Polyurethane Foams and Bio-Polyols from Liquefied Cotton Stalk Agricultural Waste



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Methods
2.2.1. Liquefaction of Cotton Stalk with PEG and Glycerin
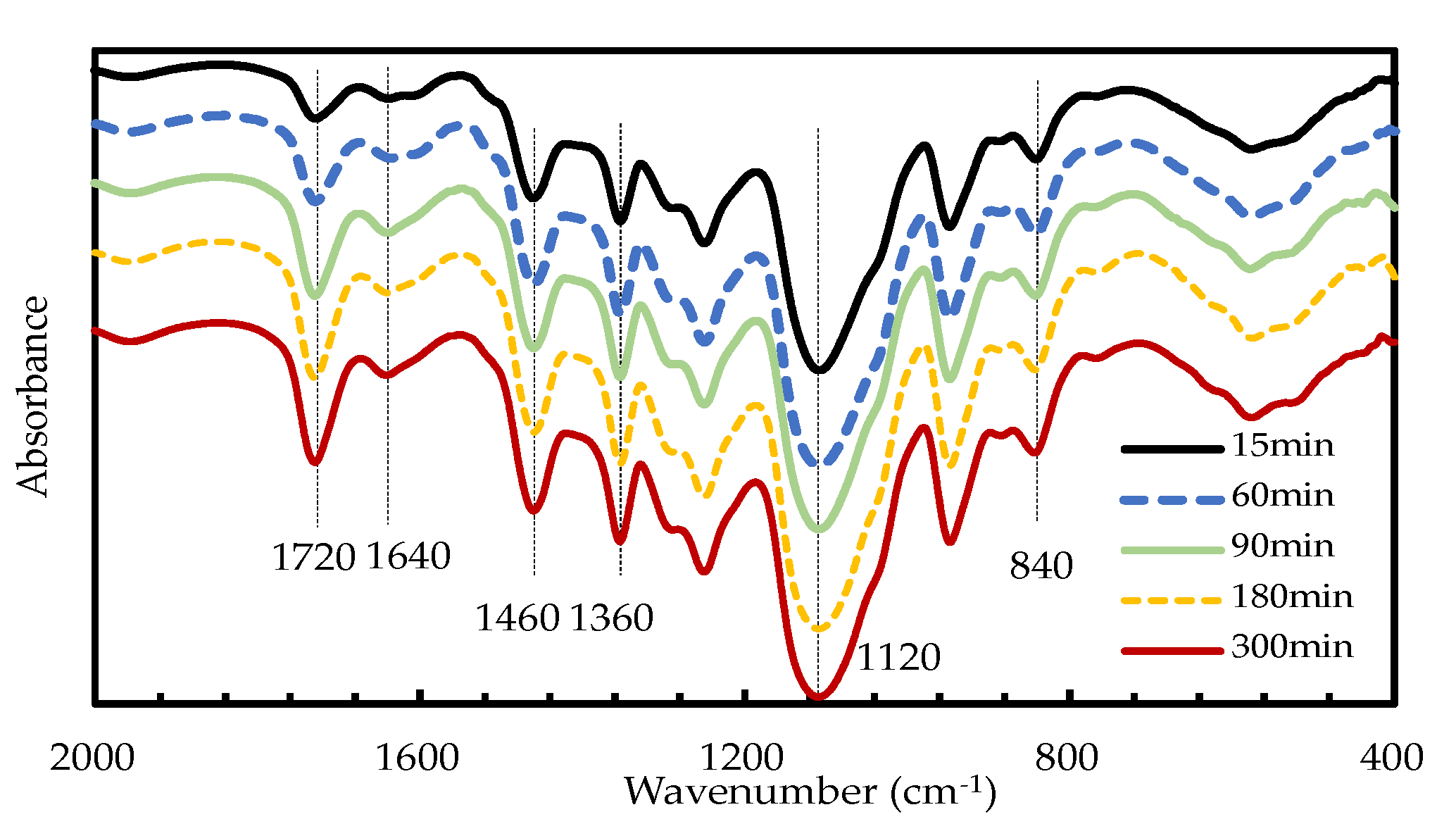
2.2.2. Characterization of Liquefaction Products
2.2.3. Preparation of Polyurethane Foams
2.2.4. Characterizations of Polyurethane Foams
2.2.5. Biodegradation of Polyurethane Foams
3. Results and Discussion
3.1. Effects of Liquefaction Conditions on the Polyol Properties
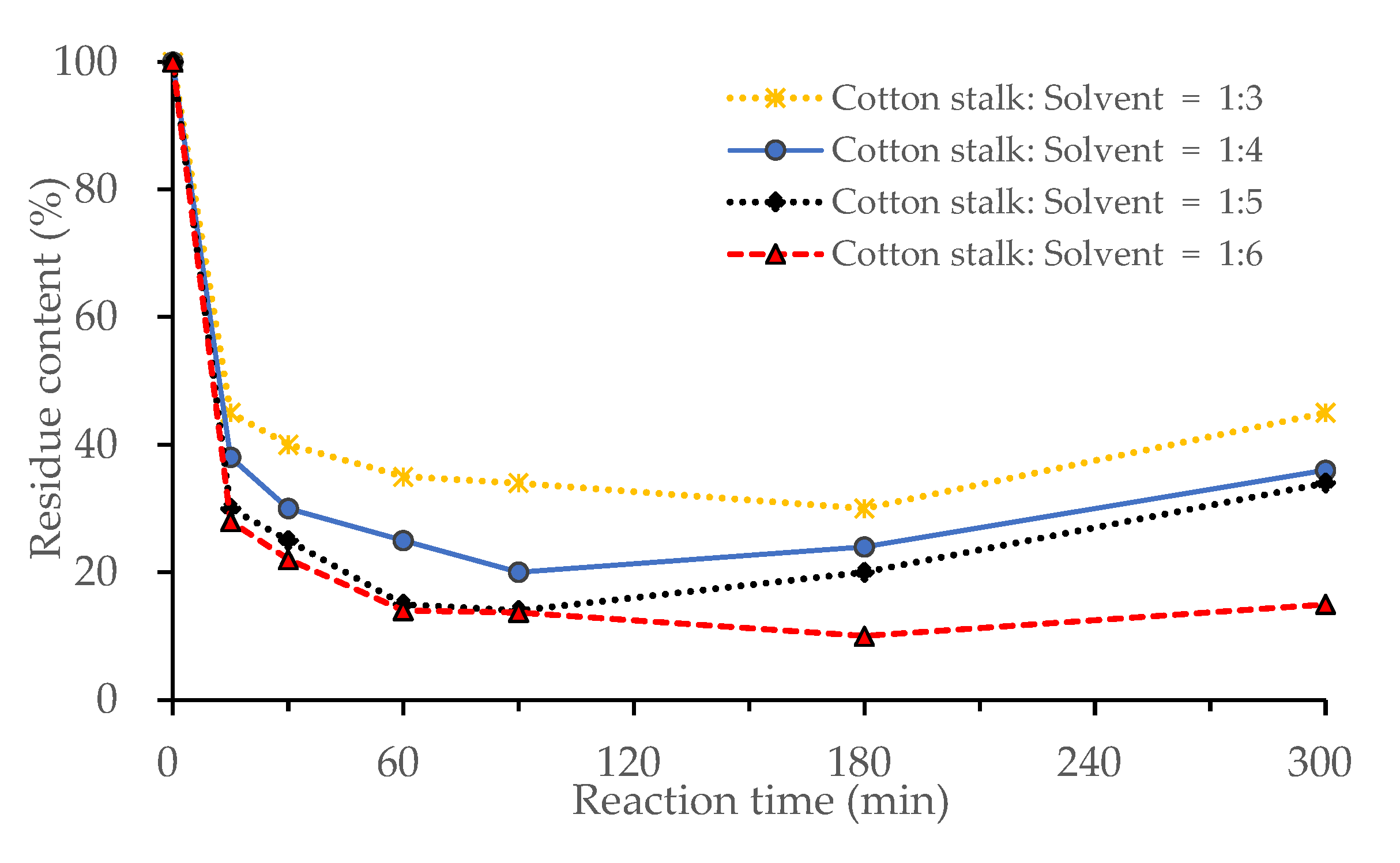
3.1.1. Effect of Biomass Loading
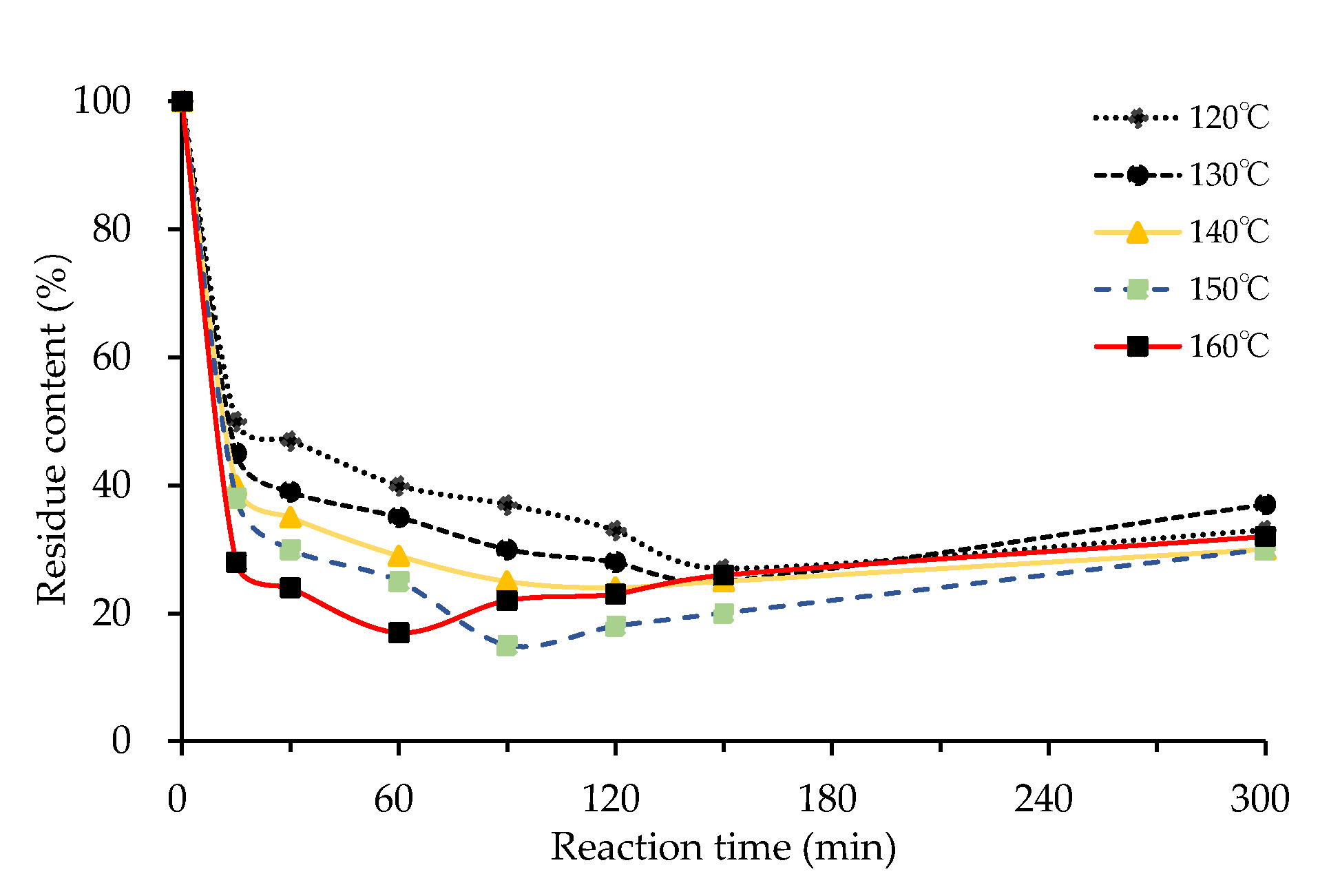
3.1.2. Effect of Reaction Temperature
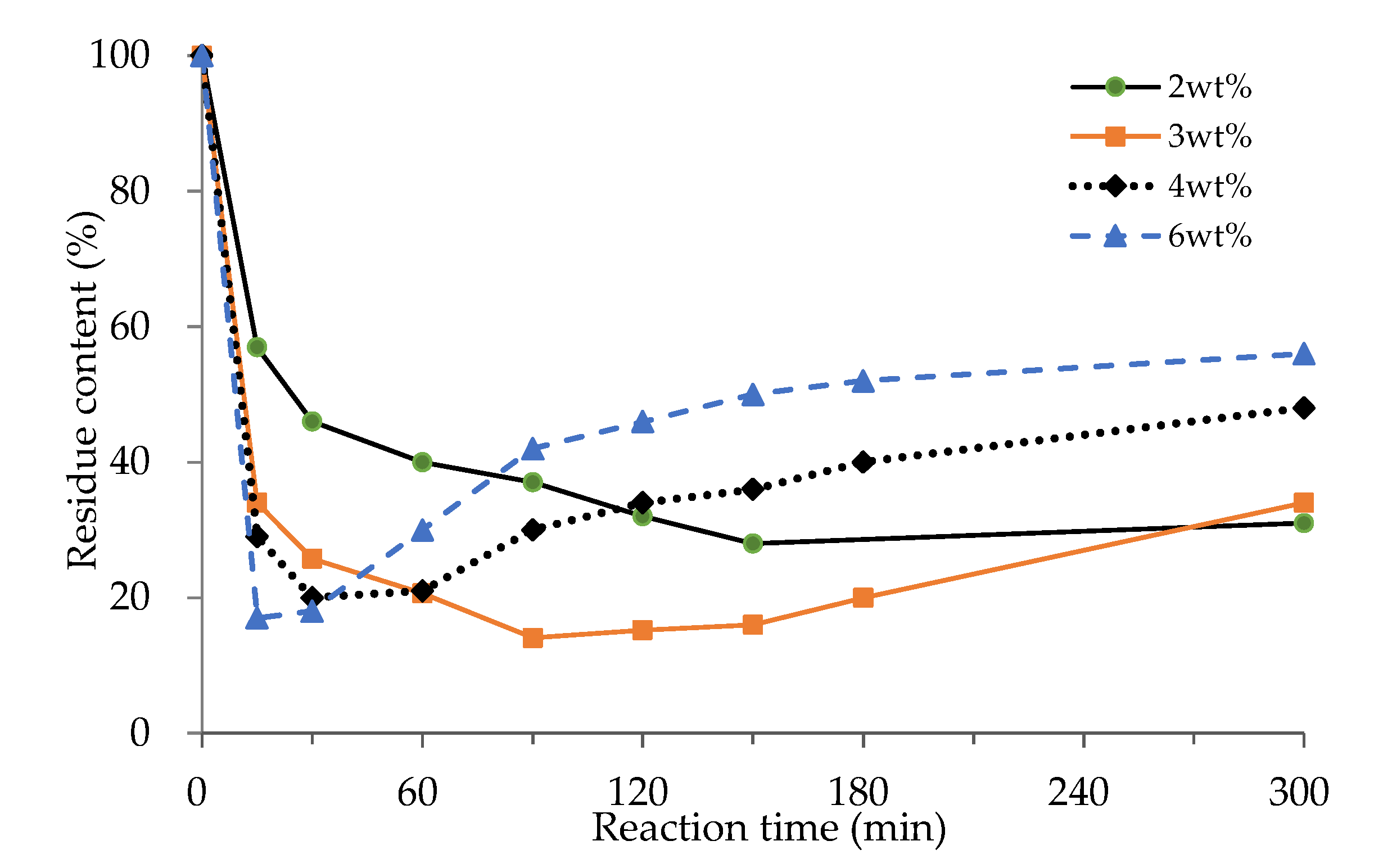
3.1.3. Effect of Catalyst Loading
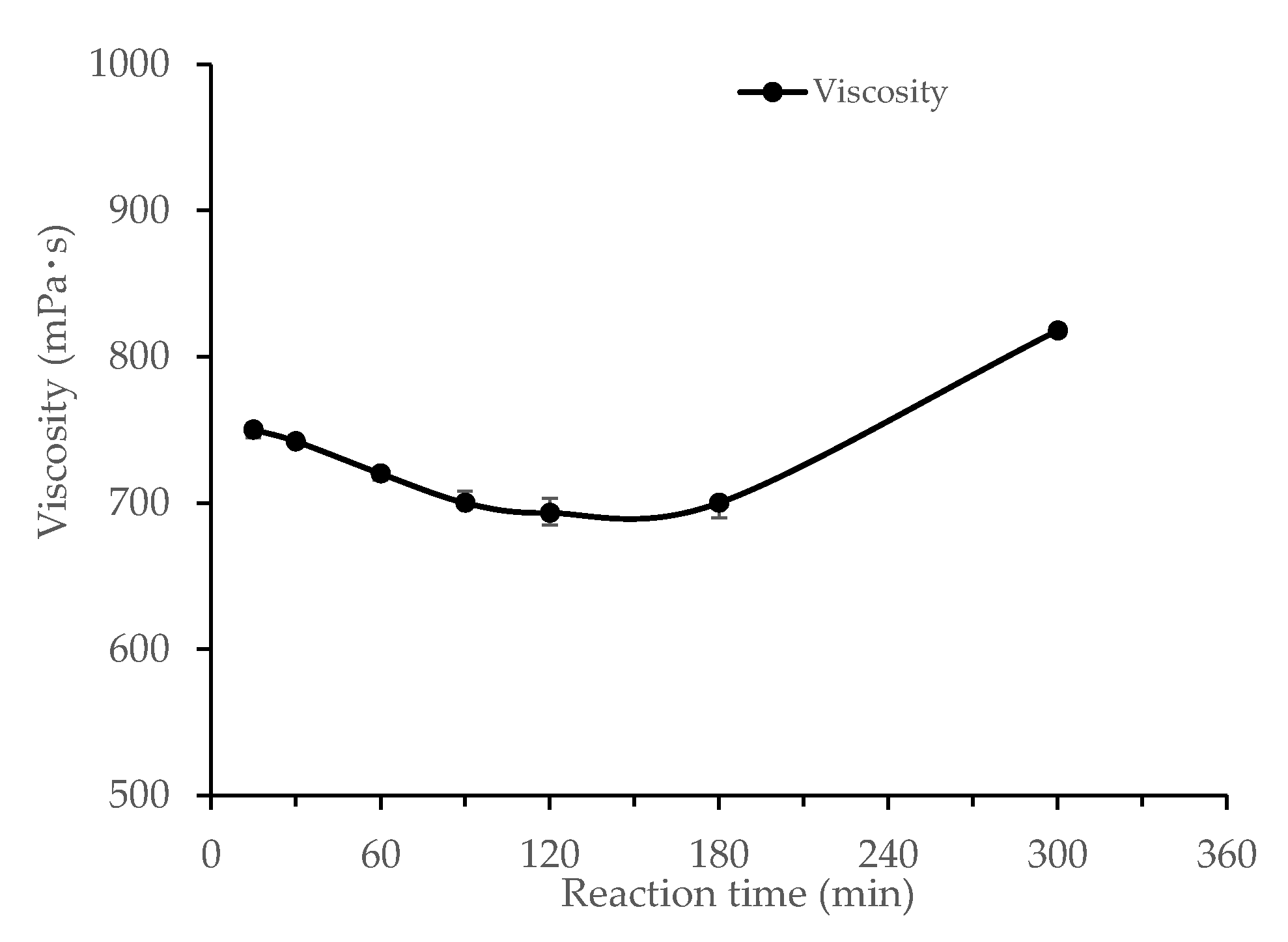
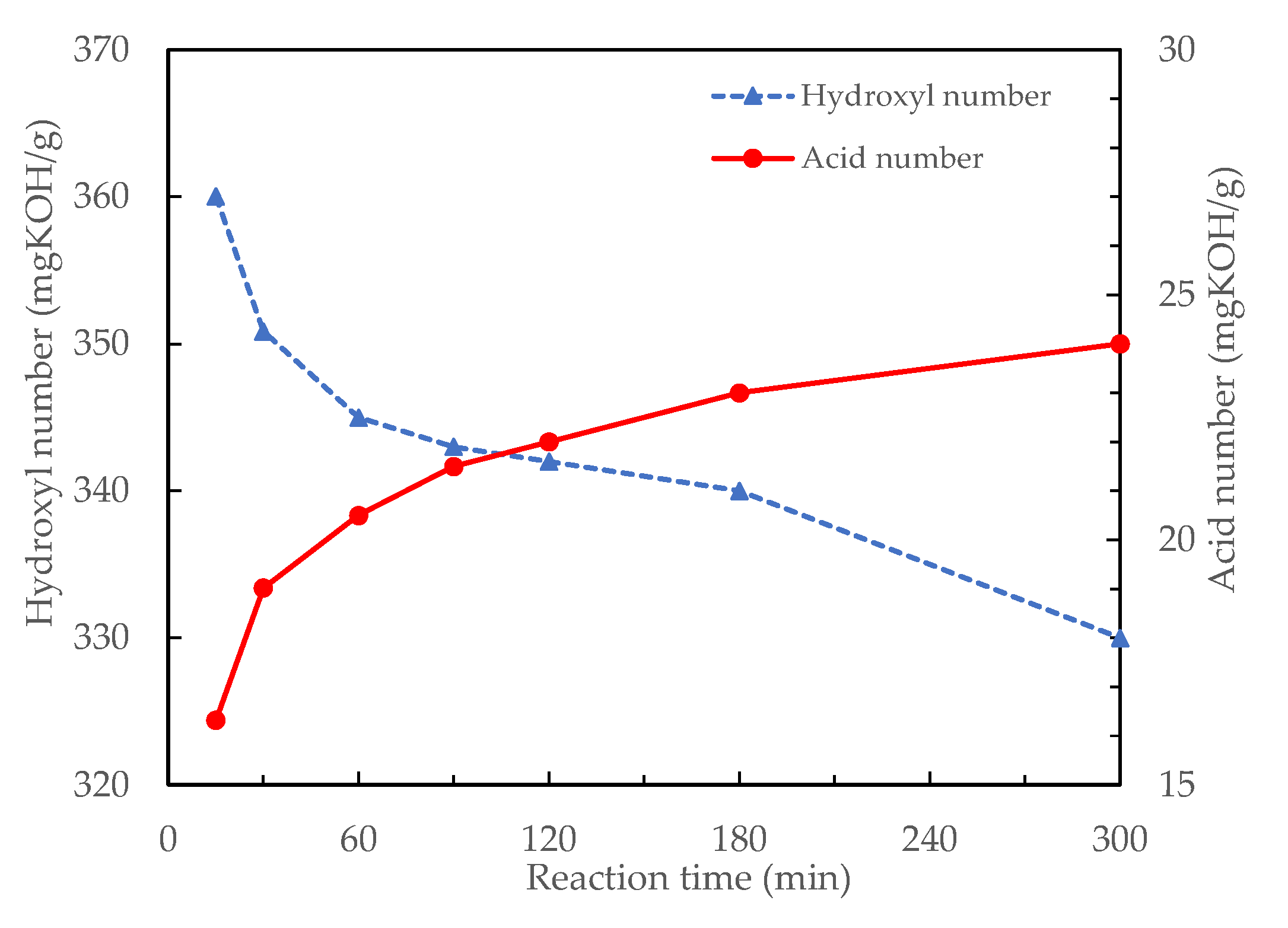
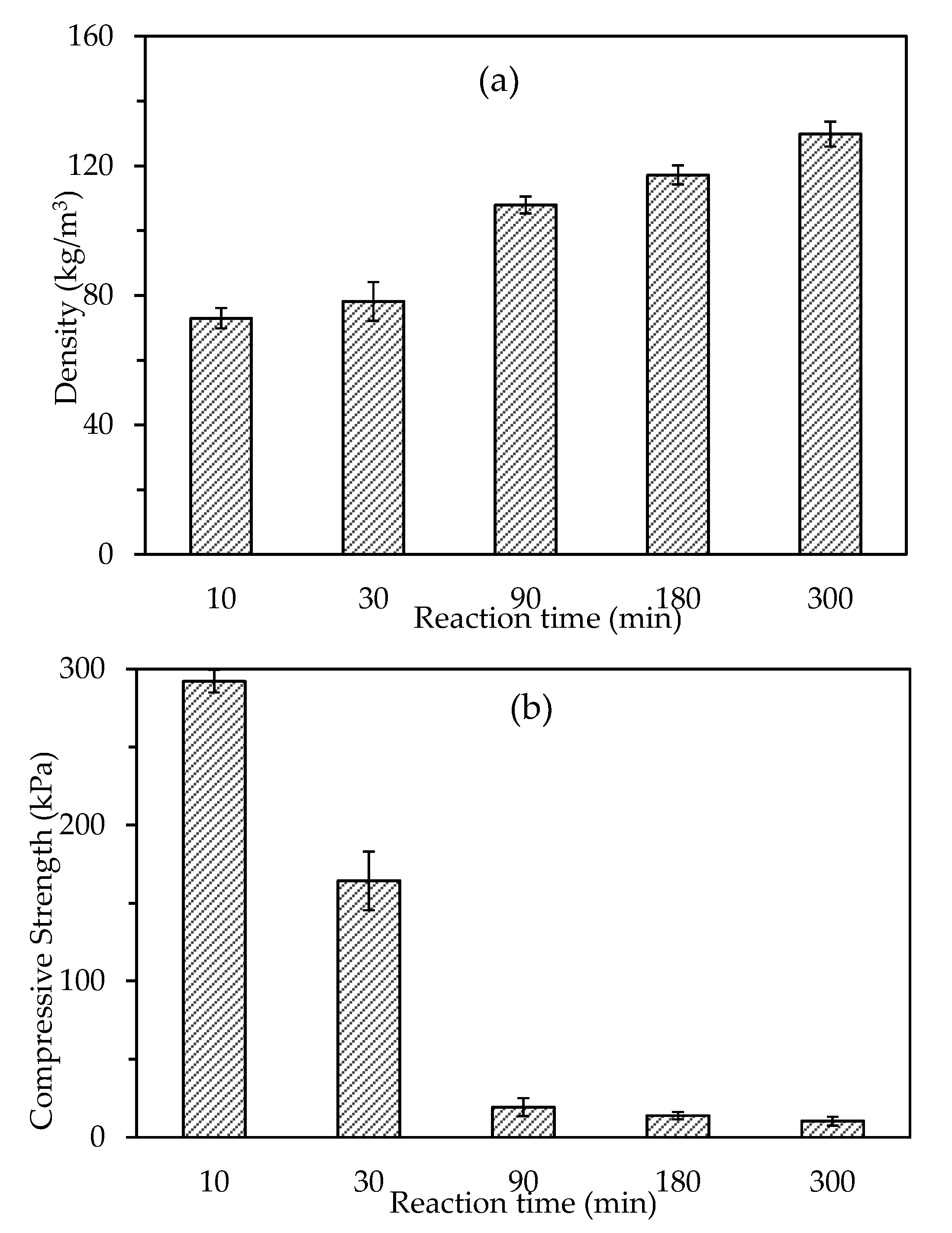
3.1.4. Effects of Reaction Time
3.2. Properties of Polyurethane Foam
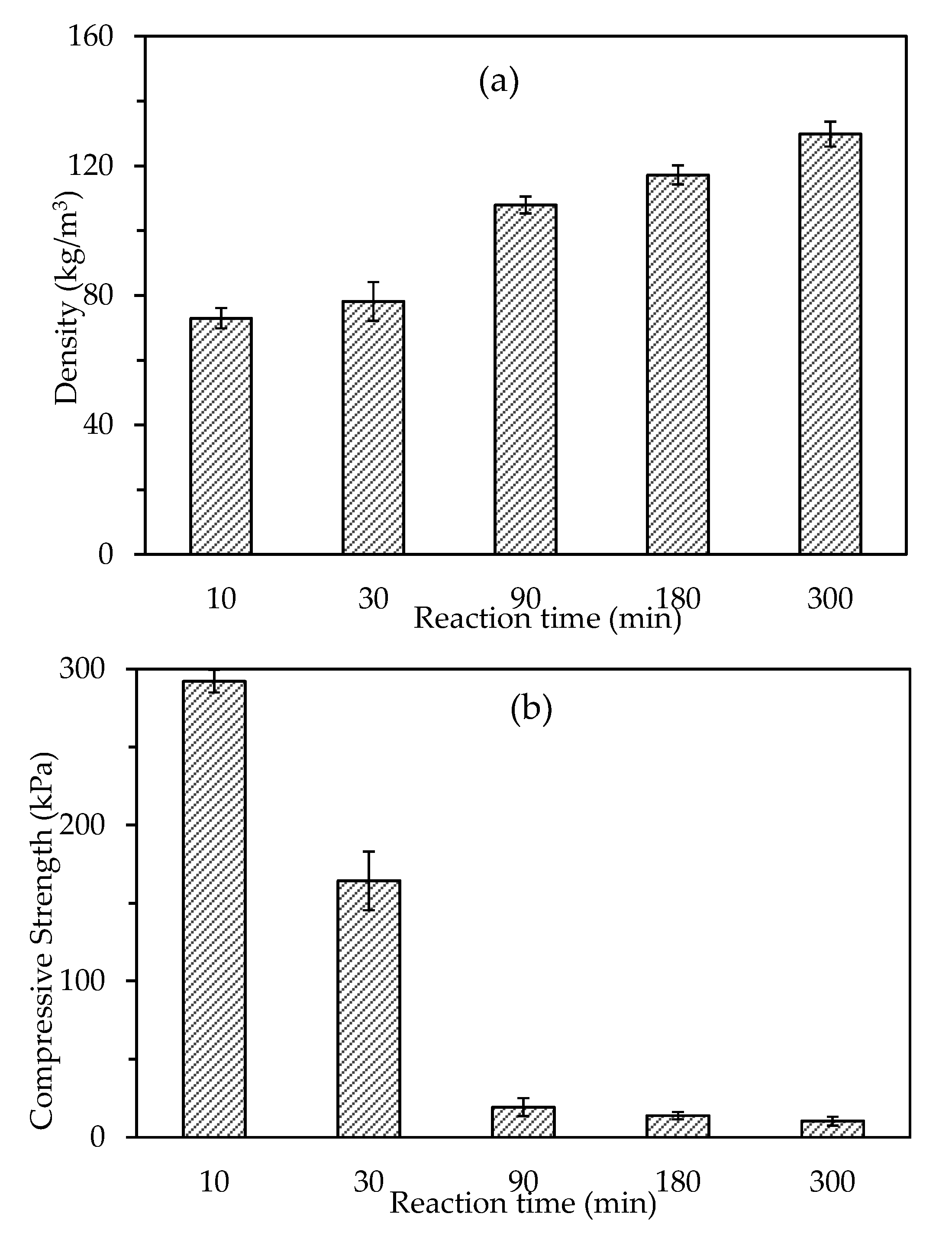
3.2.1. Density and Compressive Strength
3.2.2. Foam Morphology
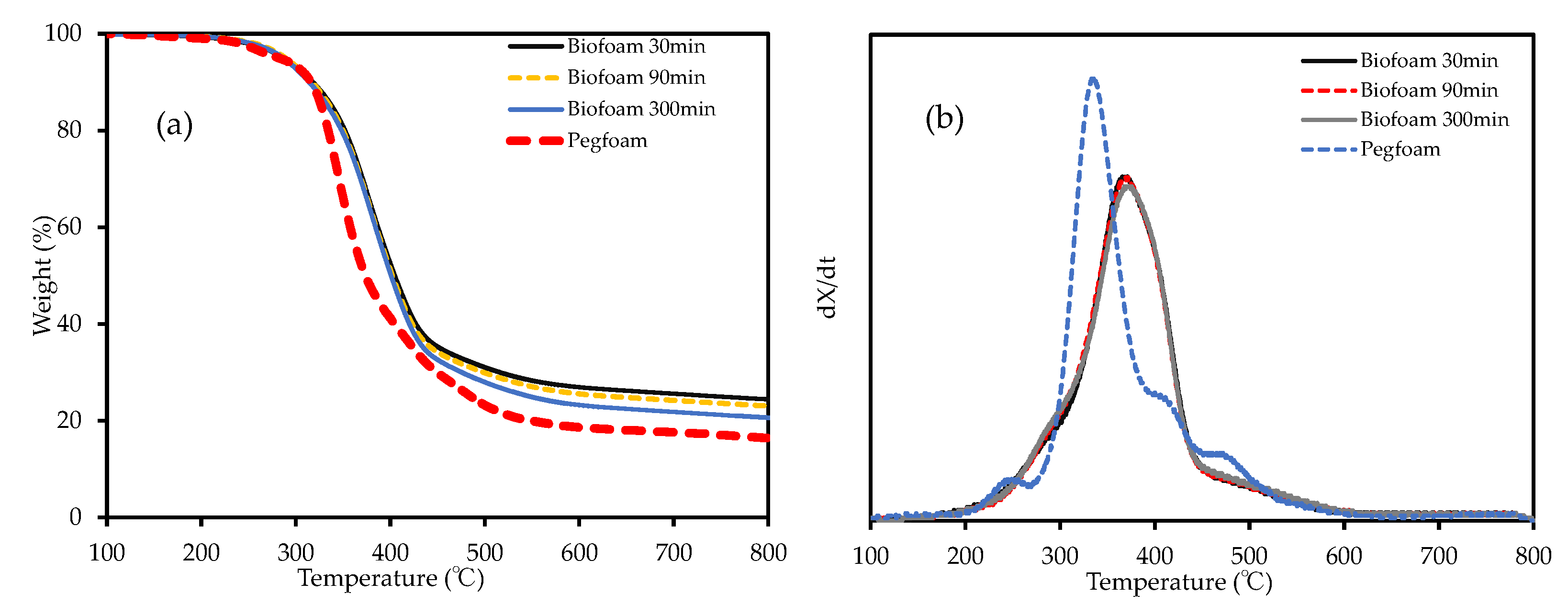
3.2.3. Thermal Stability
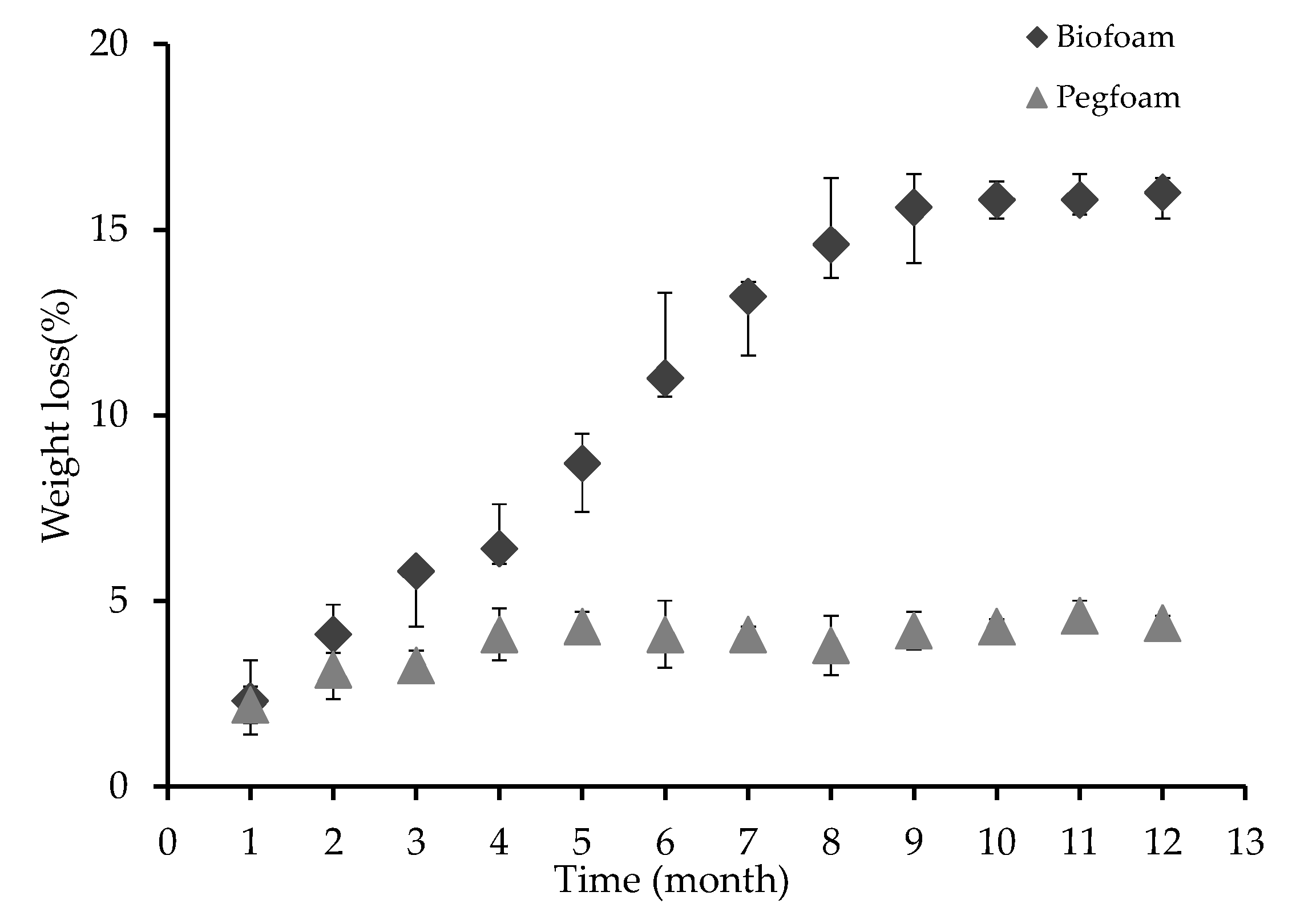
3.2.4. Biodegradation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fernando, S.; Adhikari, S.; Chandrapal, C.; Murali, N. Biorefineries: Current status, challenges, and future direction. Energy Fuels 2006, 20, 1727–1737. [Google Scholar] [CrossRef]
- Effendi, A.; Gerhauser, H.; Bridgwater, A.V. Production of renewable phenolic resins by thermochemical conversion of biomass: A review. Renew. Sustain. Energy Rev. 2008, 12, 2092–2116. [Google Scholar] [CrossRef]
- Wang, M.; Xu, C.C.; Leitch, M. Liquefaction of cornstalk in hot-compressed phenol-water medium to phenolic feedstock for the synthesis of phenol-formaldehyde resin. Bioresour. Technol. 2009, 100, 2305–2307. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Gutiérrez, C.V.; Castro-Salazar, H.T.; Ríos-Reyes, C.A. Synthesis and properties of polyurethane foams obtained from cassava starch and rice by-products. J. Mex. Chem. Soc. 2018, 62, 1–8. [Google Scholar] [CrossRef]
- Huang, X.; De Hoop, C.F.; Xie, J.; Wu, Q.; Boldor, D.; Qi, J. High bio-content polyurethane (PU) foam made from bio-polyol and cellulose nanocrystals (CNCs) via microwave liquefaction. Mater. Des. 2018, 138, 11–20. [Google Scholar] [CrossRef]
- Yang, Z.; Peng, H.; Wang, W.; Liu, T. Crystallization behavior of poly(ε-caprolactone)/layered double hydroxide nanocomposites. J. Appl. Polym. Sci. 2010, 116, 2658–2667. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.H.; Kim, D.K.; Park, C.H.; Yu, J.H.; Lee, E.Y. Crude glycerol-mediated liquefaction of empty fruit bunches saccharification residues for preparation of biopolyurethane. J. Ind. Eng. Chem. 2016, 34, 157–164. [Google Scholar] [CrossRef]
- Yamada, T.; Aratani, M.; Kubo, S.; Ono, H. Chemical analysis of the product in acid-catalyzed solvolysis of cellulose using polyethylene glycol and ethylene carbonate. J. Wood Sci. 2007, 53, 487–493. [Google Scholar] [CrossRef]
- Hassan, E.; barbary, M.; Shukry, N. Polyhydric alcohol liquefaction of some lignocellulosic agricultural residues. Ind. Crop. Prod. 2008, 27, 33–38. [Google Scholar] [CrossRef]
- Lee, S.; Yoshioka, M.; Shiraishi, N. Liquefaction of Corn Bran (CB) in the Presence of Alcohols and Preparation of Polyurethane Foam from Its Liquefied Polyol. J. Appl. Polym. Sci. 2000, 78, 319–325. [Google Scholar] [CrossRef]
- Li, H.; Feng, S.; Yuan, Z.; Wei, Q.; Xu, C.C. Highly efficient liquefaction of wheat straw for the production of bio-polyols and bio-based polyurethane foams. Ind. Crop. Prod. 2017, 109, 426–433. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Q.; Mitsumura, N.; Animesh, S. Behavior of cellulose liquefaction after pretreatment using ionic liquids with water mixtures. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Lu, X.; Wang, Y.; Zhang, Y.; Cheng, X.; Yu, Y.; Jin, Y. Preparation of bio-polyols by liquefaction of hardwood residue and their application in the modification of polyurethane foams. J. Wuhan Univ. Technol. Mater. Sci. Ed. 2016, 31, 918–924. [Google Scholar] [CrossRef]
- Wang, Q.; Kawamura, S. Decayed woody material from mushroom cultivation: Characterization of liquefaction. Wit Trans. Ecol. Environ. 2018, 217, 481–492. [Google Scholar]
- Wang, Q.; Takahashi, H.; Takahashi, Y.; Kurokawa, H.; Sekiguchi, K.; Sugiyama, K. Characterization of liquefied waste bamboo and white-rotted wood. Wit Trans. Ecol. Environ. 2015, 195, 63–74. [Google Scholar]
- Ertaş, M.; Fidan, M.S.; Alma, M.H. Preparation and characterization of biodegradable rigid polyurethane foams from the liquefied eucalyptus and pine woods. Wood Res. 2014, 59, 97–108. [Google Scholar]
- Zhu, H.; Peng, Z.; Chen, Y.; Li, G.; Wang, L.; Tang, Y.; Pang, R.; Khan, Z.U.H.; Wan, P. Preparation and characterization of flame retardant polyurethane foams containing phosphorus-nitrogen-functionalized lignin. RSC Adv. 2014, 4, 55271–55279. [Google Scholar] [CrossRef]
- Yao, Y.; Yoshioka, M.; Shiraishi, N. Water-absorbing polyurethane foams from liquefied starch. J. Appl. Polym. Sci. 1996, 60, 1939–1949. [Google Scholar] [CrossRef]
- Wang, H.; Chen, H.Z. A novel method of utilizing the biomass resource: Rapid liquefaction of wheat straw and preparation of biodegradable polyurethane foam (PUF). J. Chin. Inst. Chem. Eng. 2007, 38, 95–102. [Google Scholar] [CrossRef]
- Kobayashi, M.; Asano, T.; Kajiyama, M.; Tomita, B. Analysis on residue formation during wood liquefaction with polyhydric alcohol. J. Wood Sci. 2004, 50, 407–414. [Google Scholar] [CrossRef]
- Kurimoto, Y.; Koizumi, A.; Doi, S.; Tamura, Y.; Ono, H. Wood species effects on the characteristics of liquefied wood and the properties of polyurethane films prepared from the liquefied wood. Biomass Bioenergy 2001, 21, 381–390. [Google Scholar] [CrossRef]
- Kim, K.H.; Jo, Y.J.; Lee, C.G.; Lee, E.Y. Solvothermal liquefaction of microalgal Tetraselmis sp. biomass to prepare biopolyols by using PEG#400-blended glycerol. Algal Res. 2015, 12, 539–544. [Google Scholar]
- Dísouza, J.; Wong, S.Z.; Camargo, R.; Yan, N. Solvolytic Liquefaction of Bark: Understanding the Role of Polyhydric Alcohols and Organic Solvents on Polyol Characteristics. ACS Sustain. Chem. Eng. 2016, 4, 851–861. [Google Scholar] [CrossRef]
- Luo, X.; Mohanty, A.; Misra, M. Water-blown rigid biofoams from soy-based biopolyurethane and microcrystalline cellulose. J. Am. Oil Chem. Soc. 2012, 89, 2057–2065. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Content | Cotton Stalk |
|---|---|---|
| Composition analysis (wt.%) | Lignin | 25.3 |
| Hemicellulose | 17.4 | |
| a-Cellulose | 48.8 | |
| Ash | 6.8 |
| GPC | Conditions |
|---|---|
| Mobile phase | Tetrahydrofuran |
| Flow rate | 1 mL/min |
| Dilute concentration of sample | 0.1 w/v % |
| Tetrahydrofuran column temp. | 40 °C |
| Volume of sample loop | 100 µL |
| Reaction Time (min) | Mp1 | Mp2 | AreaP1: AreaP2 | Mn | Mw | d = Mw/Mn |
|---|---|---|---|---|---|---|
| 30 | 1340 | 1070 | 2.13 | 1020 | 1200 | 1.18 |
| 60 | 1400 | 1060 | 2.1 | 1030 | 1150 | 1.2 |
| 90 | 1450 | 1040 | 2.12 | 1040 | 740 | 1.22 |
| 300 | 1500 | 1035 | 2.13 | 1035 | 1280 | 1.23 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Tuohedi, N. Polyurethane Foams and Bio-Polyols from Liquefied Cotton Stalk Agricultural Waste. Sustainability 2020, 12, 4214. https://doi.org/10.3390/su12104214
Wang Q, Tuohedi N. Polyurethane Foams and Bio-Polyols from Liquefied Cotton Stalk Agricultural Waste. Sustainability. 2020; 12(10):4214. https://doi.org/10.3390/su12104214
Chicago/Turabian StyleWang, Qingyue, and Nuerjiamali Tuohedi. 2020. "Polyurethane Foams and Bio-Polyols from Liquefied Cotton Stalk Agricultural Waste" Sustainability 12, no. 10: 4214. https://doi.org/10.3390/su12104214
APA StyleWang, Q., & Tuohedi, N. (2020). Polyurethane Foams and Bio-Polyols from Liquefied Cotton Stalk Agricultural Waste. Sustainability, 12(10), 4214. https://doi.org/10.3390/su12104214