
Shifts in Bacterial Community Associated with Green Manure Soybean Intercropping and Edaphic Properties in a Tea Plantation


Abstract
:1. Introduction
2. Materials and Methods
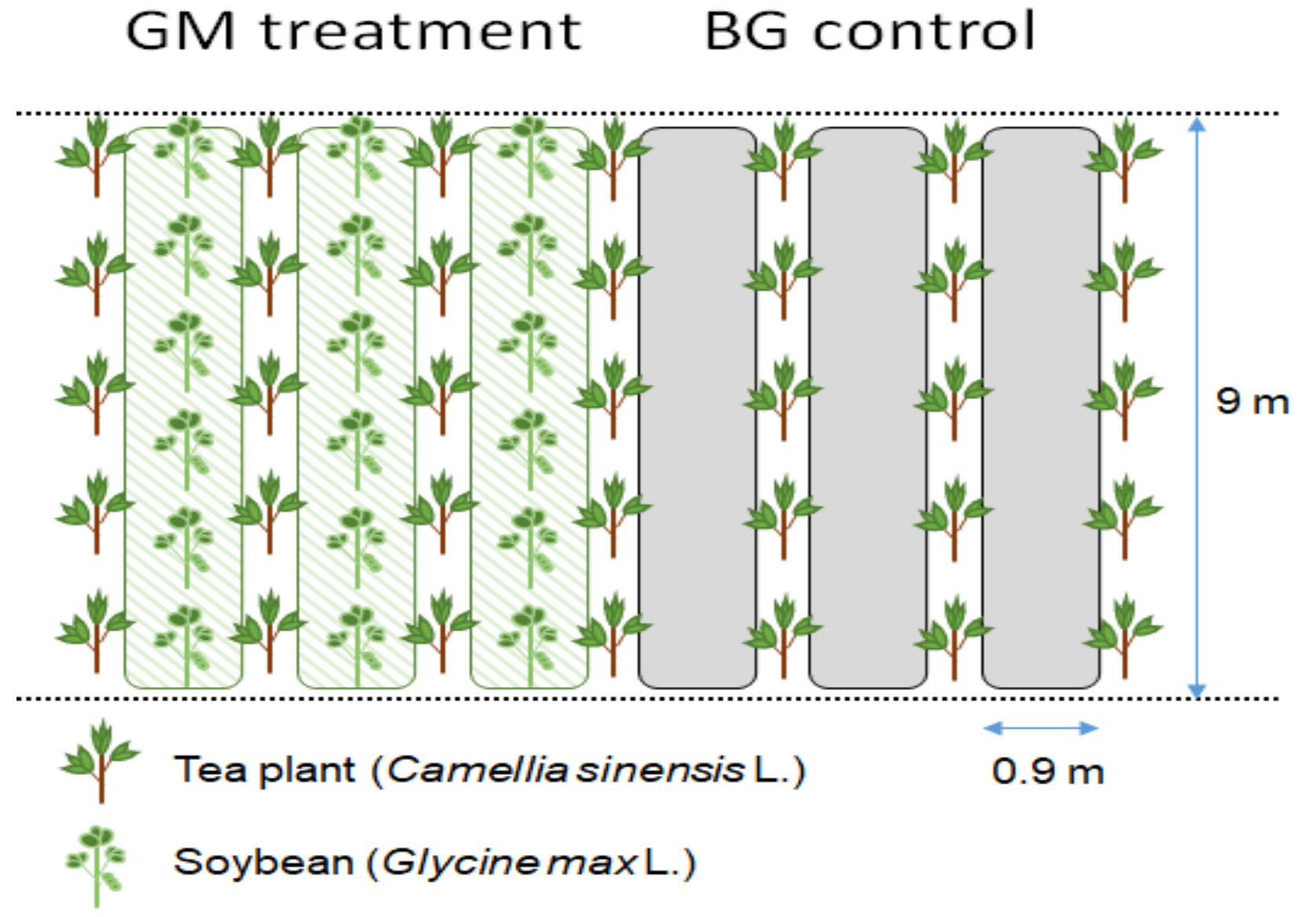
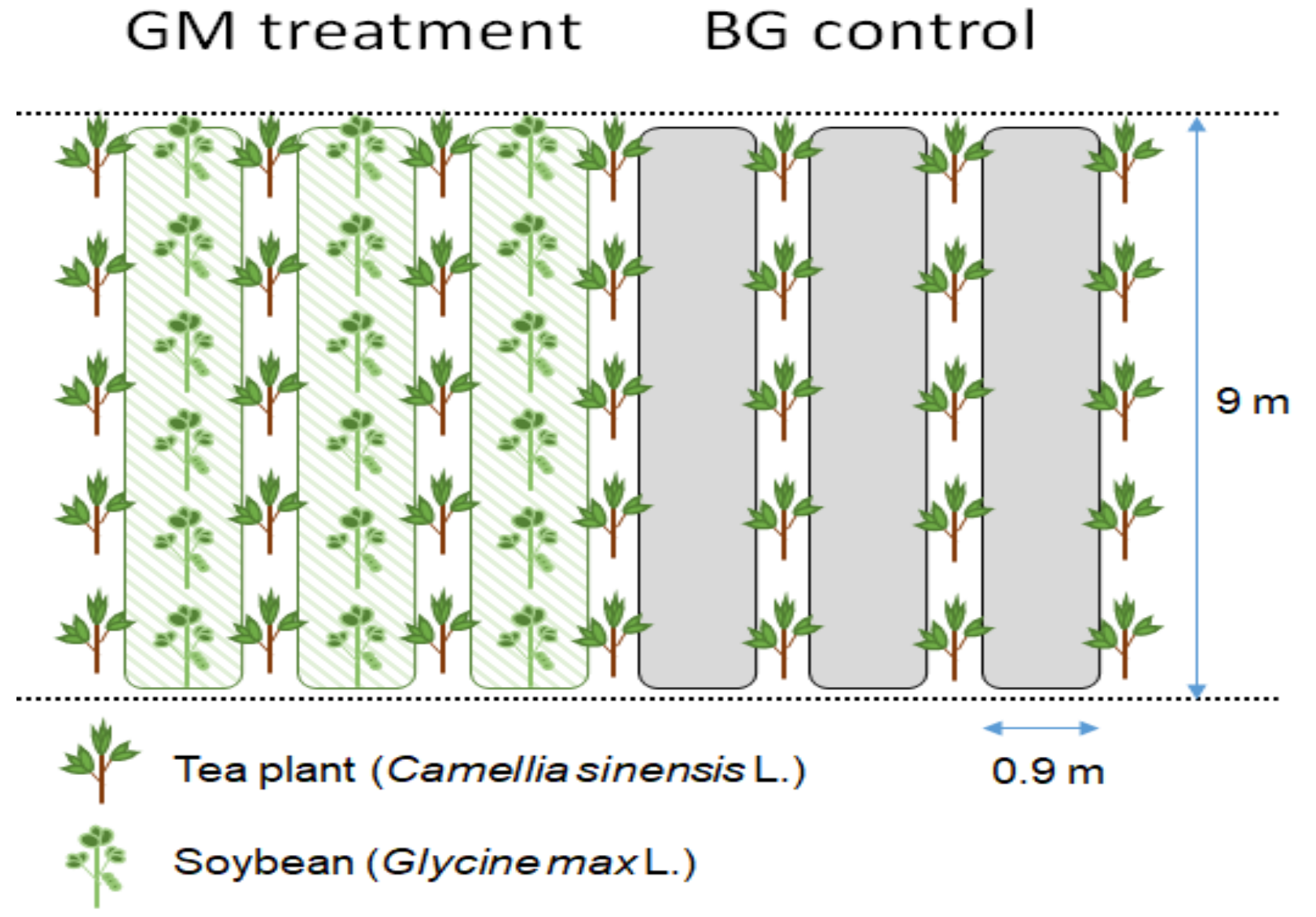
2.1. Site Descriptions and Sampling
2.2. Analyses of Soil Chemical Properties
2.3. Determination of Functional Diversity and Metabolic Potential of Bacterial Communities
2.4. Determination of Population Diversity and Structure of Bacterial Communities
2.5. Statistical Analysis
3. Results
3.1. Soil Chemical Properties
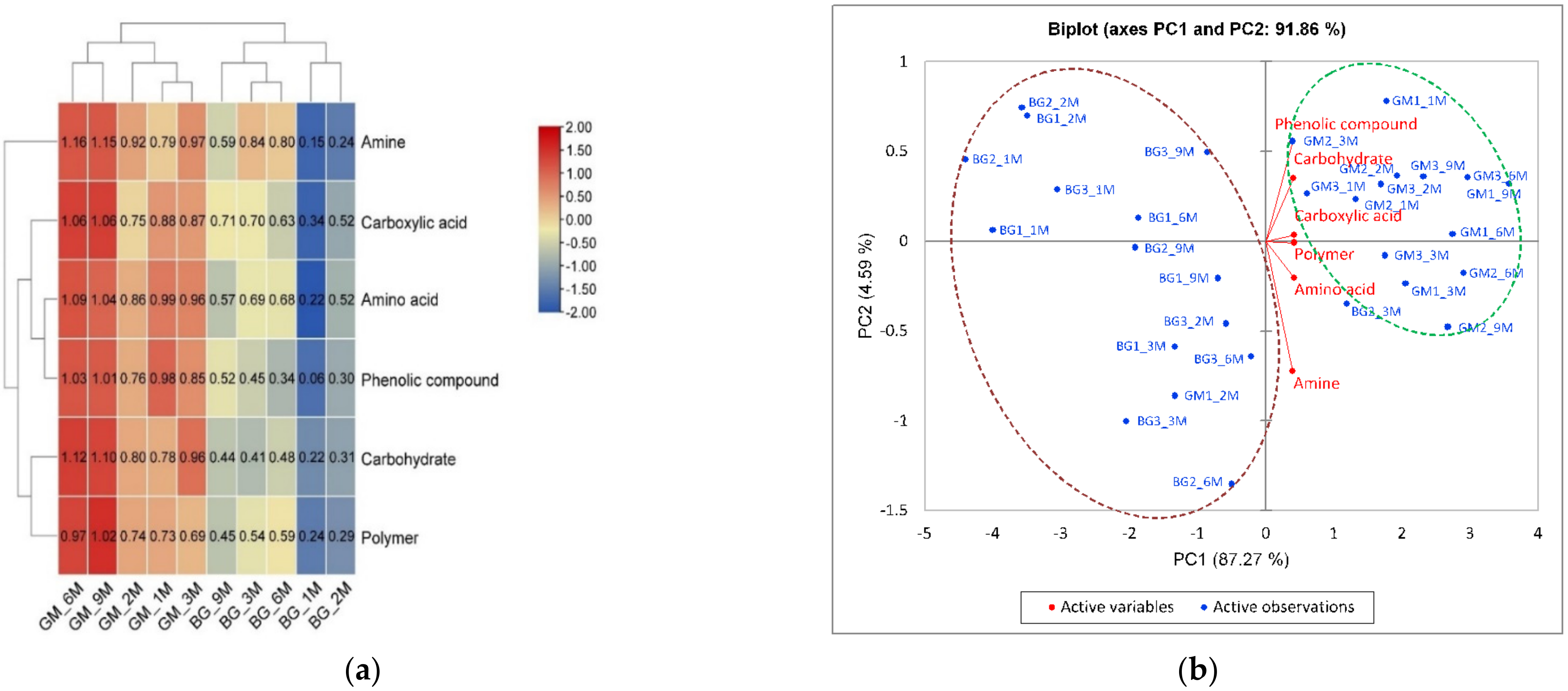
3.2. Functional Diversity and Metabolic Potential of Bacterial Communities
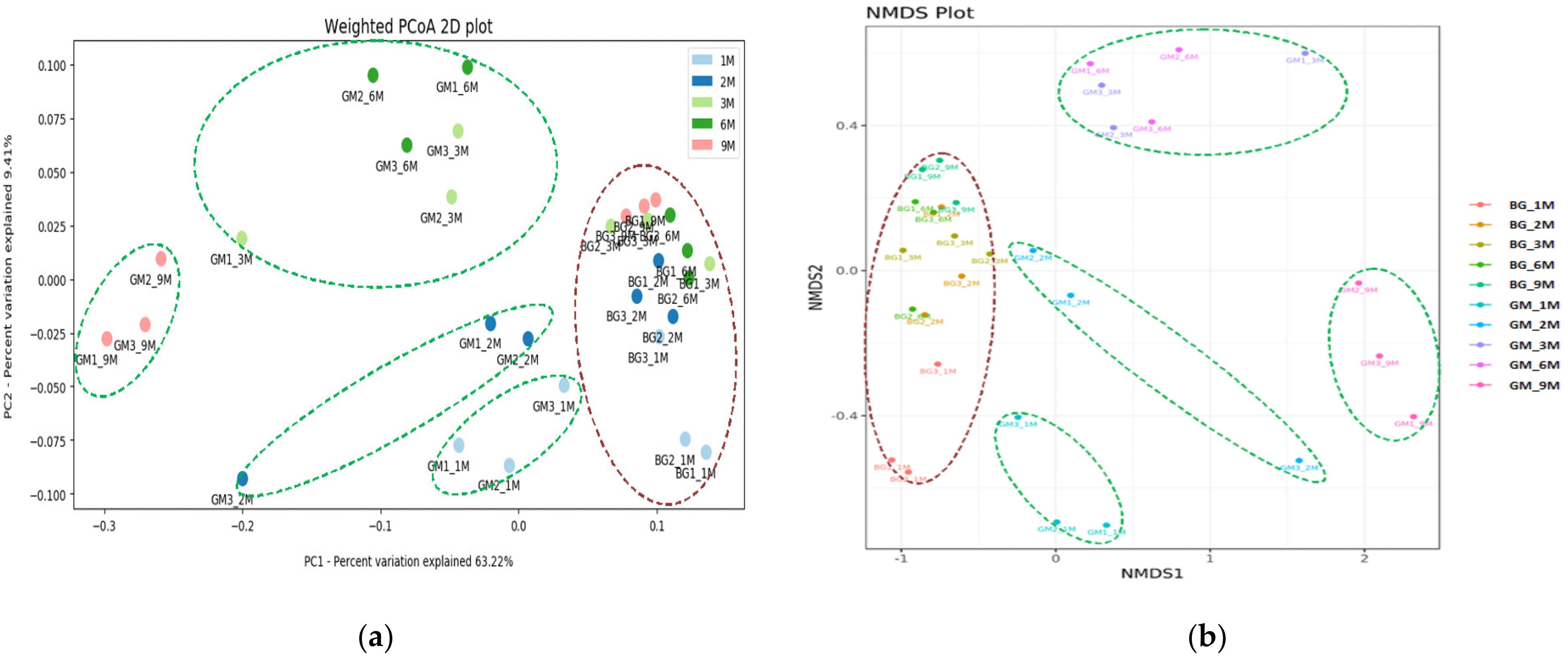
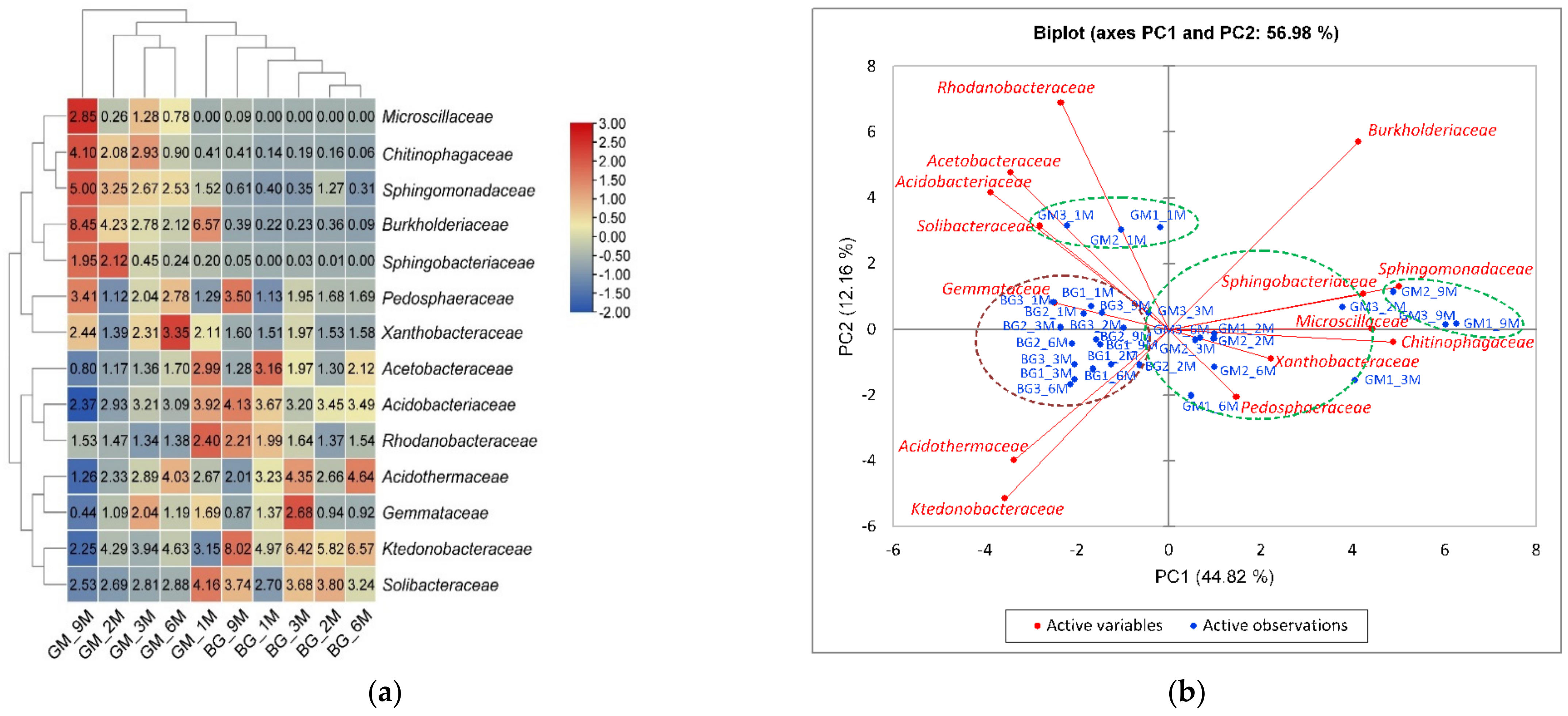
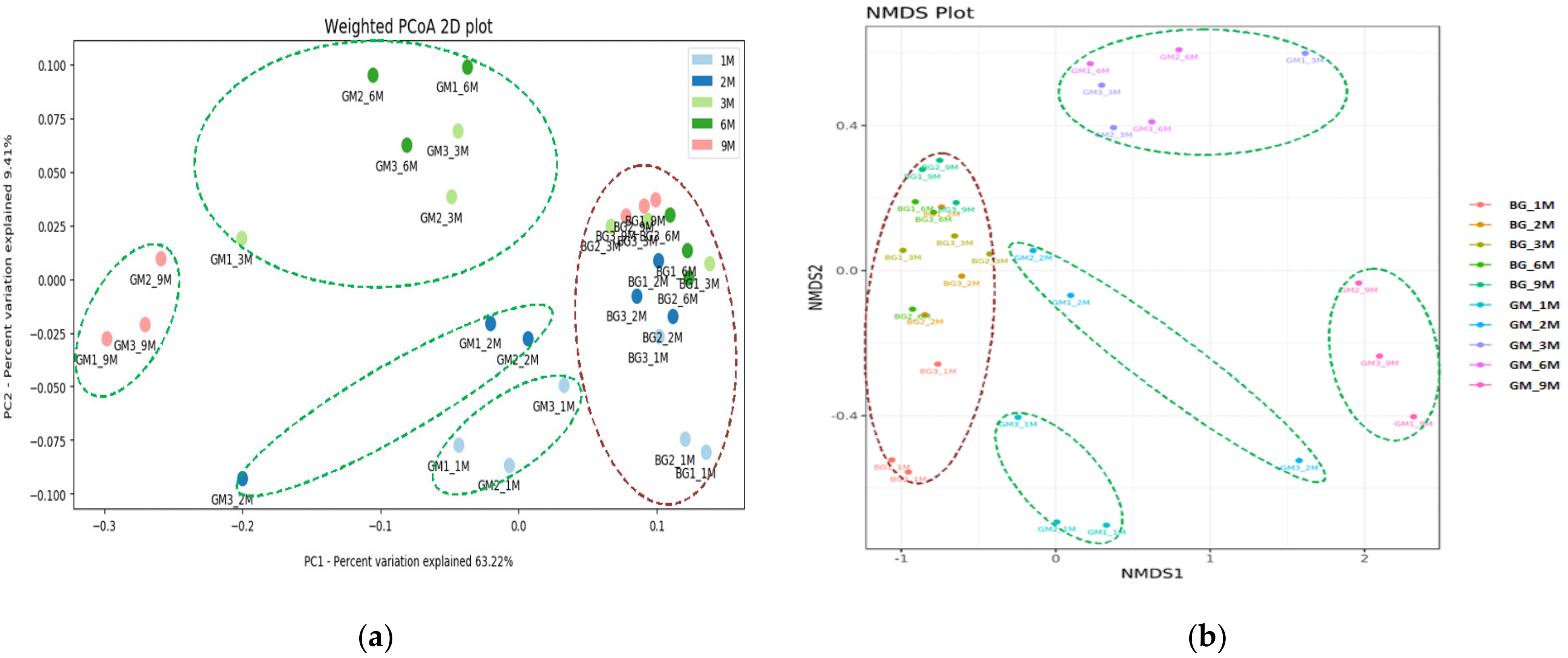
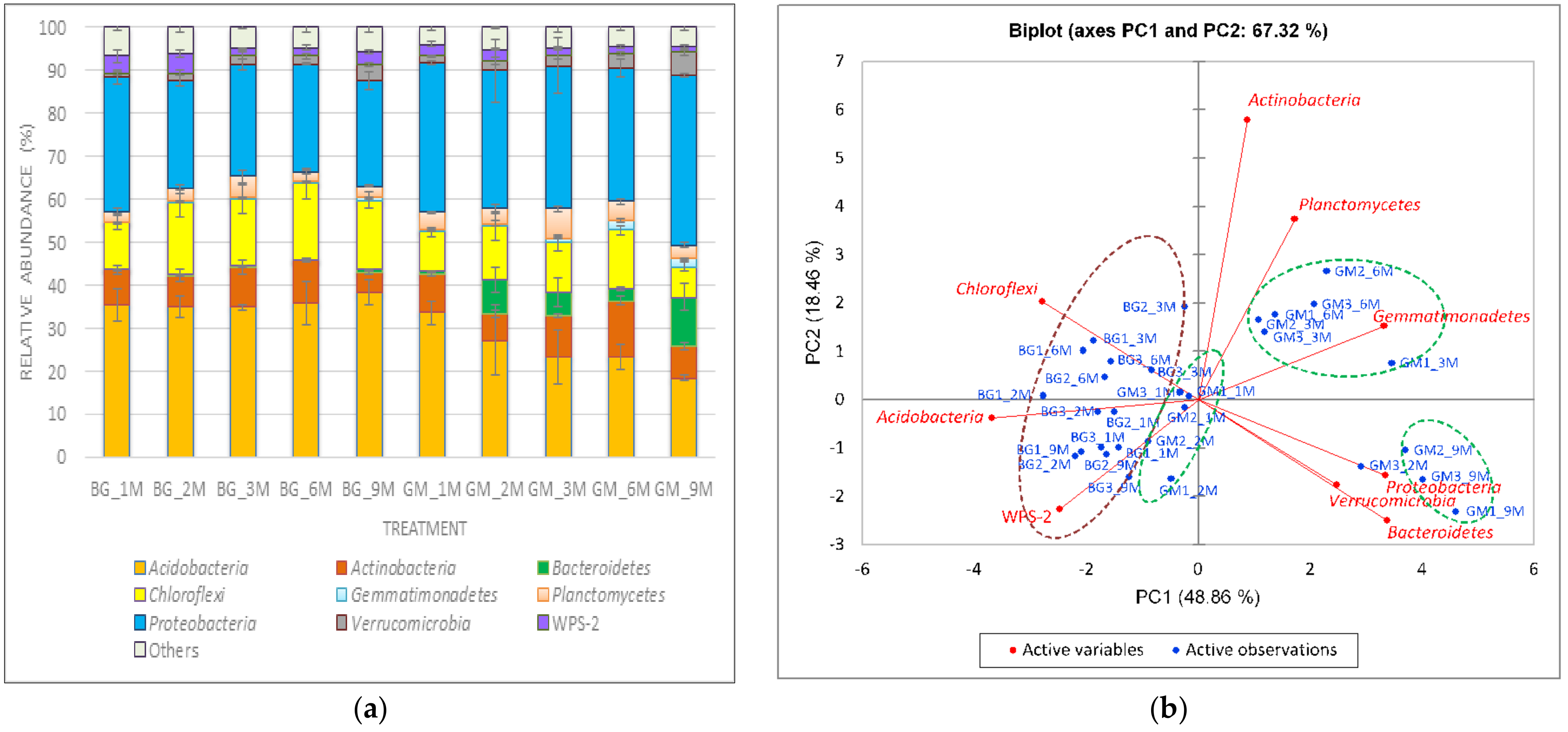
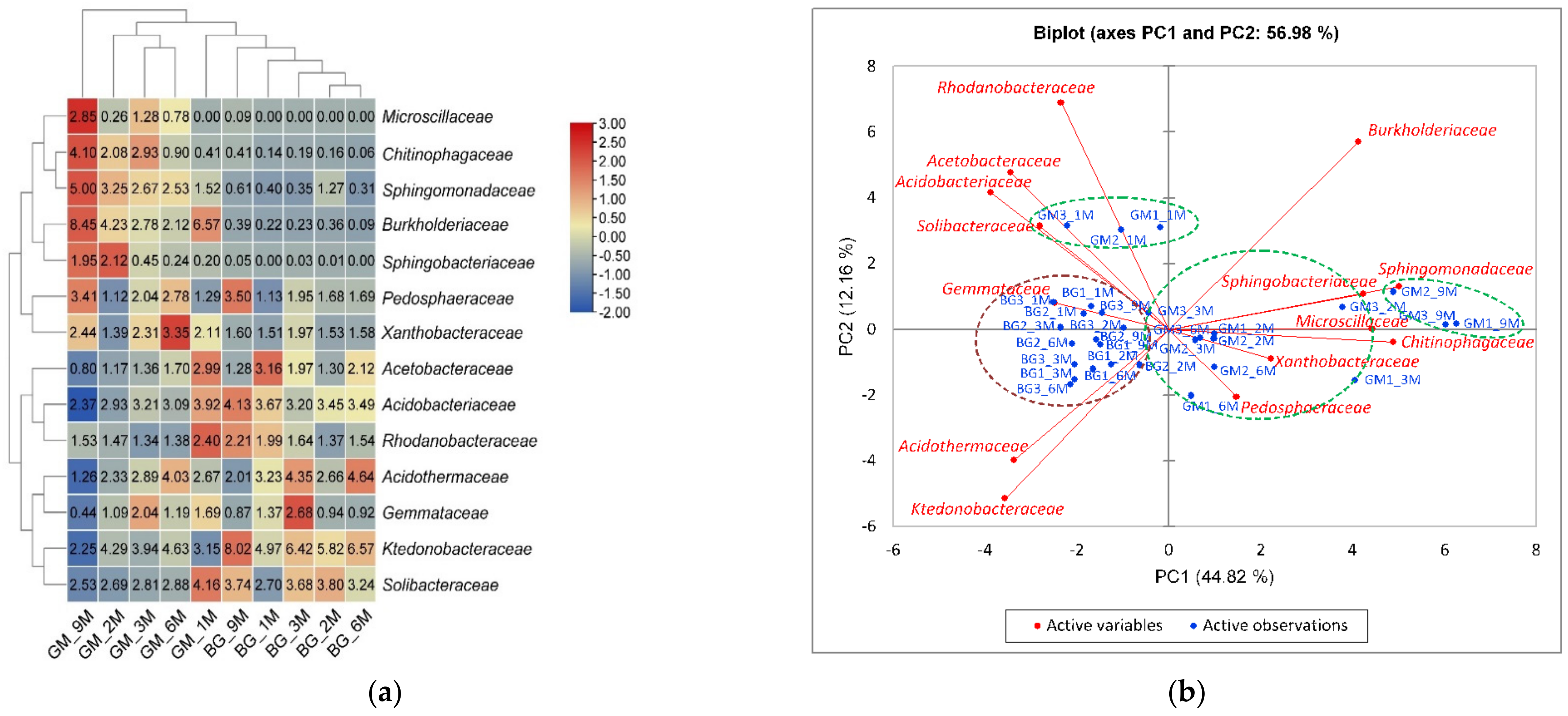
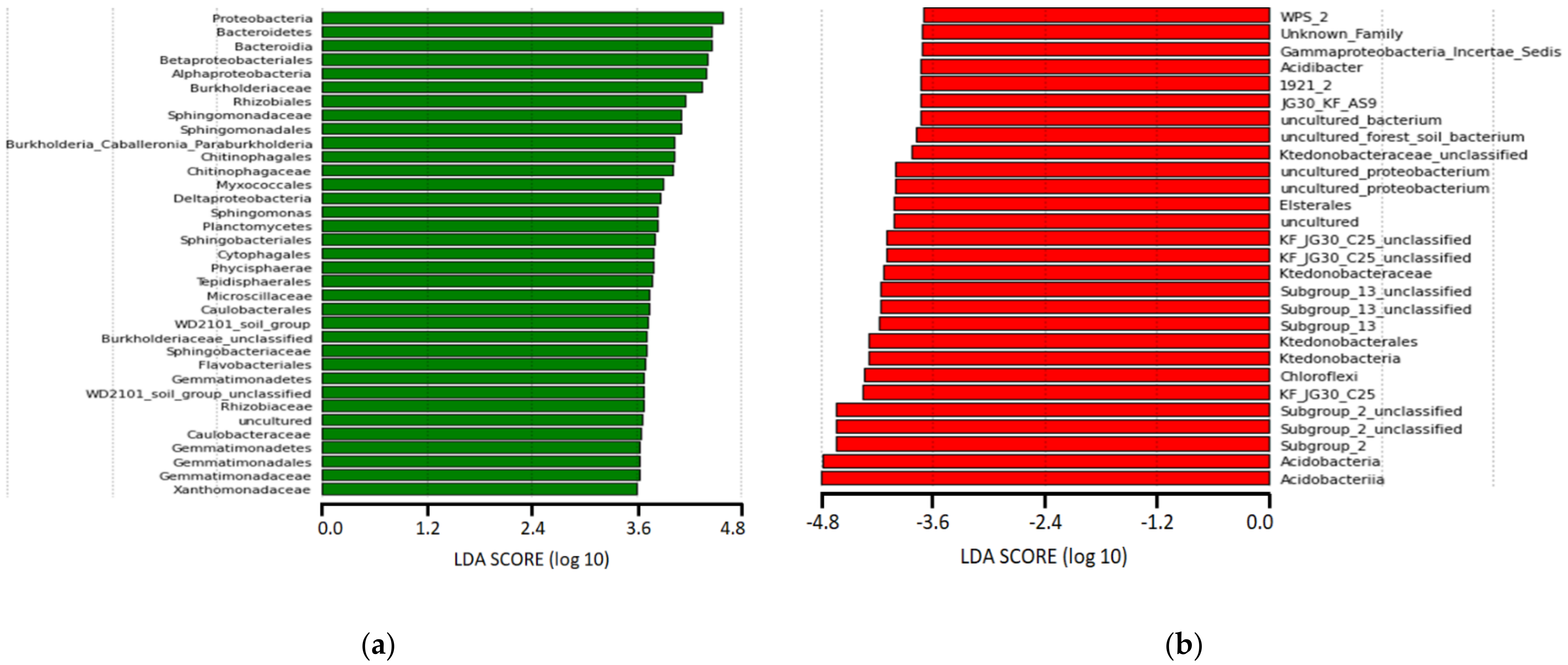
3.3. Population Diversity and Structure of Bacterial Communities
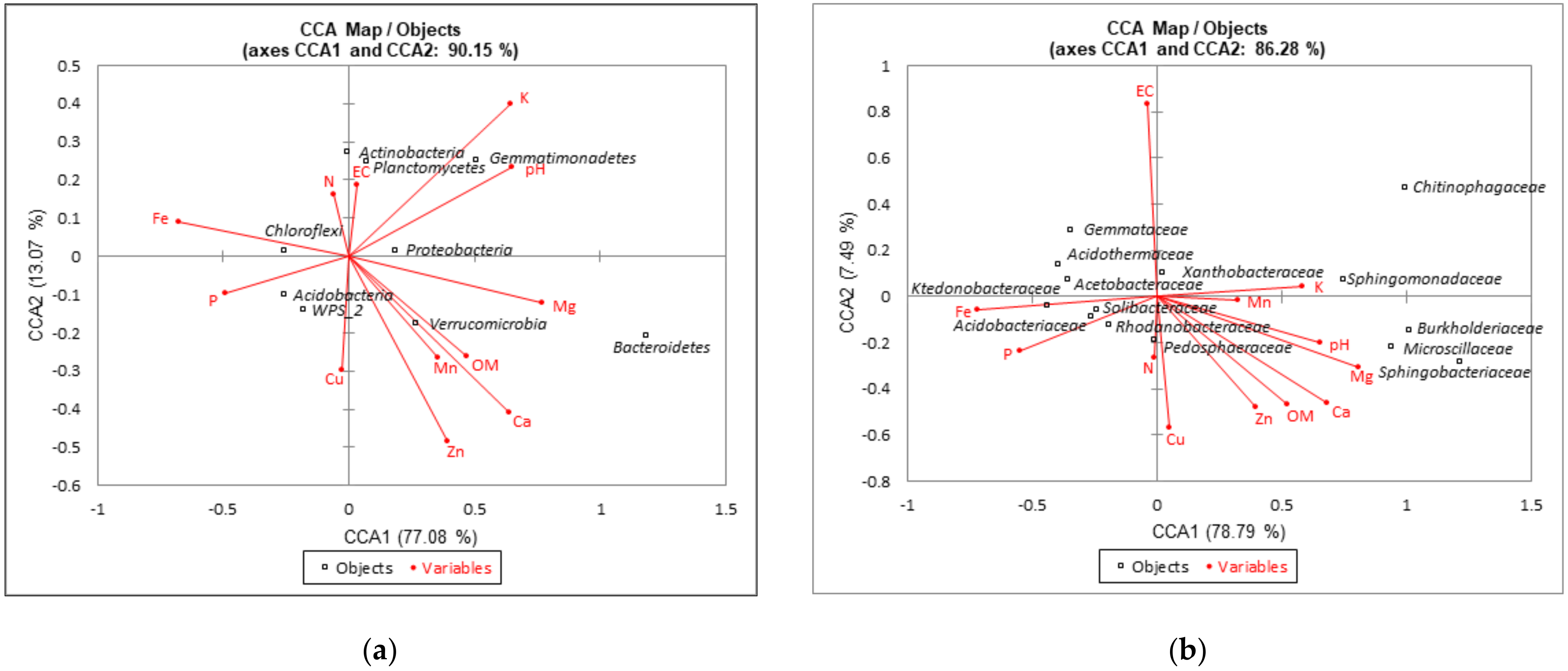
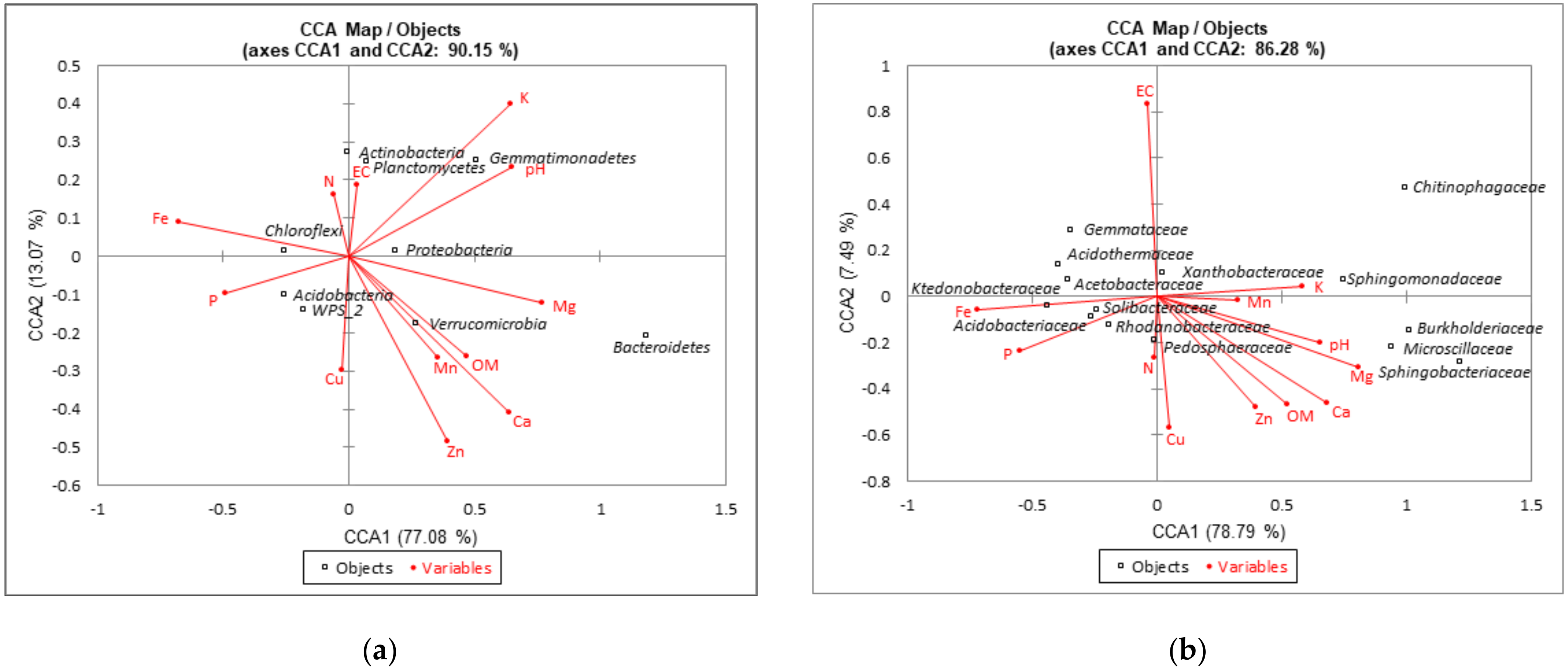
3.4. Relationships between Dominant Bacterial Taxa and Soil Properties or Metabolic Potential of Bacterial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Pii, Y.; Mimmo, T.; Tomasi, N.; Terzano, R.; Cesco, S.; Crecchio, C. Microbial interactions in the rhizosphere: Beneficial influences of plant growth-promoting rhizobacteria on nutrient acquisition process—A review. Biol. Fertil. Soils 2015, 51, 403–415. [Google Scholar] [CrossRef]
- Heo, Y.-M.; Lee, H.; Kwon, S.-L.; Yoo, Y.; Kim, D.; Han, S.-I.; Lee, A.-H.; Kim, C.; Kim, G.-H.; Kim, J.-J. Influence of tree vegetation on soil microbial communities in temperate forests and their potential as a proactive indicator of vegetation shift due to climate change. Sustainability 2020, 12, 10591. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef]
- Qiao, C.; Penton, C.R.; Xiong, W.; Liu, C.; Wang, R.; Liu, Z.; Xu, X.; Li, R.; Shen, Q. Reshaping the rhizosphere microbiome by bio-organic amendment to enhance crop yield in a maize-cabbage rotation system. Appl. Soil Ecol. 2019, 142, 136–146. [Google Scholar] [CrossRef]
- Liu, Z.; Guo, Q.; Feng, Z.; Liu, Z.; Li, H.; Sun, Y.; Liu, C.; Lai, H. Long-term organic fertilization improves the productivity of kiwifruit (Actinidia chinensis Planch.) through increasing rhizosphere microbial diversity and network complexity. Appl. Soil Ecol. 2020, 147, 103426. [Google Scholar] [CrossRef]
- Tejada, M.; Gonzalez, J.L.; García-Martínez, A.M.; Parrado, J. Application of a green manure and green manure composted with beet vinasse on soil restoration: Effects on soil properties. Bioresour. Technol. 2008, 99, 4949–4957. [Google Scholar] [CrossRef]
- Jiang, Y.-H.; Arafat, Y.; Letuma, P.; Ali, L.; Tayyab, M.; Waqas, M.; Li, Y.-C.; Lin, W.-W.; Lin, S.; Lin, W.-X. Restoration of long-term monoculture degraded tea orchard by green and goat manures application system. Sustainability 2019, 11, 1011. [Google Scholar] [CrossRef] [Green Version]
- Longa, C.M.O.; Nicola, L.; Antonielli, L.; Mescalchin, E.; Zanzotti, R.; Turco, E.; Pertot, I. Soil microbiota respond to green manure in organic vineyards. J. Appl. Microbiol. 2017, 123, 1547–1560. [Google Scholar] [CrossRef]
- Khan, M.I.; Gwon, H.S.; Alam, M.A.; Song, H.J.; Das, S.; Kim, P.J. Short term effects of different green manure amendments on the composition of main microbial groups and microbial activity of a submerged rice cropping system. Appl. Soil Ecol. 2020, 147, 103400. [Google Scholar] [CrossRef]
- Li, Y.C.; Li, Z.; Li, Z.W.; Jiang, Y.H.; Weng, B.Q.; Lin, W.X. Variations of rhizosphere bacterial communities in tea (Camellia sinensis L.) continuous cropping soil by high-throughput pyrosequencing approach. J. Appl. Microbiol. 2016, 121, 787–799. [Google Scholar] [CrossRef]
- Arafat, Y.; Wei, X.-Y.; Jiang, Y.-H.; Chen, T.; Saqib, H.S.A.; Lin, S.; Lin, W.-X. Spatial distribution patterns of root-associated bacterial communities mediated by root exudates in different aged ratooning tea monoculture systems. Int. J. Mol. Sci. 2017, 18, 1727. [Google Scholar] [CrossRef] [Green Version]
- Lynn, T.M.; Liu, Q.; Hu, Y.; Yuan, H.; Wu, X.; Khai, A.A.; Wu, J.; Ge, T. Influence of land use on bacterial and archaeal diversity and community structures in three natural ecosystems and one agricultural soil. Arch. Microbiol. 2017, 199, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, T.; Zheng, Z.; Chen, H.Y.H. Soil aggregate-associated bacterial metabolic activity and community structure in different aged tea plantations. Sci. Total Environ. 2019, 654, 1023–1032. [Google Scholar] [CrossRef]
- Chen, W.C.; Ko, C.H.; Su, Y.S.; Lai, W.A.; Shen, F.T. Metabolic potential and community structure of bacteria in an organic tea plantation. Appl. Soil Ecol. 2021, 157, 103762. [Google Scholar] [CrossRef]
- Tan, L.; Gu, S.; Li, S.; Ren, Z.; Deng, Y.; Liu, Z.; Gong, Z.; Xiao, W.; Hu, Q. Responses of microbial communities and interaction networks to different management practices in tea plantation soils. Sustainability 2019, 11, 4428. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.; Ni, K.; Wu, Z.; Zhang, J.; Yi, X.; Yang, X.; Ling, N.; You, Z.; Guo, S.; Ruan, J. Effect of organic substitution rates on soil quality and fungal community composition in a tea plantation with long-term fertilization. Biol. Fertil. Soils 2020, 56, 633–646. [Google Scholar] [CrossRef]
- Dennis, P.G.; Miller, A.J.; Hirsch, P.R. Are root exudates more important than other sources of rhizodeposits in structuring rhizosphere bacterial communities? FEMS Microbiol. Ecol. 2010, 72, 313–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Council of Agriculture. Available online: https://agrstat.coa.gov.tw/sdweb/public/inquiry/InquireAdvance.aspx (accessed on 20 September 2021).
- Soil Survey Staff. Keys to Soil Taxonomy, 12th ed.; USDA-Natural Resources Conservation Service: Washington, DC, USA, 2014.
- Wu, C.H.; Lien, T.J. The development of a new soybean variety, Tainan No. 7, for covering and green manure. Tainan Dist. Agric. Res. Ext. Stn. Bull. 2003, 42, 22. (In Chinese) [Google Scholar]
- Shen, F.T.; Lin, S.H. Priming effects of cover cropping on bacterial community in a tea plantation. Sustainability 2021, 13, 4345. [Google Scholar] [CrossRef]
- Nelson, D.W.; Sommers, L.E. Total carbon, organic carbon and organic matter. In Methods of Soil Analysis, Part 2: Chemical and Microbiological Properties, 2nd ed.; Page, A.L., Miller, R.H., Keeney, D.R., Eds.; ASA-SSSA: Madison, WI, USA, 1982; pp. 539–580. [Google Scholar]
- Kjeldahl, J. A new method for the determination of nitrogen in organic matter. Z. Anal. Chem. 1883, 22, 366–382. [Google Scholar] [CrossRef] [Green Version]
- Bremner, J.M. Nitrogen total. In Methods of Soil Analysis, Part 3: Chemical Methods; Sparks, D.L., Page, A.L., Helmke, P.A., Loeppert, R.H., Soltanpour, P.N., Tabatabai, M.A., Johnston, C.T., Sumner, M.E., Eds.; Soil Science Society of America Inc.: Madison, WI, USA, 1996; pp. 1085–1121. [Google Scholar]
- Mehlich, A. Mehlich 3 soil test extractant: A modification of Mehlich 2. Comm. Soil Sci. Plant. Anal. 1984, 15, 1409–1416. [Google Scholar] [CrossRef]
- Classen, A.T.; Boyle, S.I.; Haskins, K.E.; Overby, S.T.; Hart, S.C. Community-level physiological profiles of bacteria and fungi: Plate type and incubation temperature influences on contrasting soils. FEMS Microbiol. Ecol. 2003, 44, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Chou, Y.M.; Shen, F.T.; Chiang, S.C.; Chang, C.M. Functional diversity and dominant populations of bacteria in banana plantation soils as influenced by long-term organic and conventional farming. Appl. Soil Ecol. 2017, 110, 21–33. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Zak, J.C.; Willig, M.R.; Moorhead, D.L.; Wildmand, H.G. Functional diversity of microbial communities: A quantitative approach. Soil Biol. Biochem. 1994, 26, 1101–1108. [Google Scholar] [CrossRef]
- Manly, B.F.J. Multivariate Statistical Methods: A Primer, 2nd ed.; Chapman and Hall: London, UK, 1994. [Google Scholar]
- Nair, A.; Ngouajio, M. Soil microbial biomass, functional microbial diversity, and nematode community structure as affected by cover crops and compost in an organic vegetable production system. Appl. Soil Ecol. 2012, 58, 45–55. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Garbeva, P.; van Elsas, J.D.; van Veen, J.A. Rhizosphere microbial community and its response to plant species and soil history. Plant. Soil 2008, 302, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Berg, G.; Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Ge, Y.; Song, J.; Rensing, C. Assembly of root-associated microbial community of typical rice cultivars in different soil types. Biol. Fertil. Soils 2020, 56, 249–260. [Google Scholar] [CrossRef]
- Timotiwu, P.B.; Sakurai, N. Identification of mono-, oligo-, and polysaccharides secreted from soybean roots. J. Plant. Res. 2002, 115, 77–85. [Google Scholar] [CrossRef]
- Tawaraya, K.; Horie, R.; Shinano, T.; Wagatsuma, T.; Saito, K.; Oikawa, A. Metabolite profiling of soybean root exudates under phosphorus deficiency. Soil Sci. Plant. Nutr. 2014, 60, 679–694. [Google Scholar] [CrossRef]
- Lauber, C.L.; Strickland, M.S.; Bradford, M.A.; Fierer, N. The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol. Biochem. 2008, 40, 2407–2415. [Google Scholar] [CrossRef]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousk, J.; Bååth, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, N.; Scheu, S.; Jousset, A. Bacterial diversity stabilizes community productivity. PLoS ONE 2012, 7, e34517. [Google Scholar] [CrossRef] [Green Version]
- Sharaf, H.; Rodrigues, R.R.; Moon, J.; Zhang, B.; Mills, K.; Williams, M.A. Unprecedented bacterial community richness in soybean nodules vary with cultivar and water status. Microbiome 2019, 7, 63. [Google Scholar] [CrossRef]
- Bakari, R.; Mungai, N.; Thuita, M.; Masso, C. Impact of soil acidity and liming on soybean (Glycine max) nodulation and nitrogen fixation in Kenyan soils. Acta Agric. Scand. Sect. B Soil Plant. Sci. 2020, 70, 667–678. [Google Scholar]
- Smit, E.; Leeflang, P.; Gommans, S.; van den Broek, J.; van Mil, S.; Wernars, K. Diversity and seasonal fluctuations of the dominant members of the bacterial soil community in a wheat field as determined by cultivation and molecular methods. Appl. Environ. Microbiol. 2001, 67, 2284–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.H.; Harris, R.F. r-selection and K-selection and microbial ecology. Adv. Microb. Ecol. 1986, 9, 99–147. [Google Scholar]
- Grime, J.P.; Pierce, S. The Evolutionary Strategies That Shape Ecosystems; John Wiley & Sons: Oxford, UK, 2012. [Google Scholar]
- Ho, A.; Di Lonardo, D.P.; Bodelier, P.L.E. Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiol. Ecol. 2017, 93, fix006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramin, K.I.; Allison, S.D. Bacterial tradeoffs in growth rate and extracellular enzymes. Front. Microbiol. 2019, 10, 2956. [Google Scholar] [CrossRef]
- Davis, K.E.R.; Sangwan, P.; Janssen, P.H. Acidobacteria, Rubrobacteridae and Chloroflexi are abundant among very slow-growing and mini-colony-forming soil bacteria. Environ. Microbiol. 2011, 13, 798–805. [Google Scholar] [CrossRef]
- Kim, H.M.; Jung, J.Y.; Yergeau, E.; Hwang, C.Y.; Hinzman, L.; Nam, S.; Hong, S.G.; Kim, O.S.; Chun, J.; Lee, Y.K. Bacterial community structure and soil properties of a subarctic tundra soil in Council, Alaska. FEMS Microbiol. Ecol. 2014, 89, 465–475. [Google Scholar] [CrossRef]
- Sun, L.; Xun, W.; Huang, T.; Zhang, G.; Gao, J.; Ran, W.; Li, D.; Shen, Q.; Zhang, R. Alteration of the soil bacterial community during parent material maturation driven by different fertilization treatments. Soil Biol. Biochem. 2016, 96, 207–215. [Google Scholar] [CrossRef]
- Ivanova, A.A.; Zhelezova, A.D.; Chernov, T.I.; Dedysh, S.N. Linking ecology and systematics of Acidobacteria: Distinct habitat preferences of the Acidobacteriia and Blastocatellia in tundra soils. PLoS ONE 2020, 15, e0230157. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Long, X.E.; Yao, H.; Wang, J.; Huang, Y.; Singh, B.K.; Zhu, Y.G. Community structure and soil pH determine chemoautotrophic carbon dioxide fixation in drained paddy soils. Environ. Sci. Technol. 2015, 49, 7152–7160. [Google Scholar] [CrossRef]
- Brewer, T.E.; Aronson, E.L.; Arogyaswamy, K.; Billings, S.A.; Botthoff, J.K.; Campbell, A.N.; Dove, N.C.; Fairbanks, D.; Gallery, R.E.; Hart, S.C.; et al. Ecological and genomic attributes of novel bacterial taxa that thrive in subsurface soil horizons. mBio 2019, 10, e01318-19. [Google Scholar] [CrossRef] [Green Version]
- Ji, M.; Greening, C.; Vanwonterghem, I.; Carere, C.R.; Bay, S.K.; Steen, J.A.; Montgomery, K.; Lines, T.; Beardall, J.; van Dorst, J.; et al. Atmospheric trace gases support primary production in Antarctic desert surface soil. Nature 2017, 552, 400–403. [Google Scholar] [CrossRef]
- Holland-Moritz, H.; Stuart, J.; Lewis, L.R.; Miller, S.; Mack, M.C.; McDaniel, S.F.; Fierer, N. Novel bacterial lineages associated with boreal moss species. Environ. Microbiol. 2018, 20, 2625–2638. [Google Scholar] [CrossRef]
- Wakelin, S.A.; Condron, L.M.; Gerard, E.; Dignam, B.E.A.; Black, A.; O’Callaghan, M. Long-term P fertilization of pasture soil did not increase soil organic matter stocks but increased microbial biomass and activity. Biol. Fertil. Soils 2017, 53, 511–521. [Google Scholar] [CrossRef]
- Aanderud, Z.T.; Lennon, J.T. Validation of heavy-water stable isotope probing for the characterization of rapidly responding soil bacteria. Appl. Environ. Microbiol. 2011, 77, 4589–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schostag, M.; Priemé, A.; Jacquiod, S.; Russel, J.; Ekelund, F.; Jacobsen, C.S. Bacterial and protozoan dynamics upon thawing and freezing of an active layer permafrost soil. ISME J. 2019, 13, 1345–1359. [Google Scholar] [CrossRef] [PubMed]
- Bailey, V.L.; Fansler, S.J.; Stegen, J.C.; McCue, L.A. Linking microbial community structure to β-glucosidic function in soil aggregates. ISME J. 2013, 7, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Li, B.; Feng, G.; Zhang, C.; He, J.; Li, H.; Zhu, J. Responses of bacterial communities and organic matter degradation in surface sediment to Macrobrachium nipponense bioturbation. Sci. Total Environ. 2021, 759, 143534. [Google Scholar] [CrossRef]
- Chung, E.J.; Park, T.S.; Jeon, C.O.; Chung, Y.R. Chitinophaga oryziterrae sp. nov., isolated from the rhizosphere soil of rice (Oryza sativa L.). Int. J. Syst. Evol. Microbiol. 2012, 62, 3030–3035. [Google Scholar] [CrossRef] [Green Version]
- Madhaiyan, M.; Poonguzhali, S.; Senthilkumar, M.; Pragatheswari, D.; Lee, J.S.; Lee, K.C. Arachidicoccus rhizosphaerae gen. nov., sp. nov., a plant-growth-promoting bacterium in the family Chitinophagaceae isolated from rhizosphere soil. Int. J. Syst. Evol. Microbiol. 2015, 65, 578–586. [Google Scholar] [CrossRef]
- Hahnke, R.L.; Meier-Kolthoff, J.P.; García-López, M.; Mukherjee, S.; Huntemann, M.; Ivanova, N.N.; Woyke, T.; Kyrpides, N.C.; Klenk, H.P.; Göker, M. Genome-based taxonomic classification of Bacteroidetes. Front. Microbiol. 2016, 7, 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Liu, Y.; Xu, B.; Wang, N.; Zhao, H.; Liu, X.; Liu, F. Comparative genomic analysis reveals the environmental impacts on two Arcticibacter strains including sixteen Sphingobacteriaceae species. Sci. Rep. 2017, 7, 2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankratov, T.A.; Tindall, B.J.; Liesack, W.; Dedysh, S.N. Mucilaginibacter paludism gen. nov., sp. nov. and Mucilaginibacter gracilis sp. nov., pectin-, xylan- and laminarin-degrading members of the family Sphingobacteriaceae from acidic Sphagnum peat bog. Int. J. Syst. Evol. Microbiol. 2007, 57, 2349–2354. [Google Scholar] [CrossRef] [PubMed]
- López-Mondéjar, R.; Zühlke, D.; Becher, D.; Riedel, K.; Baldrian, P. Cellulose and hemicellulose decomposition by forest soil bacteria proceeds by the action of structurally variable enzymatic systems. Sci. Rep. 2016, 6, 25279. [Google Scholar] [CrossRef] [PubMed]
- Compant, S.; Nowak, J.; Coenye, T.; Clément, C.; Ait Barka, E. Diversity and occurrence of Burkholderia spp. in the natural environment. FEMS Microbiol. Rev. 2008, 32, 607–626. [Google Scholar] [CrossRef] [Green Version]
- Afzal, I.; Shinwari, Z.K.; Sikandar, S.; Shahzad, S. Plant beneficial endophytic bacteria: Mechanisms, diversity, host range and genetic determinants. Microbiol. Res. 2019, 221, 36–49. [Google Scholar] [CrossRef]
- Mannaa, M.; Park, I.; Seo, Y.S. Genomic features and insights into the taxonomy, virulence, and benevolence of plant-associated Burkholderia species. Int. J. Mol. Sci. 2019, 20, 121. [Google Scholar] [CrossRef] [Green Version]
- Stolz, A. Degradative plasmids from sphingomonads. FEMS Microbiol. Lett. 2014, 350, 9–19. [Google Scholar] [CrossRef]
- Gatheru Waigi, M.; Sun, K.; Gao, Y. Sphingomonads in microbe-assisted phytoremediation: Tackling soil pollution. Trends Biotechnol. 2017, 35, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Numan, M.; Khan, A.L.; Al-Harrasi, A. Sphingomonas: From diversity and genomics to functional role in environmental remediation and plant growth. Crit. Rev. Biotechnol. 2020, 40, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Shen, F.T. Bioprospecting of facultatively oligotrophic bacteria from non-rhizospheric soils. Appl. Soil Ecol. 2016, 108, 315–324. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 Months | 6 Months | 9 Months | ||||
|---|---|---|---|---|---|---|
| Bare Ground Control | Green Manure Treatment | Bare Ground Control | Green Manure Treatment | Bare Ground Control | Green Manure Treatment | |
| pH | 3.72 ± 0.13 a † | 3.95 ± 0.07 a | 3.99 ± 0.02 b | 4.62 ± 0.08 a | 3.86 ± 0.10 b | 4.50 ± 0.06 a |
| EC (μS cm−1) | 156.5 ± 26.1 a | 141.0 ± 12.2 a | 122.6 ± 8.2 a | 108.6 ± 5.5 a | 148.2 ± 33.5 a | 86.0 ± 9.6 b |
| Organic matter (%) | 0.87 ± 0.41 a | 1.40 ± 0.08 a | 1.92 ± 0.27 a | 2.11 ± 0.21 a | 1.88 ± 0.36 a | 2.43 ± 0.39 a |
| Total N (%) | 0.16 ± 0.03 a | 0.15 ± 0.02 a | 0.22 ± 0.02 a | 0.21 ± 0.02 a | 0.15 ± 0.01 b | 0.19 ± 0.00 a |
| P (mg kg−1) | 294.2 ± 40.9 a | 256.7 ± 23.7 a | 332.0 ± 22.7 a | 309.4 ± 31.1 a | 304.9 ± 19.9 a | 268.3 ± 47.8 a |
| K (mg kg−1) | 92.8 ± 1.9 b | 154.2 ± 30.4 a | 106.5 ± 6.2 b | 207.5 ± 44.1 a | 104.4 ± 17.9 a | 173.0 ± 49.5 a |
| Ca (mg kg−1) | 76.4 ± 27.9 a | 79.3 ± 32.7 a | 147.0 ± 19.6 b | 212.7 ± 28.8 a | 181.8 ± 65.5 a | 336.6 ± 77.9 a |
| Mg (mg kg−1) | 39.3 ± 8.2 a | 56.5 ± 14.2 a | 53.5 ± 6.2 b | 108.5 ± 22.1 a | 65.4 ± 31.2 b | 143.9 ± 13.7 a |
| Fe (mg kg−1) | 416.0 ± 3.2 a | 389.4 ± 6.6 b | 494.5 ± 16.8 a | 448.6 ± 24.1 a | 432.2 ± 14.2 a | 380.1 ± 18.9 b |
| Mn (mg kg−1) | 8.0 ± 2.0 a | 7.1 ± 1.0 a | 6.8 ± 1.4 a | 7.0 ± 2.2 a | 6.8 ± 3.3 a | 9.4 ± 4.2 a |
| Cu (mg kg−1) | 1.0 ± 0.1 b | 1.3 ± 0.1 a | 3.4 ± 0.1 a | 3.4 ± 0.2 a | 3.4 ± 0.3 a | 3.0 ± 0.1 a |
| Zn (mg kg−1) | 1.5 ± 0.2 a | 1.2 ± 0.1 a | 1.7 ± 0.0 a | 2.1 ± 0.7 a | 2.1 ± 0.1 a | 2.6 ± 1.1 a |
| Bare Ground Control | Green Manure Treatment | |||||||
|---|---|---|---|---|---|---|---|---|
| S† | E | H | AWCD | S | E | H | AWCD | |
| 1 month | 7 ± 3 b ‡ | 1.275 ± 0.214 a | 2.314 ± 0.231 b | 0.231 ± 0.083 b | 25 ± 3 a | 1.000 ± 0.015 b | 3.198 ± 0.096 a | 0.851 ± 0.103 a |
| 2 months | 13 ± 6 b | 1.090 ± 0.064 a | 2.621 ± 0.439 b | 0.389 ± 0.187 b | 26 ± 7 a | 0.999 ± 0.015 b | 3.220 ± 0.299 a | 0.796 ± 0.204 a |
| 3 months | 18 ± 6 b | 1.023 ± 0.050 a | 2.901 ± 0.273 b | 0.574 ± 0.189 b | 28 ± 2 a | 0.997 ± 0.007 a | 3.304 ± 0.049 a | 0.900 ± 0.113 a |
| 6 months | 17 ± 2 b | 1.014 ± 0.015 a | 2.884 ± 0.111 b | 0.580 ± 0.083 b | 30 ± 1 a | 0.996 ± 0.005 b | 3.378 ± 0.022 a | 1.077 ± 0.037 a |
| 9 months | 18 ± 1 b | 1.374 ± 0.048 a | 3.967 ± 0.066 b | 0.545 ± 0.077 b | 30 ± 1 a | 1.316 ± 0.007 a | 4.477 ± 0.024 a | 1.068 ± 0.074 a |
| Bare Ground Control | Green Manure Treatment | |||||
|---|---|---|---|---|---|---|
| Chao1 † | Shannon | InvSimpson | Chao1 | Shannon | InvSimpson | |
| 1 month | 165 ± 12 b | 5.13 ± 0.21 b | 15.82 ± 4.02 a | 201 ± 12 a | 5.52 ± 0.07 a | 21.10 ± 1.78 a |
| 2 months | 205 ± 12 b | 5.35 ± 0.09 b | 19.55 ± 2.59 a | 361 ± 41 a | 6.27 ± 0.53 a | 36.06 ± 20.96 a |
| 3 months | 226 ± 37 b | 5.42 ± 0.24 b | 19.36 ± 2.84 b | 382 ± 32 a | 6.56 ± 0.31 a | 45.45 ± 13.05 a |
| 6 months | 224 ± 23 b | 5.23 ± 0.16 b | 16.53 ± 2.75 b | 483 ± 51 a | 6.65 ± 0.27 a | 45.03 ± 12.61 a |
| 9 months | 277 ± 7 b | 5.38 ± 0.11 b | 18.45 ± 1.73 b | 574 ± 33 a | 7.16 ± 0.13 a | 68.96 ± 6.43 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, F.-T.; Lin, S.-H. Shifts in Bacterial Community Associated with Green Manure Soybean Intercropping and Edaphic Properties in a Tea Plantation. Sustainability 2021, 13, 11478. https://doi.org/10.3390/su132011478
Shen F-T, Lin S-H. Shifts in Bacterial Community Associated with Green Manure Soybean Intercropping and Edaphic Properties in a Tea Plantation. Sustainability. 2021; 13(20):11478. https://doi.org/10.3390/su132011478
Chicago/Turabian StyleShen, Fo-Ting, and Shih-Han Lin. 2021. "Shifts in Bacterial Community Associated with Green Manure Soybean Intercropping and Edaphic Properties in a Tea Plantation" Sustainability 13, no. 20: 11478. https://doi.org/10.3390/su132011478
APA StyleShen, F.-T., & Lin, S.-H. (2021). Shifts in Bacterial Community Associated with Green Manure Soybean Intercropping and Edaphic Properties in a Tea Plantation. Sustainability, 13(20), 11478. https://doi.org/10.3390/su132011478