of Water on Nucleation of CO2 Hydrate Crystals. J. Phys. Chem. B 2000, 104, 4164–4168. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, B. Memory effect on the Pressure-Temperature Condition and Induction Time of Gas Hydrate nucleation. J. Nat. Gas Chem. 2010, 19, 446–451. [Google Scholar] [CrossRef]
- Duchateau, C.; Glénat, P.; Pou, T.E.; Hidalgo, M.; Dicharry, C. Hydrate Precursor Test Method for the Laboratory Evaluation of Kinetic Hydrate Inhibitors. Energy Fuels 2010, 24, 616–623. [Google Scholar] [CrossRef]
- Zeng, H.; Wilson, L.D.; Walker, V.K.; Ripmeester, J.A. Effect of Antifreeze Proteins on the Nucleation, Growth, and the Memory Effect during Tetrahydrofuran Clathrate Hydrate Formation. J. Am. Chem. Soc. 2006, 128, 2844–2850. [Google Scholar] [CrossRef]
- Davidson, D.W.; Garg, S.K.; Gough, S.R.; Handa, Y.P.; Ratcliffe, C.I.; Ripmeester, J.A.; Tse, J.S.; Lawson, W.F. Laboratory analysis of a naturally occurring gas hydrate from sediment of the Gulf of Mexico. Geochim. Cosmochim. Acta 1986, 50, 619–623. [Google Scholar] [CrossRef]
- Sloan, E.D.; Fleyfel, F. A molecular mechanism for gas hydrate nucleation from ice. AIChE J. 1991, 37, 1281–1292. [Google Scholar] [CrossRef]
- Muller-Bongartz, B.; Wildeman, T.R.; Sloan, E.D. A hypothesis for hydrate nucleation phenomena. In Proceedings of the Second International Offshore and Polar Engineering Conference, San Francisco, CA, USA, 14–19 June 1992; International Society of Offshore and Polar Engineers: Mountain View, CA, USA, 1992. [Google Scholar]
- Gambelli, A.M. An experimental description of the double positive effect of CO2 injection in methane hydrate deposits in terms of climate change mitigation. Chem. Eng. Sci. 2021, 233, 116430. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Rossi, F. Thermodynamic and kinetic characterization of methane hydrate nucleation, growth and dissociation processes, according to the Labile Cluster Theory. Chem. Eng. J. 2021, 425, 130706. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Tinivella, U.; Giovannetti, R.; Castellani, B.; Giustiniani, M.; Rossi, A.; Zannotti, M.; Rossi, F. Observation of the Main Parameters Influencing the Formation of Gas Hydrates. Energies 2021, 14, 1803. [Google Scholar] [CrossRef]
- Gambelli, A.M. Variations in terms of CO2 capture and CH4 recovery during replacement processes in gas hydrate reservoirs, associated to the “memory effect”. J. Clean. Prod. 2022, 360, 132154. [Google Scholar] [CrossRef]
- Rossi, F.; Li, Y.; Gambelli, A.M. Thermodynamic and kinetic description of the main effects related to the memory effect during carbon dioxide hydrates formation in a confined environment. Sustainability 2021, 13, 13797. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Presciutti, A.; Rossi, F. Kinetic considerations and formation rate for carbon dioxide hydrate, formed in presence of a natural silica-based porous medium: How initial thermodynamic conditions may modify the process kinetic. Thermochim. Acta 2021, 705, 179039. [Google Scholar] [CrossRef]
- Takeya, S.; Kida, M.; Minami, H.; Sakagami, H.; Hachikubo, A.; Takahashi, N.; Shoji, H.; Soloviev, V.; Wallmann, K.; Biebow, N.; et al. Structure and thermal expansion of natural gas clathrate hydrates. Chem. Eng. Sci. 2006, 61, 2670–2674. [Google Scholar] [CrossRef]
- Aregba, A.G. Gas Hydrate—Properties, Formation and Benefits. Open J. Yangtze Oil Gas 2017, 2, 27–44. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, G.C.; Castaldi, M.J.; Zhou, Y. Large scale reactor details and results for the formation and decomposition of methane hydrates via thermal stimulation dissociation. J. Pet. Sci. Eng. 2012, 94, 19–27. [Google Scholar] [CrossRef]
- Nema, Y.; Ohmura, R.; Senaha, I.; Yasuda, K. Quadruple point determination in carbon dioxide hydrate forming system. Fluid Phase Equilibr. 2017, 441, 49–53. [Google Scholar] [CrossRef]
- Nagashima, H.D.; Fukushima, N.; Ohmura, R. Phase equilibrium condition measurements in carbon dioxide clathrate hydrate forming system from 199.1 K to 247.1 K. Fluid Phase Equilibr. 2016, 413, 53–56. [Google Scholar] [CrossRef]
- Khan, M.S.; Partoon, B.; Bavoh, C.B.; Lal, B.; Mellon, B.M. Influence of tetramethylammonium hydroxide on methane and carbon dioxide gas hydrate phase equilibrium conditions. Fluid Phase Equilibr. 2017, 440, 1–8. [Google Scholar] [CrossRef]
- Khan, M.S.; Bavoh, C.; Partoon, B.; Lal, B.; Bustam, M.A.; Shariff, A.M. Thermodynamic effect of ammonium based ionic liquids on CO2 hydrates phase boundary. J. Mol. Liq. 2017, 238, 533–539. [Google Scholar] [CrossRef]
- Sadeq, D.; Iglauer, S.; Lebedev, M.; Smith, C.; Barifcani, A. Experimental determination of hydrate phase equilibrium for different gas mixtures containing methane, carbon dioxide and nitrogen with motor current measurements. J. Nat. Gas Sci. Eng. 2017, 38, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Sun, C.; Chen, G.; Nie, Y.; Sun, Z.; Liu, Y. Measurements of Hydrate Equilibrium Conditions for CH4, CO2, and CH4+C2H6+C3H8 in Various Systems by Step-heating Method. Chin. J. Chem. Eng. 2009, 17, 635–641. [Google Scholar] [CrossRef]
- Jarrahian, A.; Nakhaee, A. Hydrate-liquid-vapor equilibrium condition for N2 + CO2 + H2O system: Measurement and modelling. Fuel 2019, 237, 769–774. [Google Scholar] [CrossRef]
- Kyung, D.; Lee, K.; Kim, H.; Lee, W. Effect of marine environmental factors on the phase equilibrium of CO2 hydrate. Int. J. Greenh. Gas Control 2014, 20, 285–292. [Google Scholar] [CrossRef]
- Seo, Y.T.; Lee, H. Multiple-phase hydrate equilibria of the ternary carbon dioxide, methane, and water mixtures. J. Phys. Chem. B 2001, 105, 10084–10090. [Google Scholar] [CrossRef]
- Yu, Y.S.; Zhou, S.D.; Li, X.S.; Wang, S.L. Effect of graphite nanoparticles on CO2 hydrate phase equilibrium. Fluid Phase Equilibr. 2016, 414, 23–28. [Google Scholar] [CrossRef]
- Pahlavanzadeh, H.; Farhoudi, A.; Manteghian, M. Experimental measurement of carbon dioxide clathrate hydrate in the presence of adamantane and other water soluble and insoluble additives. J. Chem. Thermodyn. 2019, 135, 352–358. [Google Scholar] [CrossRef]
- Herri, J.M.; Bouchemoua, A.; Kwatersky, M.; Fezoua, A.; Ouabbas, Y.; Cameirao, A. Gas hydrate equilibria for CO2-N2 and CO2-CH4 gas mixtures—Experimental studies and thermodynamic modelling. Fluid Phase Equilibr. 2011, 301, 171–190. [Google Scholar] [CrossRef] [Green Version]
- Gambelli, A.M.; Rossi, F. Experimental investigation on the possibility of defining the feasibility of CO2/CH4 exchange into a natural gas hydrate marine reservoir via fast analysis of sediment properties. Chem. Eng. Res. Des. 2021, 171, 327–339. [Google Scholar] [CrossRef]
- Gambelli, A.M. Analyses on CH4 and CO2 hydrate formation to define the optimal pressure for CO2 injection to maximize the replacement efficiency into natural gas hydrate in presence of a silica-based natural porous medium, via depressurization techniques. Chem. Eng. Process. 2021, 167, 108512. [Google Scholar] [CrossRef]
- Chaturvedi, E.; Maiti, M.; Laik, S.; Mandal, A. Mineralogical and structural characterization of the sediments of Krishna Godavari and Mahanadi Basin and their influences on hydrate formation kinetics. Adv. Powder Technol. 2021, 32, 1247–1263. [Google Scholar] [CrossRef]
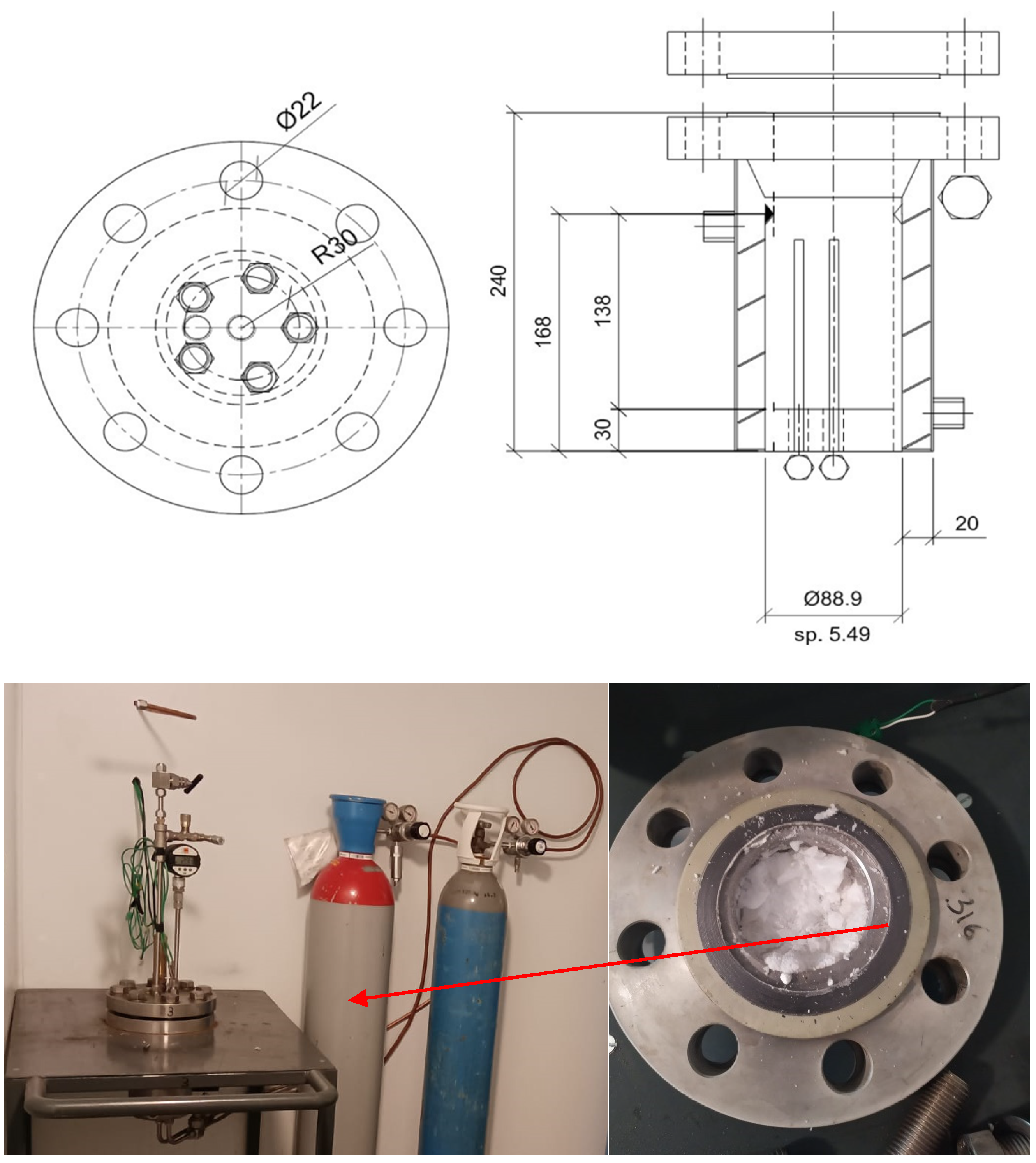







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test n° | k [mol/min] |
|---|---|
| 1 | 8.9 × 10−4 |
| 2 | 2.82 × 10−5 |
| 3 | 2.25 × 10−4 |
| 4 | 2.63 × 10−4 |
| 5 | 1.16 × 10−4 |
| 6 | 3.91 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambelli, A.M.; Filipponi, M.; Rossi, F. Sequential Formation of CO2 Hydrates in a Confined Environment: Description of Phase Equilibrium Boundary, Gas Consumption, Formation Rate and Memory Effect. Sustainability 2022, 14, 8829. https://doi.org/10.3390/su14148829
Gambelli AM, Filipponi M, Rossi F. Sequential Formation of CO2 Hydrates in a Confined Environment: Description of Phase Equilibrium Boundary, Gas Consumption, Formation Rate and Memory Effect. Sustainability. 2022; 14(14):8829. https://doi.org/10.3390/su14148829
Chicago/Turabian StyleGambelli, Alberto Maria, Mirko Filipponi, and Federico Rossi. 2022. "Sequential Formation of CO2 Hydrates in a Confined Environment: Description of Phase Equilibrium Boundary, Gas Consumption, Formation Rate and Memory Effect" Sustainability 14, no. 14: 8829. https://doi.org/10.3390/su14148829
APA StyleGambelli, A. M., Filipponi, M., & Rossi, F. (2022). Sequential Formation of CO2 Hydrates in a Confined Environment: Description of Phase Equilibrium Boundary, Gas Consumption, Formation Rate and Memory Effect. Sustainability, 14(14), 8829. https://doi.org/10.3390/su14148829