
Dexmedetomidine Attenuates Methotrexate-Induced Neurotoxicity and Memory Deficits in Rats through Improving Hippocampal Neurogenesis: The Role of miR-15a/ROCK-1/ERK1/2/CREB/BDNF Pathway Modulation

 ,
,  , and
, and
Abstract
1. Introduction
2. Results
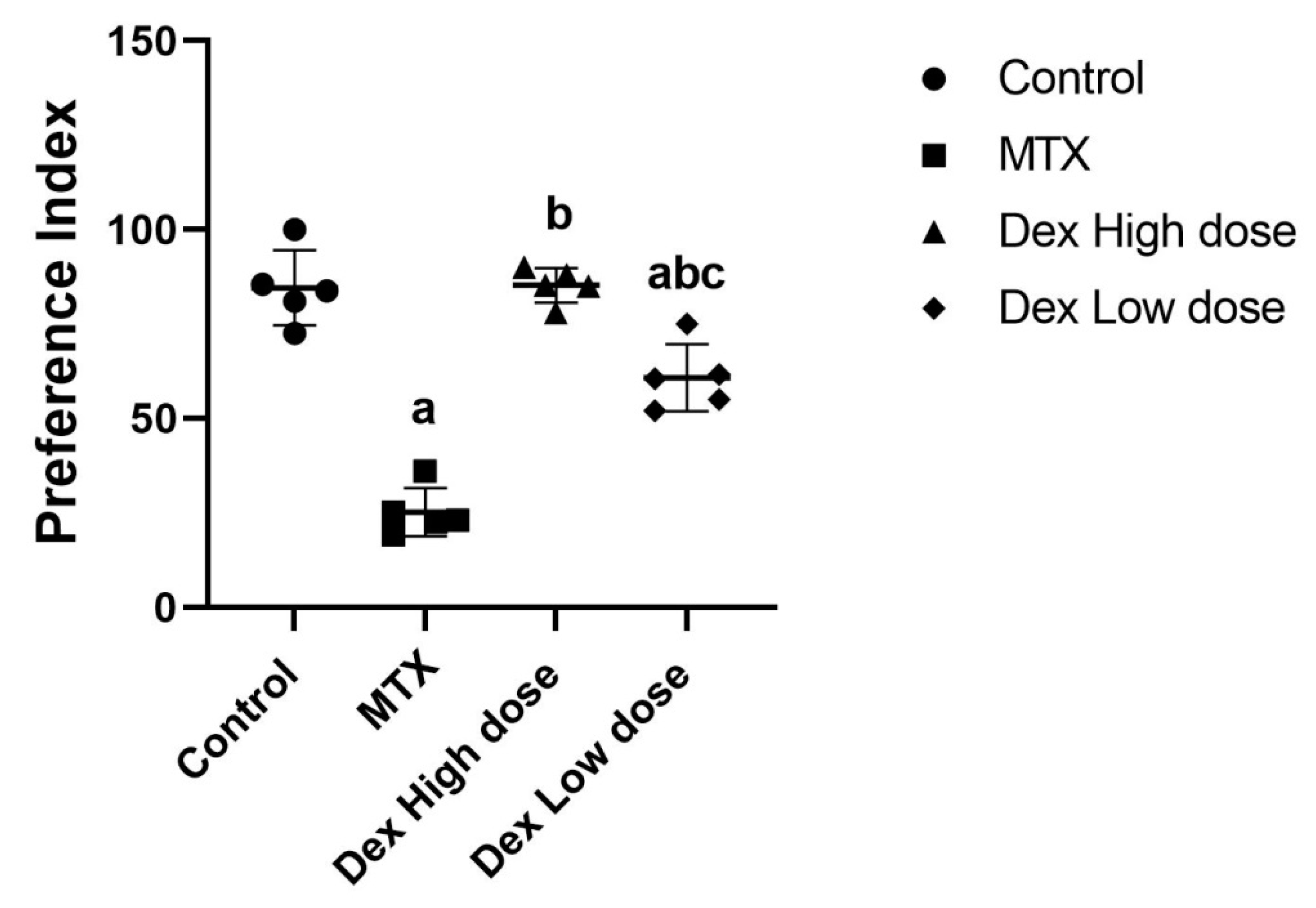
2.1. Dex Improves the Behavioral and Memory Deficits Induced by MTX Administration
2.2. Possible Molecular Mechanisms Underlie the Dex Effects on the Neurogenesis Process in MTX-Treated Rats
2.2.1. Dex Restores the BDNF Downregulation Induced by MTX
2.2.2. Dex Alleviates MTX-Induced Oxidative Stress in the Hippocampal Tissues of Rats
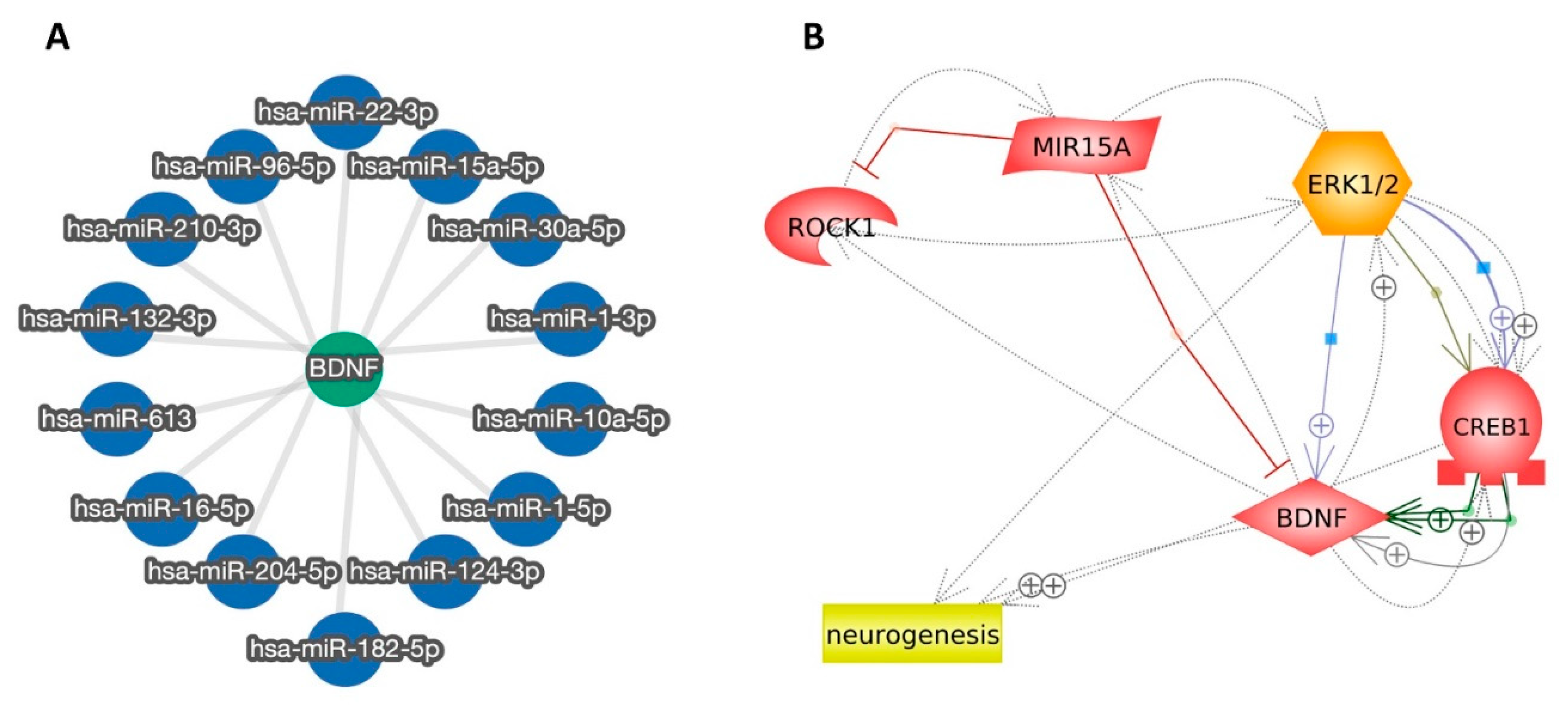
2.2.3. miR-15a Promotes Neurogenesis via Regulating ROCK-1/ERK1/2/CREB/BDNF Transcription Factors: Extensive Bioinformatic Approach
2.2.4. Modulation of miR-15a/ROCK-1/ERK1/2/CREB Signaling Pathway by Dex in MTX-Treated Rats
2.3. Possible Cellular Mechanisms Underpin the Dex Effects on the Neurogenesis Process in MTX-Treated Rats
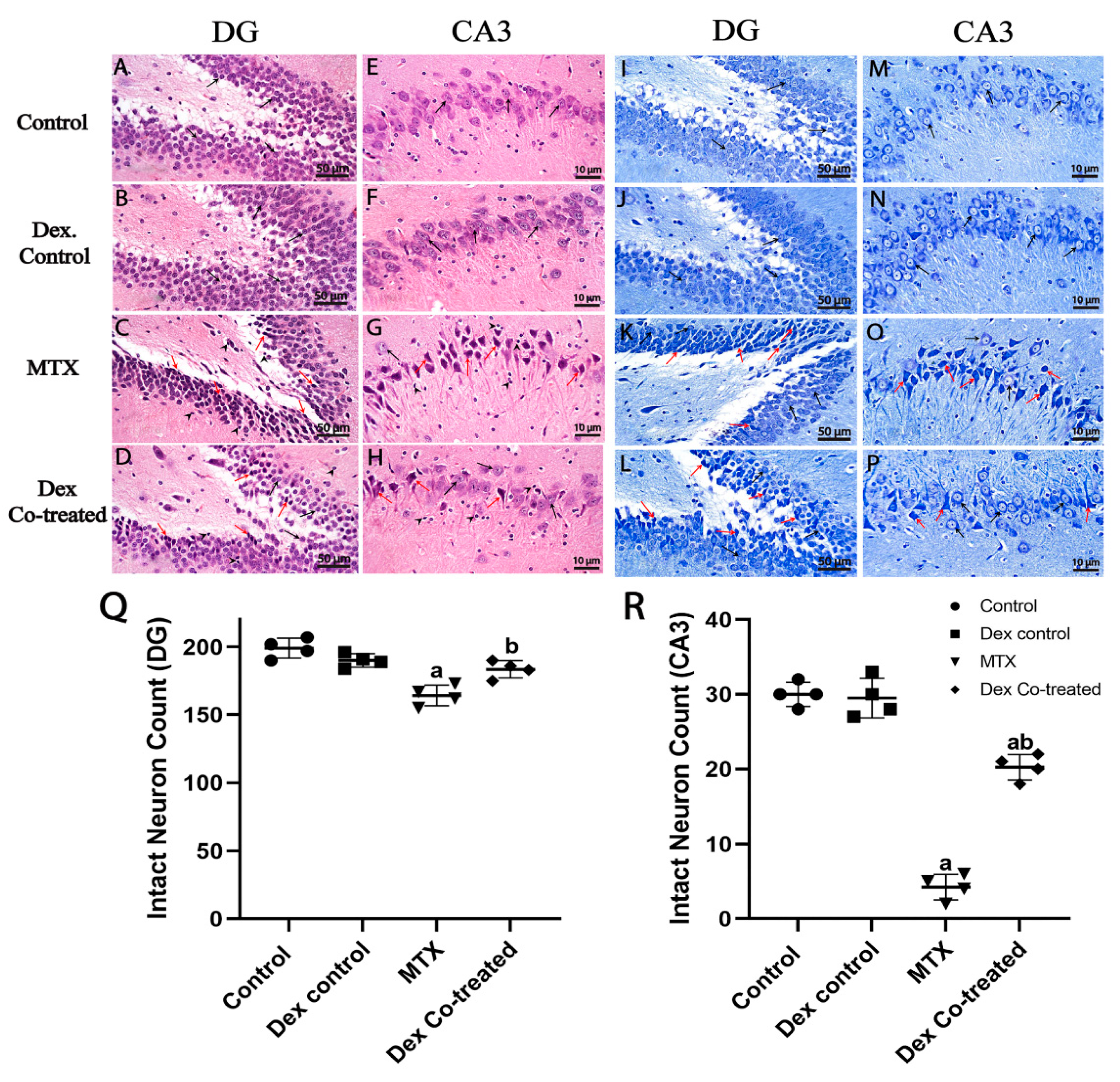
2.3.1. Dex Preserves Hippocampal Neurons from MTX-Induced Neuronal Injury
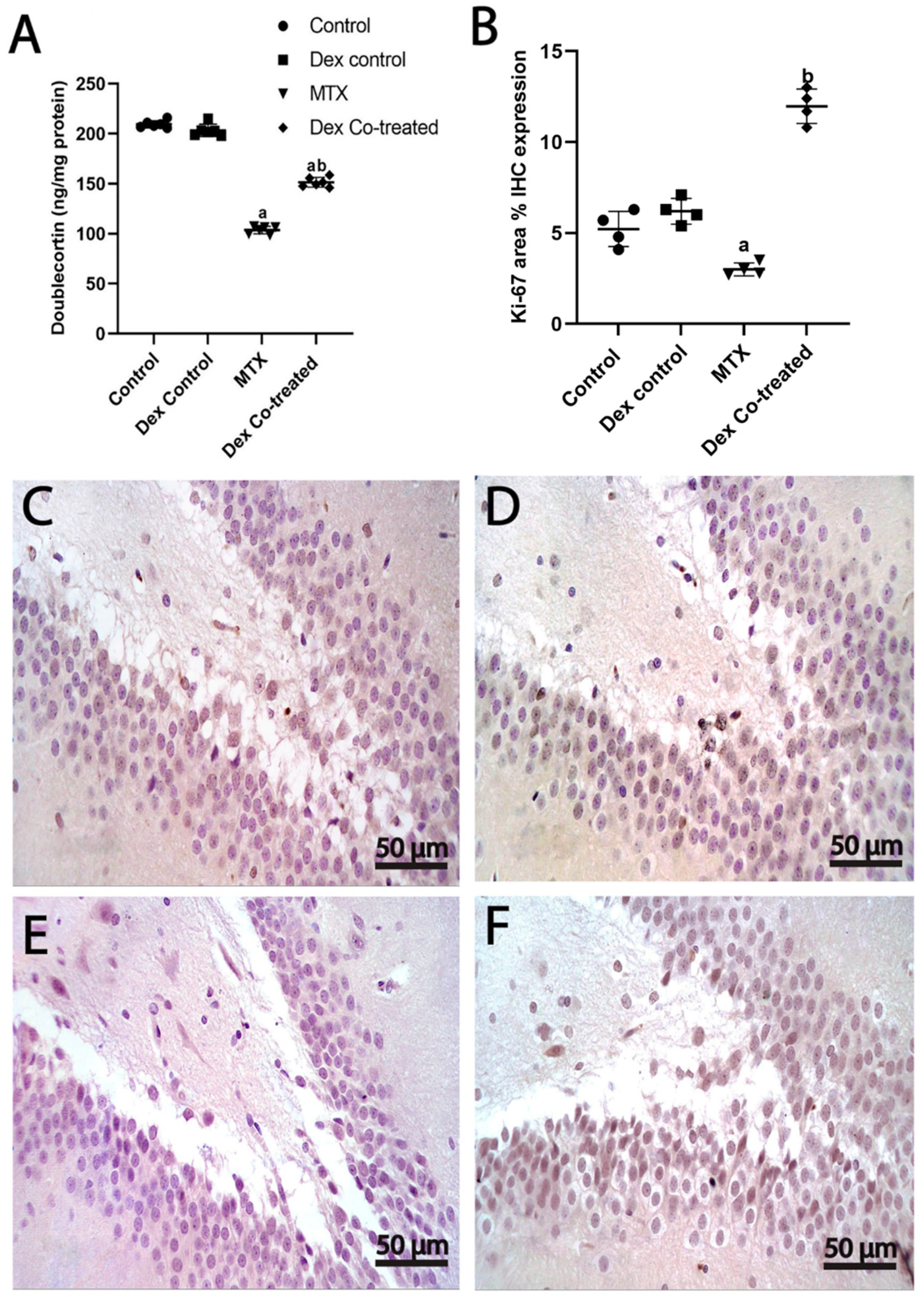
2.3.2. Dex Increases the Number of Immature Neurons in the Hippocampus of Rats Treated with MTX
2.3.3. Dex Promotes Cell Proliferation in the Hippocampus of Rats Treated with MTX
3. Discussion
Limitations of the Study
4. Materials and Methods
4.1. Experimental Animals
4.2. Drugs and Chemicals
4.3. Experimental Design
4.4. Behavioral Tests
4.4.1. Novel Object Recognition (NOR) Test
4.4.2. Morris Water Maze (MWM) Test
4.5. Bioinformatic Analysis
4.6. Biochemical Assays
4.6.1. Colorimetric Assay
4.6.2. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6.3. Reverse Transcriptase-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.6.4. Western Blotting
4.7. Histology and Immunohistochemistry
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALL | Acute lymphoblastic leukemia |
| BDNF | Brain-derived neurotrophic factor |
| CA | Cornu Ammonis |
| CRE | cAMP response element |
| CREB | cAMP-responsive element binding protein |
| DCX | Doublecortin |
| Dex | Dexmedetomidine |
| DG | Dentate gyrus |
| DI | Discrimination Index |
| ELISA | Enzyme-linked immunosorbent essay |
| ERK1/2 | Extracellular signal-regulated kinase ½ |
| FFPE | Formalin-fixed, paraffin-embedded |
| HMDD | Human microRNA Disease Database |
| LCV | Leucovorin |
| MDA | Malondialdehyde |
| miR-15a | MicroRNA-15a |
| MTX | Methotrexate |
| MWM | Morris water maze |
| NOR | Novel object recognition |
| PI | Preference index |
| ROCK-1 | Rho-associated protein kinase 1 |
| RT-qPCR | Reverse transcriptase-quantitative polymerase chain reaction |
| SGZ | Subgranular zone |
| SOD | Superoxide dismutase |
| SVZ | Subventricular zone |
| TrkB | Tropomyosin receptor kinase B |
References
- Mager, D.R. Methotrexate. Home Healthc. Now 2015, 33, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.T.; Choi, S.K. Mechanisms and implications of dual-acting methotrexate in folate-targeted nanotherapeutic delivery. Int. J. Mol. Sci. 2015, 16, 1772–1790. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.B.; DeAngelis, L.M. Cancer-treatment-induced neurotoxicity—Focus on newer treatments. Nat. Rev. Clin. Oncol. 2016, 13, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Board, P.P.T.E. Childhood acute lymphoblastic leukemia treatment (PDQ®). In PDQ Cancer Information Summaries; National Cancer Institute (US): Rockville, MD, USA, 2021. [Google Scholar]
- Geurten, C.; Forget, P.; Leroy, P.; Barrea, C.; Hoyoux, C. Methotrexate-induced Acute Myelopathy in a Teenager With High-risk Acute Lymphoblastic Leukemia. J Pediatr Hematol Oncol 2020, 42, e262–e264. [Google Scholar] [CrossRef]
- Liao, P.F.; Chan, C.H.; Peng, T.R.; Kao, W.Y. High-Dose Aminophylline for Methotrexate-Induced Neurotoxicity in a Patient With Brain Metastasis. Am. J. Ther. 2021, 28, e729–e730. [Google Scholar] [CrossRef] [PubMed]
- Youssef, A.A.; Raafat, T.A.; Madney, Y. Child with acute methotrexate related neurotoxicity: Can diffusion weighted MRI help? Egypt. J. Radiol. Nucl. Med. 2015, 46, 1149–1153. [Google Scholar] [CrossRef][Green Version]
- Rijmenams, I.; Moechars, D.; Uyttebroeck, A.; Radwan, A.; Blommaert, J.; Deprez, S.; Sunaert, S.; Segers, H.; Gillebert, C.R.; Lemiere, J.; et al. Age- and Intravenous Methotrexate-Associated Leukoencephalopathy and Its Neurological Impact in Pediatric Patients with Lymphoblastic Leukemia. Cancers 2021, 13, 1939. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Maxwell, R.R.; Wolf, A.J.; Spira, M.; Gulinello, M.E.; Cole, P.D. Methotrexate causes persistent deficits in memory and executive function in a juvenile animal model. Neuropharmacology 2018, 139, 76–84. [Google Scholar] [CrossRef]
- Shuper, A.; Stark, B.; Kornreich, L.; Cohen, I.J.; Aviner, S.; Steinmetz, A.; Stein, J.; Goshen, Y.; Yaniv, I. Methotrexate treatment protocols and the central nervous system: Significant cure with significant neurotoxicity. J. Child Neurol. 2000, 15, 573–580. [Google Scholar] [CrossRef]
- Ding, J.; Li, Q.Y.; Wang, X.; Sun, C.H.; Lu, C.Z.; Xiao, B.G. Fasudil protects hippocampal neurons against hypoxia-reoxygenation injury by suppressing microglial inflammatory responses in mice. J. Neurochem. 2010, 114, 1619–1629. [Google Scholar] [CrossRef]
- Yang, M.; Kim, J.S.; Kim, J.; Kim, S.H.; Kim, J.C.; Kim, J.; Wang, H.; Shin, T.; Moon, C. Neurotoxicity of methotrexate to hippocampal cells in vivo and in vitro. Biochem. Pharmacol. 2011, 82, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Sirichoat, A.; Krutsri, S.; Suwannakot, K.; Aranarochana, A.; Chaisawang, P.; Pannangrong, W.; Wigmore, P.; Welbat, J.U. Melatonin protects against methotrexate-induced memory deficit and hippocampal neurogenesis impairment in a rat model. Biochem. Pharmacol. 2019, 163, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Naewla, S.; Sirichoat, A.; Pannangrong, W.; Chaisawang, P.; Wigmore, P.; Welbat, J.U. Hesperidin Alleviates Methotrexate-Induced Memory Deficits via Hippocampal Neurogenesis in Adult Rats. Nutrients 2019, 11, 936. [Google Scholar] [CrossRef] [PubMed]
- Abrous, D.N.; Wojtowicz, J.M. Interaction between neurogenesis and hippocampal memory system: New vistas. Cold Spring Harb. Perspect. Biol. 2015, 7, a018952. [Google Scholar] [CrossRef]
- Begega, A.; Alvarez-Suarez, P.; Sampedro-Piquero, P.; Cuesta, M. Effects of physical activity on the cerebral networks. In Physical Activity and the Aging Brain; Elsevier: Amsterdam, The Netherlands, 2017; Volume 25, pp. 3–11. [Google Scholar]
- Seigers, R.; Schagen, S.B.; Beerling, W.; Boogerd, W.; van Tellingen, O.; van Dam, F.S.; Koolhaas, J.M.; Buwalda, B. Long-lasting suppression of hippocampal cell proliferation and impaired cognitive performance by methotrexate in the rat. Behav. Brain Res. 2008, 186, 168–175. [Google Scholar] [CrossRef]
- Ortega-Martínez, S. A new perspective on the role of the CREB family of transcription factors in memory consolidation via adult hippocampal neurogenesis. Front. Mol. Neurosci. 2015, 8, 46. [Google Scholar] [CrossRef]
- Waterhouse, E.G.; An, J.J.; Orefice, L.L.; Baydyuk, M.; Liao, G.-Y.; Zheng, K.; Lu, B.; Xu, B. BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. J. Neurosci. 2012, 32, 14318–14330. [Google Scholar] [CrossRef]
- Sun, J.; Nan, G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target (Review). Int. J. Mol. Med. 2017, 39, 1338–1346. [Google Scholar] [CrossRef]
- Delghandi, M.P.; Johannessen, M.; Moens, U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell. Signal. 2005, 17, 1343–1351. [Google Scholar] [CrossRef]
- Catlow, B.J.; Jalloh, A.; Sanchez-Ramos, J. Hippocampal neurogenesis: Effects of psychedelic drugs. In Neuropathology of Drug Addictions and Substance Misuse; Elsevier: Amsterdam, The Netherlands, 2016; pp. 821–831. [Google Scholar]
- Stappert, L.; Klaus, F.; Brustle, O. MicroRNAs Engage in Complex Circuits Regulating Adult Neurogenesis. Front. Neurosci. 2018, 12, 707. [Google Scholar] [CrossRef]
- Hébert, S.S.; Papadopoulou, A.S.; Smith, P.; Galas, M.-C.; Planel, E.; Silahtaroglu, A.N.; Sergeant, N.; Buée, L.; De Strooper, B. Genetic ablation of Dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum. Mol. Genet. 2010, 19, 3959–3969. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Kino, Y.; Niida, S. MicroRNA-Seq Data Analysis Pipeline to Identify Blood Biomarkers for Alzheimer’s Disease from Public Data. Biomark. Insights 2015, 10, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Pan, J.; Liu, W.; Li, Y.; Li, F.; Liu, M. MicroRNA-15a-5p serves as a potential biomarker and regulates the viability and apoptosis of hippocampus neuron in children with temporal lobe epilepsy. Diagn. Pathol. 2020, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, S.-W.; Xi-Ling, L.; Yu, F.-Y.; Cong, H.-M. Knockdown of long non-coding RNA TUG1 depresses apoptosis of hippocampal neurons in Alzheimer’s disease by elevating microRNA-15a and repressing ROCK1 expression. Inflamm. Res. 2020, 69, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jiang, Q.; Shi, K.; Luo, H.; Yang, Y.; Xu, C. RhoA modulates functional and physical interaction between ROCK1 and Erk1/2 in selenite-induced apoptosis of leukaemia cells. Cell Death Dis. 2013, 4, e708. [Google Scholar] [CrossRef] [PubMed]
- Bhana, N.; Goa, K.L.; McClellan, K.J. Dexmedetomidine. Drugs 2000, 59, 263–268; discussion 269–270. [Google Scholar] [CrossRef]
- Tu, Y.; Liang, Y.; Xiao, Y.; Lv, J.; Guan, R.; Xiao, F.; Xie, Y.; Xiao, Q. Dexmedetomidine attenuates the neurotoxicity of propofol toward primary hippocampal neurons in vitro via Erk1/2/CREB/BDNF signaling pathways. Drug Des. Dev. Ther. 2019, 13, 695–706. [Google Scholar] [CrossRef]
- Sifringer, M.; von Haefen, C.; Krain, M.; Paeschke, N.; Bendix, I.; Buhrer, C.; Spies, C.D.; Endesfelder, S. Neuroprotective effect of dexmedetomidine on hyperoxia-induced toxicity in the neonatal rat brain. Oxidative Med. Cell. Longev. 2015, 2015, 530371. [Google Scholar] [CrossRef]
- Unchiti, K.; Leurcharusmee, P.; Samerchua, A.; Pipanmekaporn, T.; Chattipakorn, N.; Chattipakorn, S.C. The potential role of dexmedetomidine on neuroprotection and its possible mechanisms: Evidence from in vitro and in vivo studies. Eur. J. Neurosci. 2021, 54, 7006–7047. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, Q.; Wu, Z.; Xue, H.; Liu, B.; Zhao, P. Dexmedetomidine Promotes Hippocampal Neurogenesis and Improves Spatial Learning and Memory in Neonatal Rats. Drug Des. Dev. Ther. 2019, 13, 4439–4449. [Google Scholar] [CrossRef]
- Hu, S.; Zhao, J.; Wang, W.; Liu, Y.; Wu, H.; Chen, C.; Yu, L.; Gui, J. Dexmedetomidine increases acetylation level of histone through ERK1/2 pathway in dopamine neuron. Hum. Exp. Toxicol. 2017, 36, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Niu, J.; Zhou, B.; Deng, W.; Deng, F.; Zhou, Z.; Xu, G. Dexmedetomidine attenuates cisplatin-induced cognitive impairment by modulating miR-429-3p expression in rats. 3 Biotech 2020, 10, 244. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Li, C.; Ding, H.; Ye, J.; Xia, Z. Dexmedetomidine reverses MTX-induced neurotoxicity and inflammation in hippocampal HT22 cell lines via NCOA4-mediated ferritinophagy. Aging 2021, 13, 6182–6193. [Google Scholar] [CrossRef] [PubMed]
- Moloudizargari, M.; Hekmatirad, S.; Mofarahe, Z.S.; Asghari, M.H. Exosomal microRNA panels as biomarkers for hematological malignancies. Curr. Probl. Cancer 2021, 45, 100726. [Google Scholar] [CrossRef]
- Zhao, B.M.; Cheng, F.H.; Cai, L. Long noncoding RNA AFAP1-AS1 promoted osteosarcoma proliferation and invasion via upregulating BDNF. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8624. [Google Scholar] [CrossRef]
- Castillo, R.L.; Ibacache, M.; Cortinez, I.; Carrasco-Pozo, C.; Farias, J.G.; Carrasco, R.A.; Vargas-Errazuriz, P.; Ramos, D.; Benavente, R.; Torres, D.H.; et al. Dexmedetomidine Improves Cardiovascular and Ventilatory Outcomes in Critically Ill Patients: Basic and Clinical Approaches. Front. Pharmacol. 2019, 10, 1641. [Google Scholar] [CrossRef]
- Sritawan, N.; Prajit, R.; Chaisawang, P.; Sirichoat, A.; Pannangrong, W.; Wigmore, P.; Welbat, J.U. Metformin alleviates memory and hippocampal neurogenesis decline induced by methotrexate chemotherapy in a rat model. Biomed. Pharmacother. 2020, 131, 110651. [Google Scholar] [CrossRef]
- Antunes, N.L.; Souweidane, M.M.; Lis, E.; Rosenblum, M.K.; Steinherz, P.G. Methotrexate leukoencephalopathy presenting as Klüver-Bucy syndrome and uncinate seizures. Pediatr. Neurol. 2002, 26, 305–308. [Google Scholar] [CrossRef]
- Lutfy, S.M.O.; Shehata, A.S.A.; Ahmed, S.M.; Hassan, M.Z.M. Toxic Effects of Methotrexate on Cerebellar Cortex. Egypt. J. Hosp. Med. 2022, 89, 4566–4569. [Google Scholar] [CrossRef]
- Uzar, E.; Koyuncuoglu, H.R.; Uz, E.; Yilmaz, H.R.; Kutluhan, S.; Kilbas, S.; Gultekin, F. The activities of antioxidant enzymes and the level of malondialdehyde in cerebellum of rats subjected to methotrexate: Protective effect of caffeic acid phenethyl ester. Mol. Cell. Biochem. 2006, 291, 63–68. [Google Scholar] [CrossRef]
- Yang, M.; Moon, C. Neurotoxicity of cancer chemotherapy. Neural Regen. Res. 2013, 8, 1606. [Google Scholar] [PubMed]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophic Factor and Glucocorticoid Stress in Neurogenesis. Int. J. Mol. Sci. 2017, 18, 2312. [Google Scholar] [CrossRef] [PubMed]
- Volosin, M.; Song, W.; Almeida, R.D.; Kaplan, D.R.; Hempstead, B.L.; Friedman, W.J. Interaction of survival and death signaling in basal forebrain neurons: Roles of neurotrophins and proneurotrophins. J. Neurosci. 2006, 26, 7756–7766. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Kumar, A.; Singh, D. Dietary Flavonoids Interaction with CREB-BDNF Pathway: An Unconventional Approach for Comprehensive Management of Epilepsy. Curr. Neuropharmacol. 2019, 17, 1158–1175. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Sun, Y.; Chen, N.; Chen, S.; Xiu, M.; Zhang, X. Interaction of oxidative stress and BDNF on executive dysfunction in patients with chronic schizophrenia. Psychoneuroendocrinology 2020, 111, 104473. [Google Scholar] [CrossRef]
- Senousy, M.A.; Hanafy, M.E.; Shehata, N.; Rizk, S.M. Erythropoietin and Bacillus Calmette–Guérin Vaccination Mitigate 3-Nitropropionic Acid-Induced Huntington-like Disease in Rats by Modulating the PI3K/Akt/mTOR/P70S6K Pathway and Enhancing the Autophagy. ACS Chem. Neurosci. 2022, 13, 721–732. [Google Scholar] [CrossRef]
- Motawi, T.K.; Al-Kady, R.H.; Abdelraouf, S.M.; Senousy, M.A. Empagliflozin alleviates endoplasmic reticulum stress and augments autophagy in rotenone-induced Parkinson’s disease in rats: Targeting the GRP78/PERK/eIF2alpha/CHOP pathway and miR-211-5p. Chem. Biol. Interact. 2022, 362, 110002. [Google Scholar] [CrossRef]
- Welbat, J.U.; Naewla, S.; Pannangrong, W.; Sirichoat, A.; Aranarochana, A.; Wigmore, P.J.B.P. Neuroprotective effects of hesperidin against methotrexate-induced changes in neurogenesis and oxidative stress in the adult rat. Biochem. Pharmacol. 2020, 178, 114083. [Google Scholar] [CrossRef]
- Chan, J.P.; Cordeira, J.; Calderon, G.A.; Iyer, L.K.; Rios, M. Depletion of central BDNF in mice impedes terminal differentiation of new granule neurons in the adult hippocampus. Mol. Cell. Neurosci. 2008, 39, 372–383. [Google Scholar] [CrossRef]
- Galvão, R.P.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Brain-derived neurotrophic factor signaling does not stimulate subventricular zone neurogenesis in adult mice and rats. J. Neurosci. 2008, 28, 13368–13383. [Google Scholar] [CrossRef]
- Zhang, Z.; Mu, X.; Zhou, X. Dexmedetomidine Alleviates Inflammatory Response and Oxidative Stress Injury of Vascular Smooth Muscle Cell via α2AR/PI3K/AKT Pathway in Intracranial Aneurysm. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Zhou, Y.; Dong, X.; Zhang, L.J.T.T. Dexmedetomidine Can Reduce the Level of Oxidative Stress and Serum miR-10a in Patients with Lung Cancer after Surgery. Thorac. Cardiovasc. Surg. 2022. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, B.; Li, G.; Zhao, H.; Tian, X.; Yu, J.; Yin, Y.; Meng, C. Dexmedetomidine Alleviates Lung Oxidative Stress Injury Induced by Ischemia-Reperfusion in Diabetic Rats via the Nrf2-Sulfiredoxin1 Pathway. Biomed. Res. Int. 2022, 2022, 5584733. [Google Scholar] [CrossRef]
- Hensel, N.; Stockbrügger, I.; Rademacher, S.; Broughton, N.; Brinkmann, H.; Grothe, C.; Claus, P. Bilateral crosstalk of rho-and extracellular-signal-regulated-kinase (ERK) pathways is confined to an unidirectional mode in spinal muscular atrophy (SMA). Cell. Signal. 2014, 26, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Hensel, N.; Rademacher, S.; Claus, P. Chatting with the neighbors: Crosstalk between Rho-kinase (ROCK) and other signaling pathways for treatment of neurological disorders. Front. Neurosci. 2015, 9, 198. [Google Scholar] [CrossRef][Green Version]
- Xu, B.; Ju, Y.; Song, G. Role of p38, ERK1/2, focal adhesion kinase, RhoA/ROCK and cytoskeleton in the adipogenesis of human mesenchymal stem cells. J. Biosci. Bioeng. 2014, 117, 624–631. [Google Scholar] [CrossRef]
- Montminy, M.R.; Bilezikjian, L.M. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature 1987, 328, 175–178. [Google Scholar] [CrossRef]
- Fathi, D.; Abulsoud, A.I.; Saad, M.A.; Nassar, N.N.; Maksimos, M.M.; Rizk, S.M.; Senousy, M.A. Agomelatine attenuates alcohol craving and withdrawal symptoms by modulating the Notch1 signaling pathway in rats. Life Sci. 2021, 284, 119904. [Google Scholar] [CrossRef]
- Zhang, X.-h.; Feng, Z.-h.; Wang, X.-Y. The ROCK pathway inhibitor Y-27632 mitigates hypoxia and oxidative stress-induced injury to retinal Müller cells. Neural Regen. Res. 2018, 13, 549. [Google Scholar]
- Dahmani, S.; Paris, A.; Jannier, V.; Hein, L.; Rouelle, D.; Scholz, J.; Gressens, P.; Mantz, J. Dexmedetomidine Increases Hippocampal Phosphorylated Extracellular Signal–regulated Protein Kinase 1 and 2 Content by an α2-Adrenoceptor–independent Mechanism: Evidence for the Involvement of Imidazoline I1 Receptors. J. Am. Soc. Anesthesiol. 2008, 108, 457–466. [Google Scholar] [CrossRef]
- Sun, T.; Gong, Q.; Wu, Y.; Shen, Z.; Zhang, Y.; Ge, S.; Duan, J.-S. Dexmedetomidine alleviates cardiomyocyte apoptosis and cardiac dysfunction may be associated with inhibition of RhoA/ROCK pathway in mice with myocardial infarction. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wu, Y.; Zhuang, Y.; Zhang, J. Dexmedetomidine reduces dextran sulfate sodium (DSS)-induced NCM460 cell inflammation and barrier damage by inhibiting RhoA/ROCK signaling pathway. Allergol. Immunopathol. 2022, 50, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, E.; Miles, R.M. The CA3 region of the hippocampus: How is it? What is it for? How does it do it? Front. Cell. Neurosci. 2015, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.S.; Dhikav, V. Hippocampus in health and disease: An overview. Ann. Indian Acad. Neurol. 2012, 15, 239–246. [Google Scholar] [CrossRef]
- Seigers, R.; Pourtau, L.; Schagen, S.B.; Van Dam, F.S.; Koolhaas, J.M.; Konsman, J.P.; Buwalda, B. Inhibition of hippocampal cell proliferation by methotrexate in rats is not potentiated by the presence of a tumor. Brain Res. Bull. 2010, 81, 472–476. [Google Scholar] [CrossRef]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process. 2012, 13, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, N.J.; Squire, L.R.; Clark, R.E. Spatial memory, recognition memory, and the hippocampus. Proc. Natl. Acad. Sci. USA 2004, 101, 14515–14520. [Google Scholar] [CrossRef] [PubMed]
- Gehring, T.V.; Luksys, G.; Sandi, C.; Vasilaki, E. Detailed classification of swimming paths in the Morris Water Maze: Multiple strategies within one trial. Sci. Rep. 2015, 5, 14562. [Google Scholar] [CrossRef] [PubMed]
- Kern, F.; Aparicio-Puerta, E.; Li, Y.; Fehlmann, T.; Kehl, T.; Wagner, V.; Ray, K.; Ludwig, N.; Lenhof, H.P.; Meese, E.; et al. miRTargetLink 2.0-interactive miRNA target gene and target pathway networks. Nucleic Acids Res. 2021, 49, W409–W416. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef]
- Culling, C.F.A. Handbook of Histopathological and Histochemical Techniques: Including Museum Techniques; Butterworth-Heinemann: Oxford, UK, 2013. [Google Scholar]
- Abbas, H.; Gad, H.A.; Khattab, M.A.; Mansour, M.J.P. The Tragedy of Alzheimer’s Disease: Towards Better Management via Resveratrol-Loaded Oral Bilosomes. Pharmaceutics 2021, 13, 1635. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Day | Trial 1 | Trial 2 | Trial 3 | Trial 4 |
|---|---|---|---|---|
| Day 1 | N | W | SW | NE |
| Day 2 | SW | N | NE | W |
| Day 3 | NE | SW | W | N |
| Day 4 | Probe trial started at NW | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taha, M.; Eldemerdash, O.M.; Elshaffei, I.M.; Yousef, E.M.; Senousy, M.A. Dexmedetomidine Attenuates Methotrexate-Induced Neurotoxicity and Memory Deficits in Rats through Improving Hippocampal Neurogenesis: The Role of miR-15a/ROCK-1/ERK1/2/CREB/BDNF Pathway Modulation. Int. J. Mol. Sci. 2023, 24, 766. https://doi.org/10.3390/ijms24010766
Taha M, Eldemerdash OM, Elshaffei IM, Yousef EM, Senousy MA. Dexmedetomidine Attenuates Methotrexate-Induced Neurotoxicity and Memory Deficits in Rats through Improving Hippocampal Neurogenesis: The Role of miR-15a/ROCK-1/ERK1/2/CREB/BDNF Pathway Modulation. International Journal of Molecular Sciences. 2023; 24(1):766. https://doi.org/10.3390/ijms24010766
Chicago/Turabian StyleTaha, Mohamed, Omar Mohsen Eldemerdash, Ismail Mohamed Elshaffei, Einas Mohamed Yousef, and Mahmoud A. Senousy. 2023. "Dexmedetomidine Attenuates Methotrexate-Induced Neurotoxicity and Memory Deficits in Rats through Improving Hippocampal Neurogenesis: The Role of miR-15a/ROCK-1/ERK1/2/CREB/BDNF Pathway Modulation" International Journal of Molecular Sciences 24, no. 1: 766. https://doi.org/10.3390/ijms24010766
APA StyleTaha, M., Eldemerdash, O. M., Elshaffei, I. M., Yousef, E. M., & Senousy, M. A. (2023). Dexmedetomidine Attenuates Methotrexate-Induced Neurotoxicity and Memory Deficits in Rats through Improving Hippocampal Neurogenesis: The Role of miR-15a/ROCK-1/ERK1/2/CREB/BDNF Pathway Modulation. International Journal of Molecular Sciences, 24(1), 766. https://doi.org/10.3390/ijms24010766