Genetic Diversity of South African Indigenous Goat Population from Four Provinces Using Genome-Wide SNP Data

 , , ,
, , ,  ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
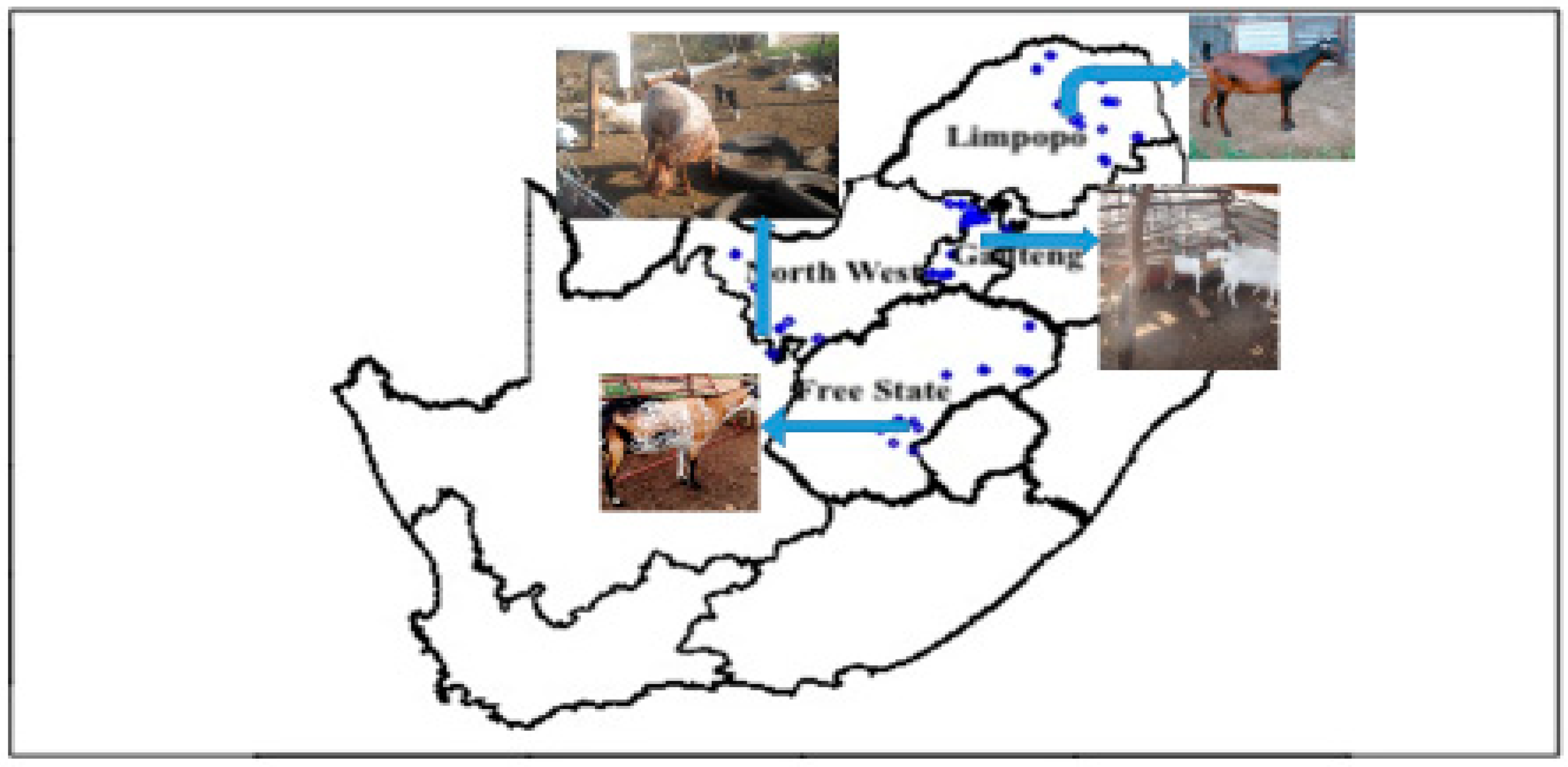
2.1. Population Description and Sampling
2.2. Genotyping and Quality Control
2.3. Genetic Diversity
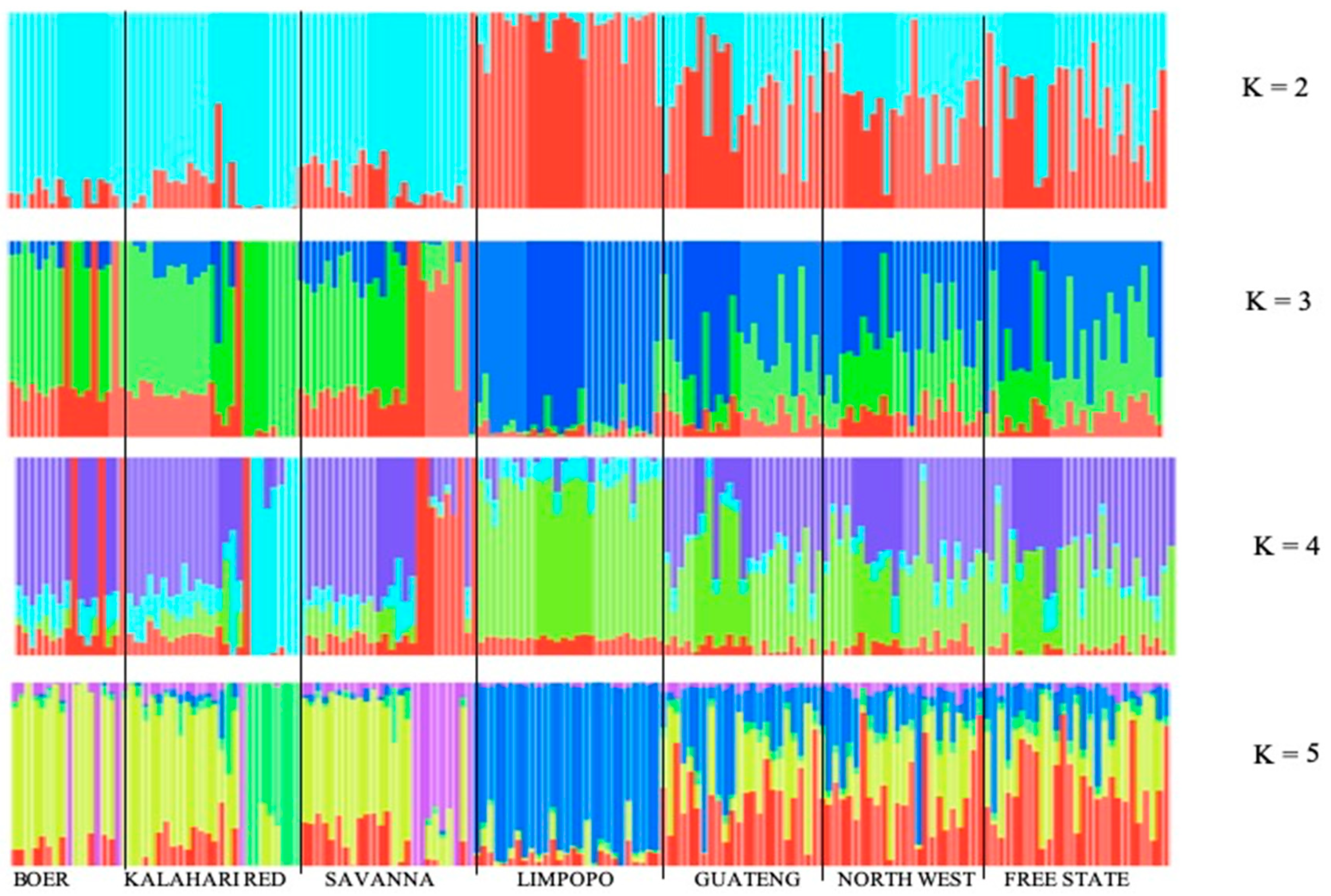
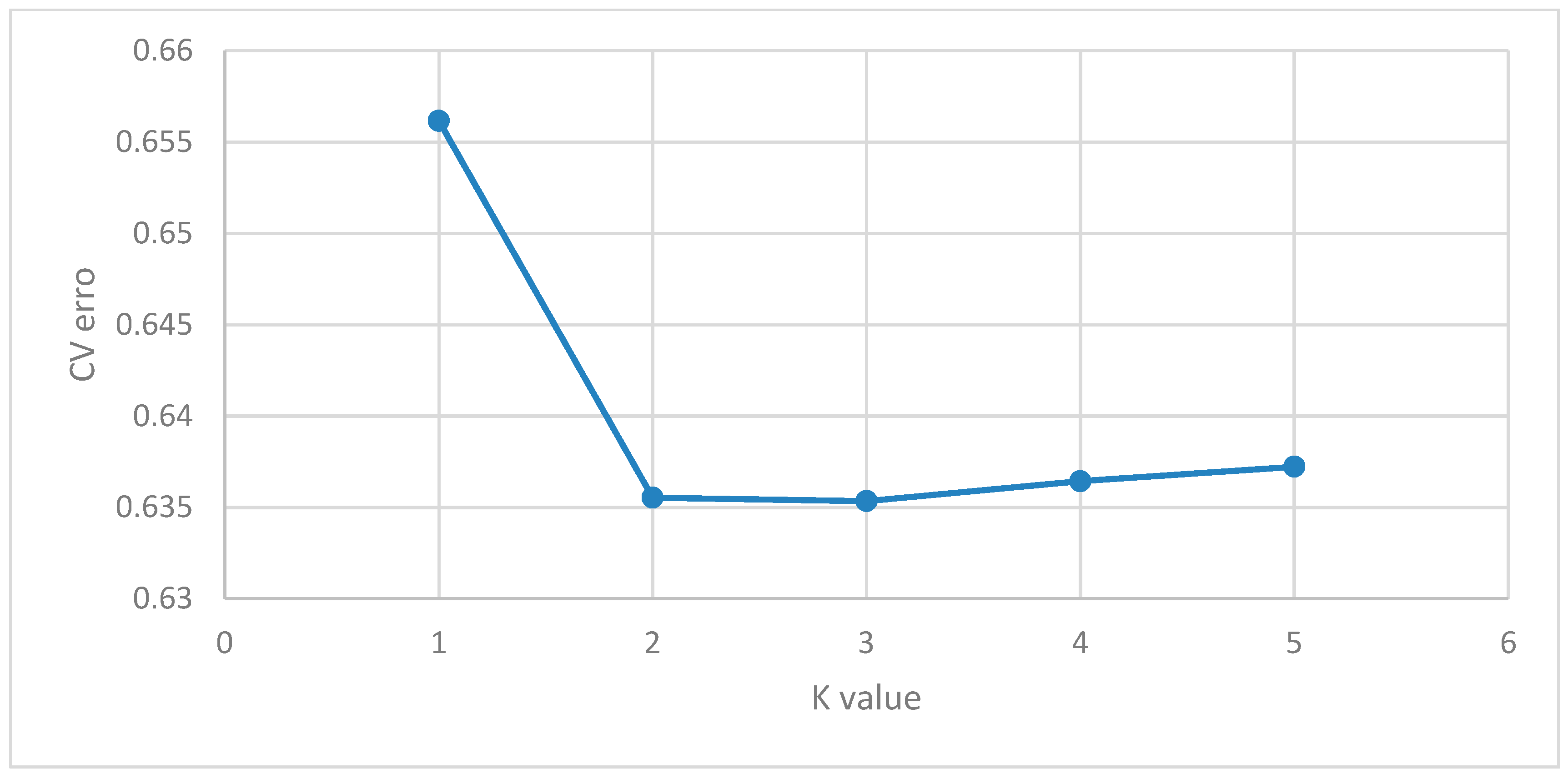
2.4. Population Structure Analysis
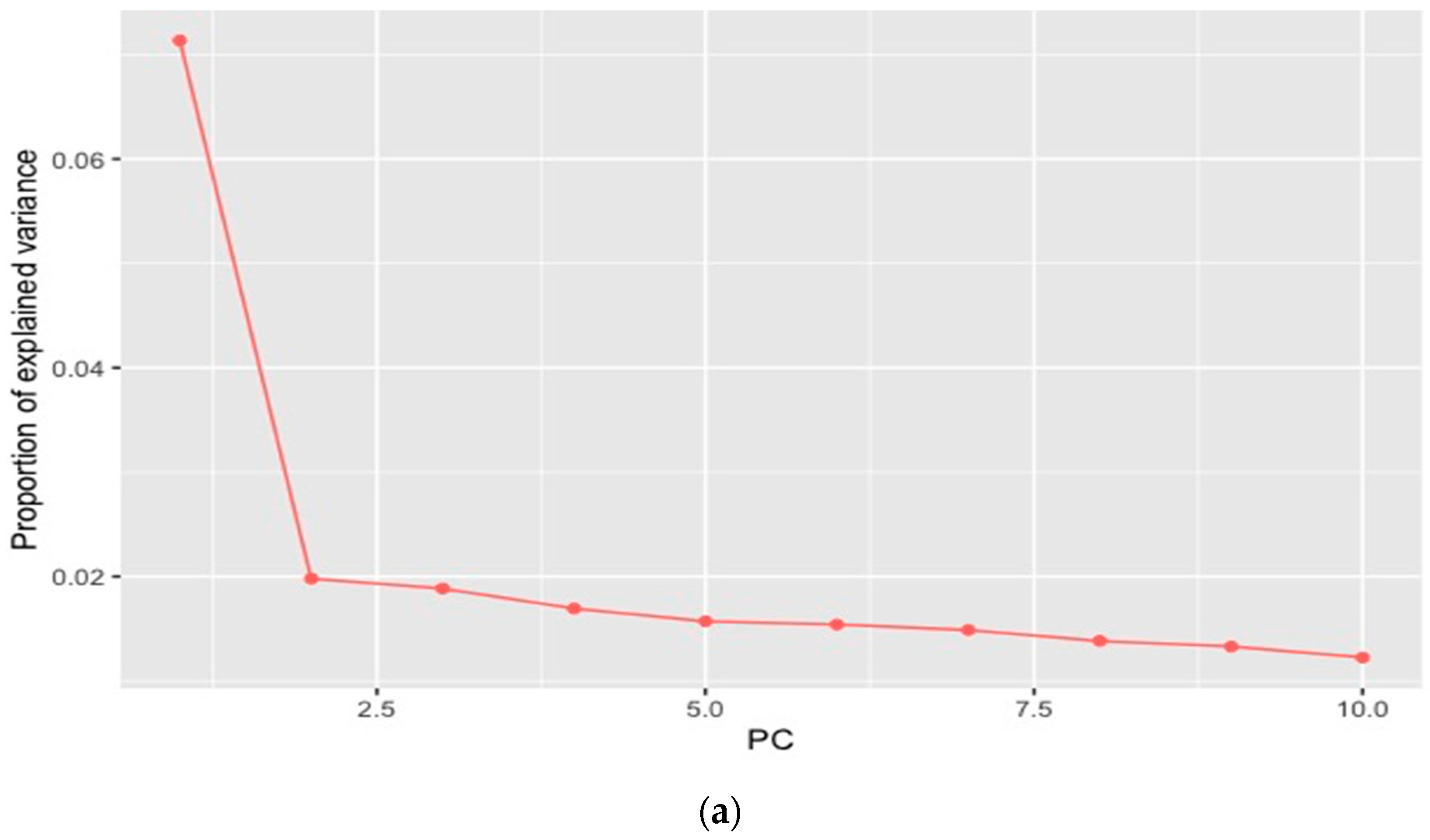
2.5. Identification of Signatures of Selection
3. Results
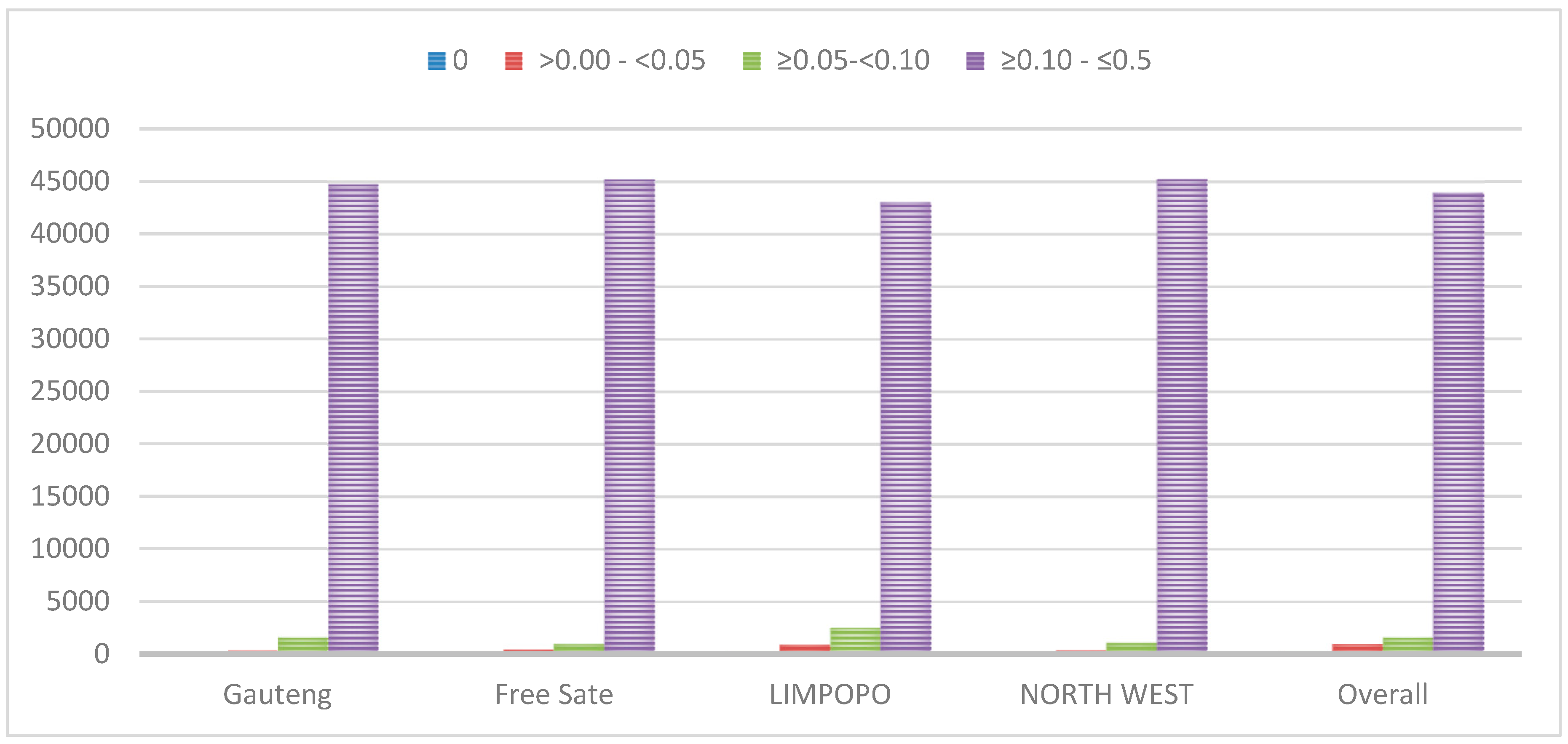
3.1. Quality Control and Genetic Diversity
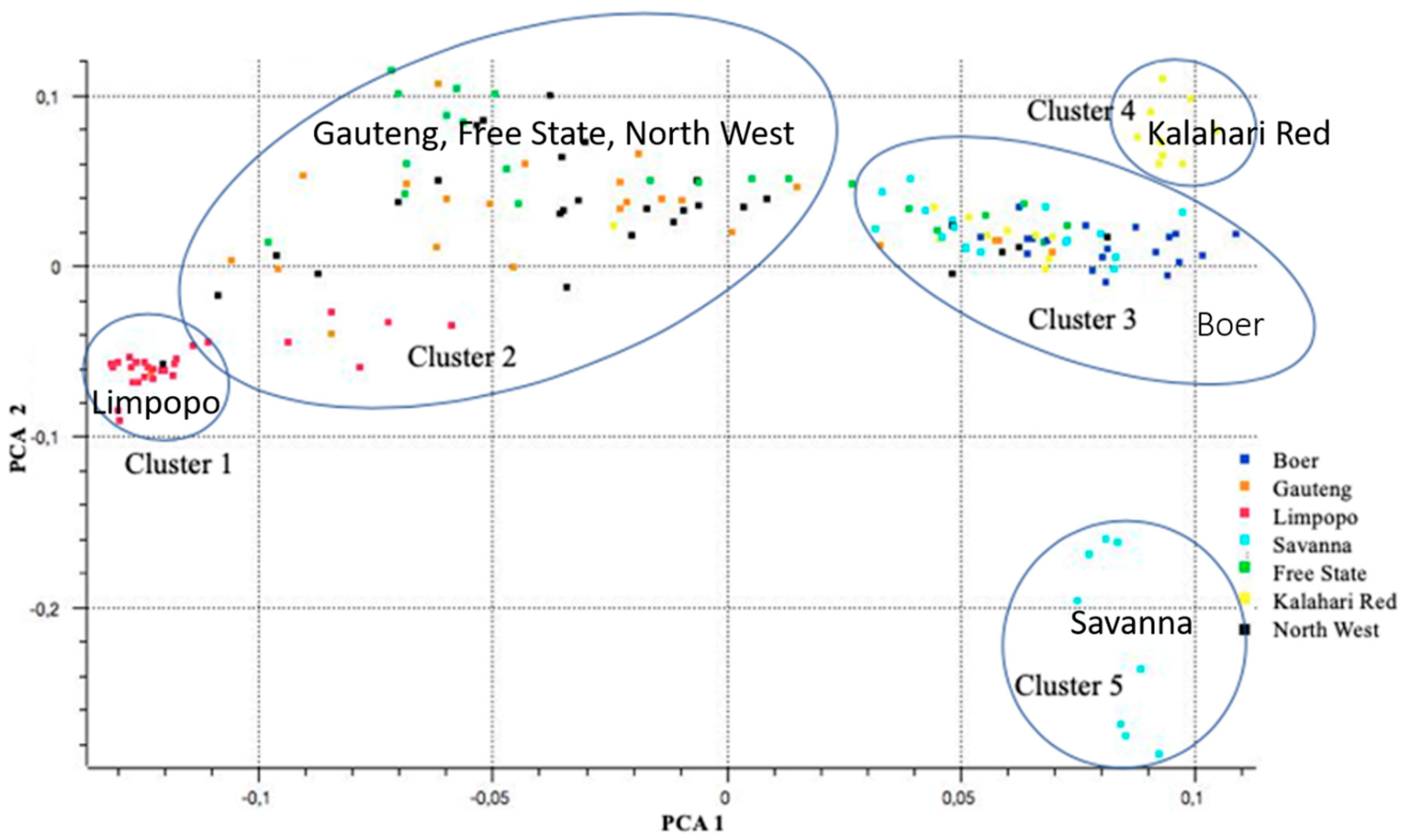
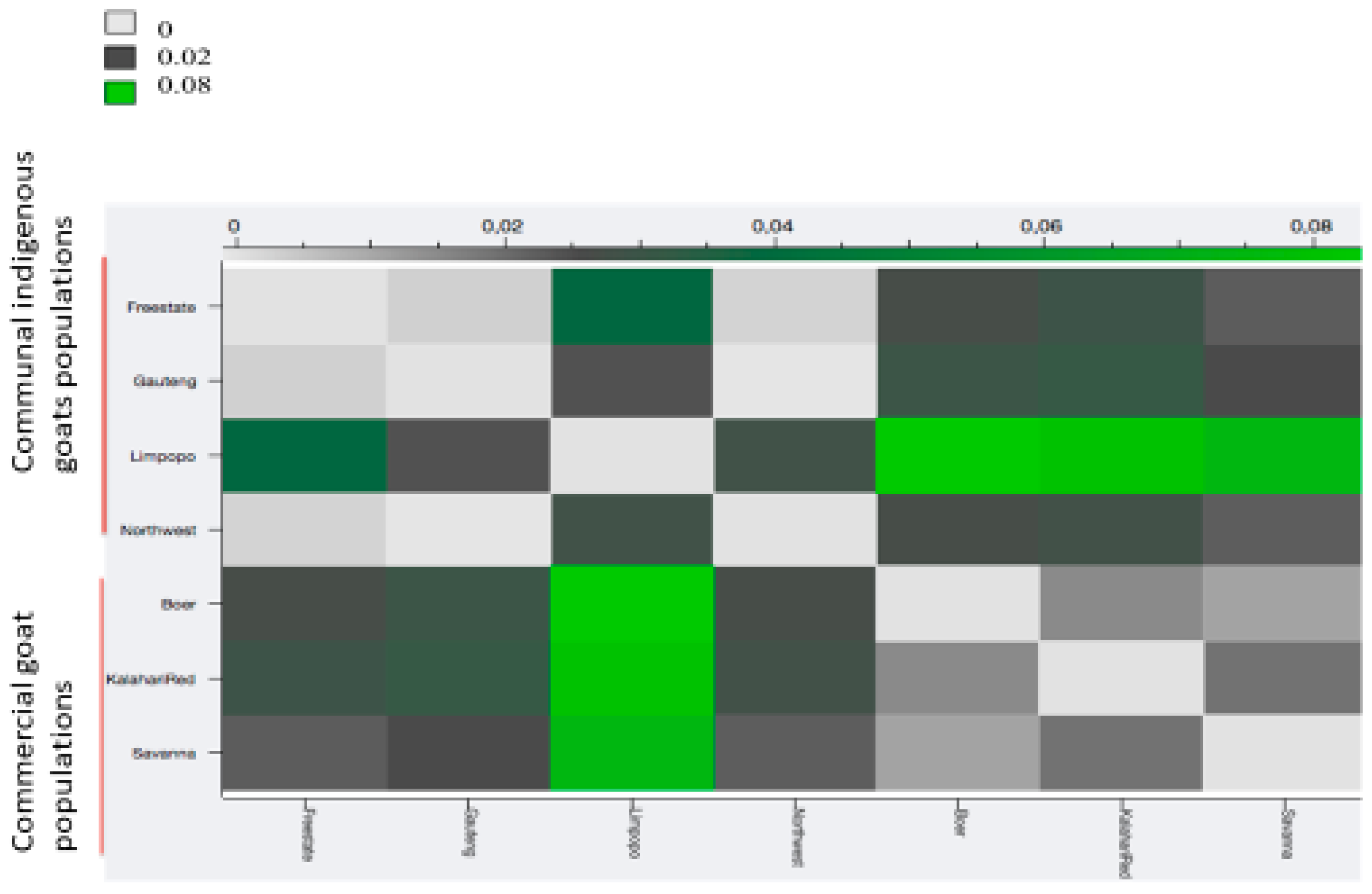
3.2. Source of Variation and Population Structure
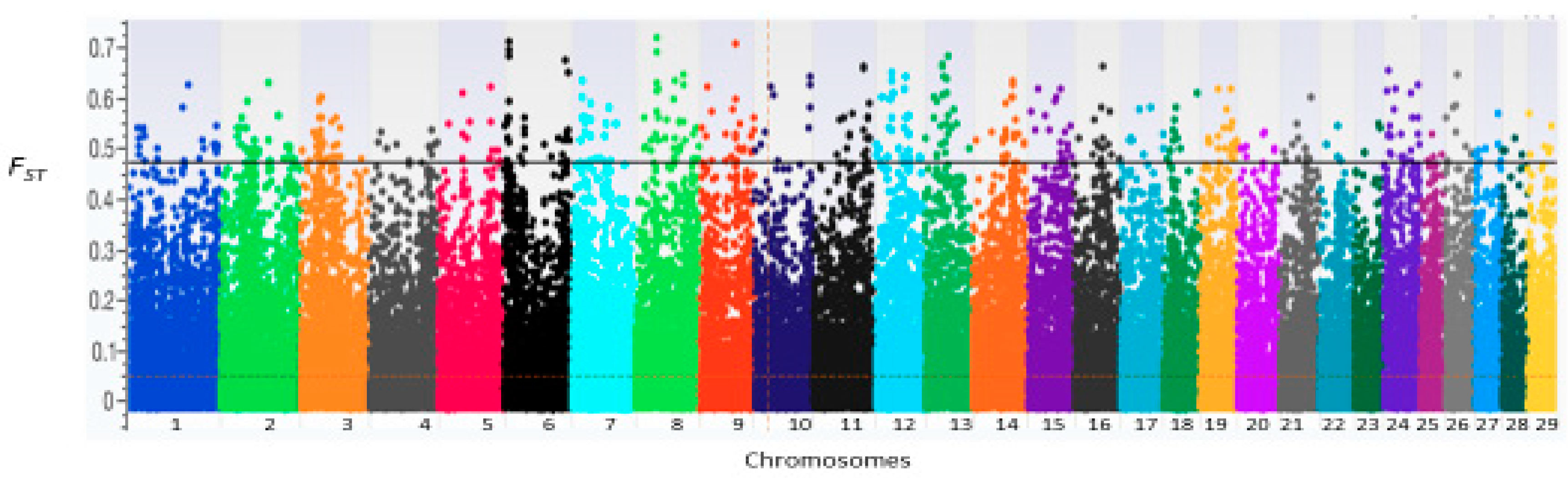
3.3. Candidate Genes under Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Compliance with Ethical Standards
References
- Department of Agriculture, Forestry and Fisheries (DAFF). Newsletter: Livestock Statistics, Estimated Livestock Numbers in RSA. 2014. Available online: http://www.daff.gov.za/daffweb3/Resource-Centre (accessed on 13 June 2018).
- Matshaba, B.; Schwalbach, L.M.; Mphaphathi, M.; Nemes, C.; Ngwane, C.; Greyling, J.P.; Váradi, É.; Nedambale, T.L. Characterization of semen from the unimproved South African indigenous goats. In Proceedings of the Second RUFORUM Biennial Meeting, Entebbe, Uganda, 20–24 September 2010. [Google Scholar]
- Chokoe, T.C.; Matelele, T.C.; Maqhashu, A.; Ramukhithi, F.V.; Mphahlele, T.D.; Mpofu, T.J.; Nephawe, K.A.; Mtileni, B. Phenotypic Diversity of South African Indigenous Goat Population in Selected Rural Areas. Am. J. Anim. Vet. Sci. 2020, 15, 59–66. [Google Scholar] [CrossRef]
- Gwaze, F.R.; Chimonyo, M.; Dzama, K. Communal goat production in Southern Africa: A review. Trop. Anim. Health Prod. 2009, 41, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Mahanjana, A.M.; Cronjé, P.B. Factors affecting goat production in a communal farming system in the Eastern Cape region of South Africa. S. Afr. J. Anim. Sci. 2000, 30, 149–154. [Google Scholar] [CrossRef]
- Monau, P.I.; Raphaka, K.; Zvinorova-Chimboza, P.; Gondwe, T. Sustainable Utilization of Indigenous Goats in Southern Africa. Diversity 2020, 12, 20. [Google Scholar] [CrossRef]
- Mdladla, K.; Dzomba, E.F.; Muchadeyi, F.C. Landscape genomics and pathway analysis to understand genetic adaptation of South African indigenous goat populations. Heredity 2018, 120, 369–378. [Google Scholar] [CrossRef]
- Casey, N.; Van Niekerk, W. The Boer goat. I. Origin, adaptability, performance testing, reproduction and milk production. Small Rumin. Res. 1988, 1, 291–302. [Google Scholar] [CrossRef]
- Campbell, Q.P. The origin and description of southern Africa’s indigenous goats. S. Afr. J. Anim. Sci. 2003, 4, 18–22. [Google Scholar]
- Onzima, R.B.; Upadhyay, M.R.; Doekes, H.P.; Brito, L.F.; Bosse, M.; Kanis, E.; Groenen, M.A.M.; Crooijmans, R.P.M.A. Genome-Wide Characterization of Selection Signatures and Runs of Homozygosity in Ugandan Goat Breeds. Front. Genet. 2018, 9, 318. [Google Scholar] [CrossRef]
- Stella, A.; Nicolazzi, E.L.; Van Tassell, C.P.; Rothschild, M.; Colli, L.; Rosen, B.D. AdaptMap: Exploring goat diversity and adaptation. Genet. Sel. Evol. 2018. [Google Scholar] [CrossRef]
- Kim, E.; Elbeltagy, A.; Aboul-Naga, A.; Rischkowsky, B.; Sayre, B.; Mwacharo, J.; Rothschild, M. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity 2016, 116, 255–264. [Google Scholar] [CrossRef]
- Mwacharo, J.M.; Kim, E.-S.; Elbeltagy, A.R.; Aboul-Naga, A.M.; Rischkowsky, B.A.; Rothschild, M.F. Genomic footprints of dryland stress adaptation in Egyptian fat-tail sheep and their divergence from East African and western Asia cohorts. Sci. Rep. 2017, 7, 17647. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tao, H.; Li, P.; Li, L.; Zhong, T.; Wang, L. Whole-genome sequencing reveals selection signatures associated with important traits in six goat breeds. Sci. Rep. 2018, 8, 10405. [Google Scholar] [CrossRef] [PubMed]
- Tosser-Klopp, G.; Bardou, P.; Bouchez, O.; Cabau, C.; Crooijmans, R.; Dong, Y.; Donnadieu-Tonon, C.; Eggen, A.; Heuven, H.C.; Jamli, S. Design and characterization of a 52K SNP chip for goats. PLoS ONE 2014, 9, e86227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; McParland, S.; Kearney, F.; Du, L.; Berry, D.P. Detection of selection signaturesin dairy and beef cattle using high-density genomic information. Genet. Sel. Evol. 2015, 47, 49. [Google Scholar] [CrossRef] [PubMed]
- Makina, S.O.; Muchadeyi, F.C.; Marle-Kster, E.; Taylor, J.F.; Makgahlela, M.L.; Maiwashe, A. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet. Sel. Evol. 2015, 47, 92. [Google Scholar] [CrossRef] [PubMed]
- Moradi, M.H.; Nejati-Javaremi, A.; Moradi-Shahrbabak, M.; Dodds, K.G.; McEwan, J.C. Genomic scan of selective sweeps in thin and fat tail sheep breeds for identifying of candidate regions associated with fat deposition. BMC Genom. 2012, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- McRae, K.M.; McEwan, J.C.; Dodds, K.G.; Gemmell, N.J. Signatures of selection in sheep bred for resistance or susceptibility to gastrointestinal nematodes. BMC Genom. 2014, 15, 637. [Google Scholar] [CrossRef]
- Stainton, J.J.; Haley, C.S.; Charlesworth, B.; Kranis, A.; Watson, K.; Wiener, P. Detecting signatures of selection in nine distinct lines of broiler chickens. Anim. Genet. 2015, 46, 37–49. [Google Scholar] [CrossRef]
- Ai, H.; Huang, L.; Ren, J. Genetic diversity, linkage disequilibrium and selection signatures in Chinese and Western pigs revealed by genome-wide SNP markers. PLoS ONE 2013, 8, e56001. [Google Scholar] [CrossRef]
- Ai, H.; Yang, B.; Li, J.; Xie, X.; Chen, H.; Ren, J. Population history and genomic signaturesfor high-altitude adaptation in Tibetan pigs. BMC Genom. 2014, 15, 834. [Google Scholar] [CrossRef]
- Nicoloso, L.; Bomba, L.; Colli, L.; Negrini, R.; Milanesi, M.; Mazza, R.; Sechi, T.; Frattini, S.; Talenti, A.; Coizet, B. Genetic diversity of Italian goat breeds assessed with a medium-density SNP chip. Genet. Sel. Evol. 2015, 47, 62. [Google Scholar] [CrossRef] [PubMed]
- Benjelloun, B.; Alberto, F.J.; Streeter, I.; Boyer, F.; Coissac, E.; Stucki, S. Characterizing neutral genomic diversity and selection signatures in indigenous populations of moroccan goats (Capra hircus) using WGS data. Front. Genet. 2015, 6, 107. [Google Scholar] [CrossRef]
- Lashmar, S.; Visser, C.; van Marle-Köster, E. Validation of the 50k Illumina goat SNP chip in the South African Angora goat. S. Afr. J. Anim. Sci. 2015, 45, 56–59. [Google Scholar] [CrossRef]
- Colli, L.; Milanesi, M.; Talenti, A.; Bertolini, F.; Chen, M.; Chen, M.; Crisa, A.; Daly, K.G.; Del Covo, M.; Guldbrandtsen, B.; et al. Genome-wide SNP profiling of worldwide goat populations reveals strong partitioning of diversity and highlights post-domestication migration routes. Genet. Sel. Evol. 2018, 50, 58. [Google Scholar] [CrossRef] [PubMed]
- Glowatzki-Mullis, M.L.; Muntwyler, J.; Baumle, E.; Gaillard, C. Genetic diversity measures of Swiss goat breeds as decision-making support for conservation policy. Small Rumin. Res. 2008, 74, 202–211. [Google Scholar] [CrossRef]
- Ağaoağlu, O.K.; Ertuğrul, O. Assessment of genetic diversity, genetic relationship and bottleneck using microsatellites in some native Turkish goat breeds. Small Rumin. Res. 2012, 105, 53–60. [Google Scholar] [CrossRef]
- Mukhongo, D.M.; Mwai, O.; Tapio, M.; Muigai, A. Genetic diversity and population structure of the indigenous sheep in Kenya based on microsatellite analysis: Implications for their conservation. J. Livest. Sci. 2014, 5, 65–78. [Google Scholar]
- Xu, S.-S.; Gao, L.; Xie, X.-L.; Ren, Y.-L.; Shen, Z.-Q.; Wang, F.; Shen, M.; Eyϸórsdóttir, E.; Hallsson, J.H.; Kiseleva, T. Genome-Wide Association Analyses Highlight the Potential for Different Genetic Mechanisms for Litter Size Among Sheep Breeds. Front. Genet. 2018, 9, 118. [Google Scholar] [CrossRef]
- Mdladla, K.; Dzomba, E.F.; Muchadeyi, F.C. Characterization of the village goat production systems in the rural communities of the Eastern Cape, KwaZulu-Natal, Limpopo and North West Provinces of South Africa. Trop. Anim. Health Prod. 2017, 49, 515–527. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Mdladla, K. Landscape Genomic Approach to Investigate Genetic Adaptation in South African Indigenous Goat Populations. Ph.D. Thesis, University of Kwa-Zulu Natal, Durban, South Africa, 2016. [Google Scholar]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry inunrelated individuals. Genet. Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, R.; Hazelhurst, S. Genesis Manual; University of the Witwatersrand: Johannesburg, South Africa, 2014. [Google Scholar]
- Luu, K.; Bazin, E.; Blum, M.G.B. pcadapt: An R package to perform genome scans for selection based on principal component analysis. Mol. Ecol. Res. 2017, 17, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Tittensor, D.P.; Walpole, M.; Hill, S.L.L.; Boyce, D.G.; Britten, G.L.; Burgess, N.D. A mid-term analysis of progress toward international biodiversity targets. Science 2014, 346, 241–244. [Google Scholar] [CrossRef]
- Morrison, J.W. A guide to the Identification of the Natural Indigenous Goats of Southern Africa. 2007. Available online: http://www.landbou.com/wp.../03/f2297405-a93f-4399-bdb7-6f3de538d75d.pdf (accessed on 20 April 2018).
- Kalinowski, S.T. Counting alleles with rarefaction: Private alleles and hierarchical sampling designs. Conserv. Genet. 2004, 5, 539–543. [Google Scholar] [CrossRef]
- Liu, J.B.; Wang, F.; Lang, X. Analysis of geographic and pairwise distances among chinese cashmere goat populations. Asian Australas. J. Anim. Sci. 2013, 26, 323–333. [Google Scholar] [CrossRef][Green Version]
- Brito, L.F.; Kijas, J.W.; Ventura, R.V.; Sargolzaei, M.; Porto-Neto, L.R.; Cánovas, A. Genetic diversity and signatures of selection in various goat breeds revealed by genome-wide SNP markers. BMC Genom. 2017, 18, 229. [Google Scholar] [CrossRef]
- Zhao, Y.X.; Yang, J.; Lv, F.H.; Hu, X.J.; Xie, X.L.; Zhang, M. Genomic reconstruction of the history of native sheep reveals the peopling patterns of nomads and the expansion of early pastoralism in East Asia. Mol. Biol. Evol. 2017, 34, 2380–2395. [Google Scholar] [CrossRef]
- Tapio, M.; Ozerov, M.; Tapio, I.; Toro, M.A.; Marzanov, N.; Cinkulov, M. Microsatellite- based genetic diversity and population structure of domestic sheep in northern Eurasia. BMC Genet. 2010, 11, 76. [Google Scholar] [CrossRef]
- Vahidi, S.M.F.; Tarang, A.R.; Naqvi, A.N.; Anbaran, M.F.; Boettcher, P.; Joost, S. Investigation of the genetic diversity of domestic Capra hircus breeds reared within an early goat domestication area in Iran. Genet. Sel. Evol. 2014, 46, 27. [Google Scholar] [CrossRef]
- Zvinorova, P.I. A Genome-Wide Association Study on Mechanisms Underlying Genetic Resistance to Gastrointestinal Parasites in Goats. Ph.D. Thesis, Stellenbosch University, Stellenbosch, South Africa, 2017. [Google Scholar]
- Ozoje, M. Incidence and relative effects of qualitative traits in West African Dwarf goat. Small Rumin. Res. 2002, 43, 97–100. [Google Scholar] [CrossRef]
- Pieters, A. Genetic Characterization of Commercial Goat Populations in South Africa. Master’s Thesis, University of Pretoria, Pretoria, South Africa, 2007. [Google Scholar]
- Ahmed, Z.; Babar, M.E.; Hussain, T.; Nadeem, A.; Awan, F.I.; Wajid, A.; Ali, M.M. Genetic diversity analysis of Kail sheep by using microsatellite markers. J. Anim. Plant. Sci. 2014, 24, 1329–1333. [Google Scholar]
- Yang, J.; Li, W.R.; Lv, F.H.; He, S.G.; Tian, S.L.; Peng, W.F.; Sun, Y.W.; Zhao, Y.X.; Tu, X.L.; Zhang, M. Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Mol. Biol. Evol. 2016, 33, 2576–2592. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, F.; Servin, B.; Talenti, A.; Rochat, E.; Kim, E.S.; Oget, C.; Palhière, I.; Crisà, A.; Catillo, G.; Steri, R.; et al. Signatures of selection and environmental adaptation across the goat genome post-domestication. Genet. Sel. Evol. 2018, 50, 57. [Google Scholar] [CrossRef]
- Islam, R.; Li, Y.; Liu, X.; Berihulay, H.; Abied, A.; Gebreselassie, G.; Ma, Q.; Ma, Y. Genome-Wide Runs of Homozygosity, Effective Population Size, and Detection of Positive Selection Signatures in Six Chinese Goat Breeds. Genes 2019, 10, 938. [Google Scholar] [CrossRef]
- Rahmatalla, S.A.; Arends, D.; Reissmann, M.; Ahmed, A.S.; Wimmers, K.; Reyer, H.; Brockmann, G.A. Whole genome population genetics analysis of Sudanese goats identifies regions harboring genes associated with major traits. BMC Genet. 2017, 18, 92. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
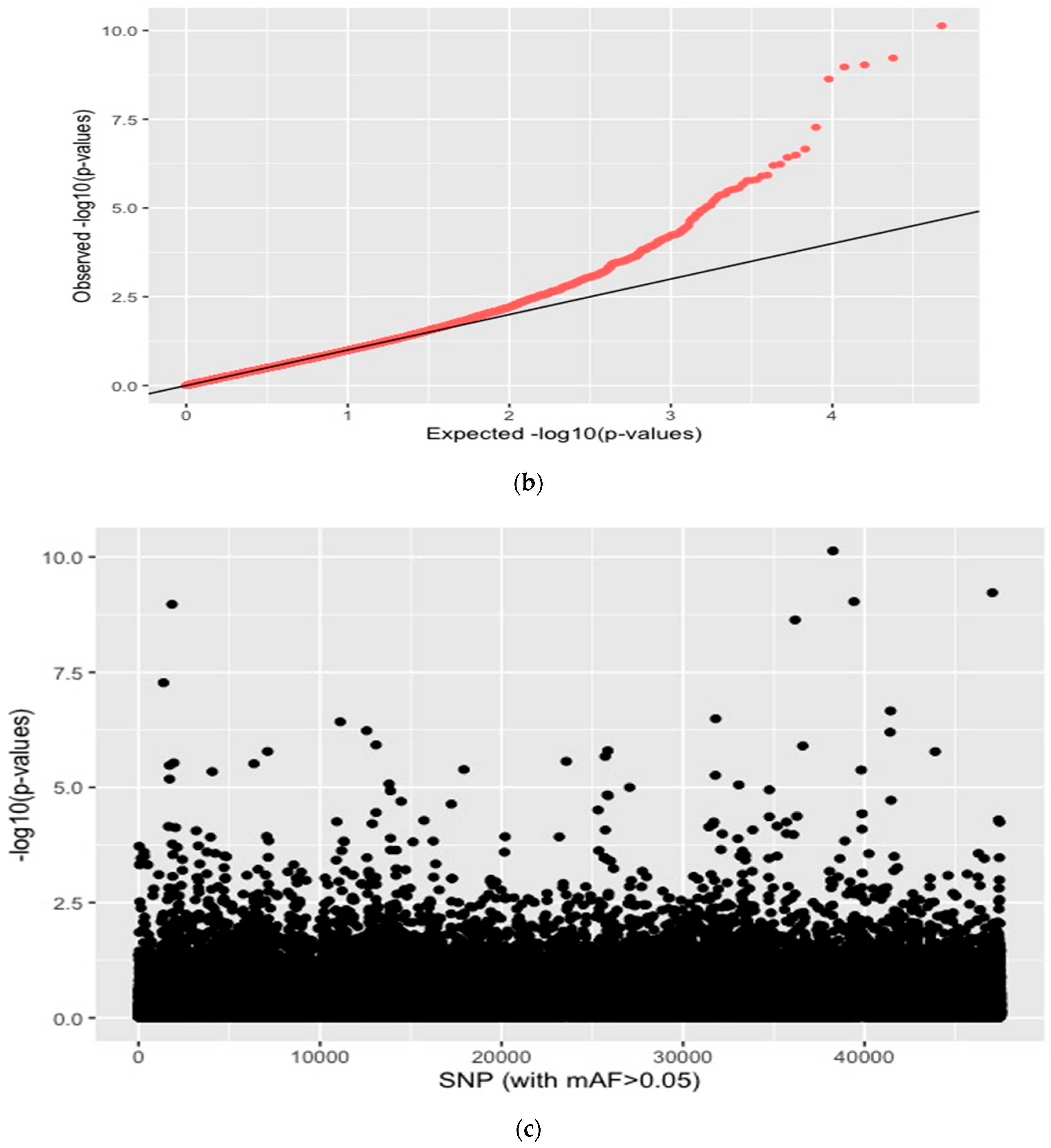{kind=link}
| Source of Variation | Degrees of Difference (d.f.) | Sum of Squares | Variance Components | Percentage of Variation |
|---|---|---|---|---|
| Among populations | 1 | 80,565.150 | 341.26752 Va | 3.39 |
| Among population within groups | 5 | 99,867.875 | 185.19042 Vb | 1.84 |
| Among individuals within populations | 182 | 1,814,879.906 | 416.85973 Vc | 4.12 |
| Within populations | 189 | 1,727,110.000 | 9138.14815 Vd | 90.64 |
| Marker | CHR | Position | FST | Gene | Gene Description |
|---|---|---|---|---|---|
| snp28078-scaffold300-3412212 | 8 | 38197294 | 0.72 | UHRF2 | ubiquitin like with PHD and ring finger domains 2 |
| snp19368-scaffold1958-104157 | 6 | 8034538 | 0.71 | - | |
| snp3556-scaffold1110-106176 | 9 | 59365070 | 0.70 | - | |
| snp19366-scaffold1958-28303 | 6 | 8110392 | 0.69 | - | |
| snp28077-scaffold300-3369813 | 8 | 38154895 | 0.69 | GLDC | glycine decarboxylase |
| snp51703-scaffold760-153052 | 6 | 7438269 | 0.68 | NDST3 | N-deacetylase and N-sulfotransferase 3 |
| snp32526-scaffold371-1443673 | 13 | 39230563 | 0.68 | CFAP61 | cilia and flagella associated protein 61 |
| snp48555-scaffold690-432877 | 6 | 104631767 | 0.67 | - | |
| snp49002-scaffold7-1603772 | 13 | 30788222 | 0.67 | CUBN | cubilin |
| snp49005-scaffold7-1739012 | 13 | 30652982 | 0.66 | CUBN | cubilin |
| Marker | Chromosome | Position | PC |
|---|---|---|---|
| snp2141-scaffold1065-308545 | 10 | 74803720 | 2 |
| snp32432-scaffold37-603475 | 15 | 12584499 | 2 |
| snp32448-scaffold37-1273016 | 15 | 13254040 | 2 |
| snp34005-scaffold40-2680622 | 20 | 50848470 | 2 |
| snp15603-scaffold165-514510 | 21 | 53972419 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chokoe, T.C.; Mdladla-Hadebe, K.; Muchadeyi, F.; Dzomba, E.; Matelele, T.; Mphahlele, T.; Mpofu, T.J.; Nephawe, K.; Mtileni, B. Genetic Diversity of South African Indigenous Goat Population from Four Provinces Using Genome-Wide SNP Data. Sustainability 2020, 12, 10361. https://doi.org/10.3390/su122410361
Chokoe TC, Mdladla-Hadebe K, Muchadeyi F, Dzomba E, Matelele T, Mphahlele T, Mpofu TJ, Nephawe K, Mtileni B. Genetic Diversity of South African Indigenous Goat Population from Four Provinces Using Genome-Wide SNP Data. Sustainability. 2020; 12(24):10361. https://doi.org/10.3390/su122410361
Chicago/Turabian StyleChokoe, Tlou Caswell, Khanyisile Mdladla-Hadebe, Farai Muchadeyi, Edgar Dzomba, Tlou Matelele, Tumudi Mphahlele, Takalani J. Mpofu, Khathutshelo Nephawe, and Bohani Mtileni. 2020. "Genetic Diversity of South African Indigenous Goat Population from Four Provinces Using Genome-Wide SNP Data" Sustainability 12, no. 24: 10361. https://doi.org/10.3390/su122410361
APA StyleChokoe, T. C., Mdladla-Hadebe, K., Muchadeyi, F., Dzomba, E., Matelele, T., Mphahlele, T., Mpofu, T. J., Nephawe, K., & Mtileni, B. (2020). Genetic Diversity of South African Indigenous Goat Population from Four Provinces Using Genome-Wide SNP Data. Sustainability, 12(24), 10361. https://doi.org/10.3390/su122410361