
Bone Marrow Aplasia and Neutropenic Fever Following Azathioprine Dose Escalation in a TPMT-Deficient Patient with Crohn’s Disease and Psoriatic Arthritis—A CARE–Compliant Case

 ,
,
Abstract
1. Introduction
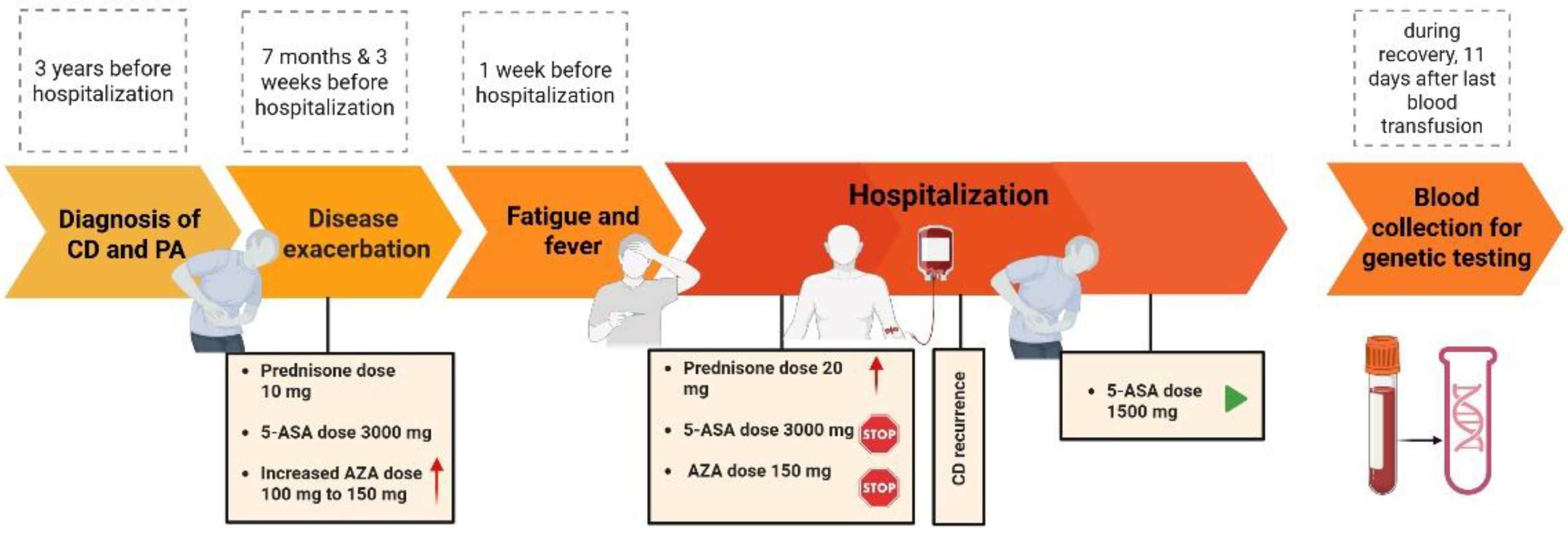
2. Case Description
3. Materials and Methods
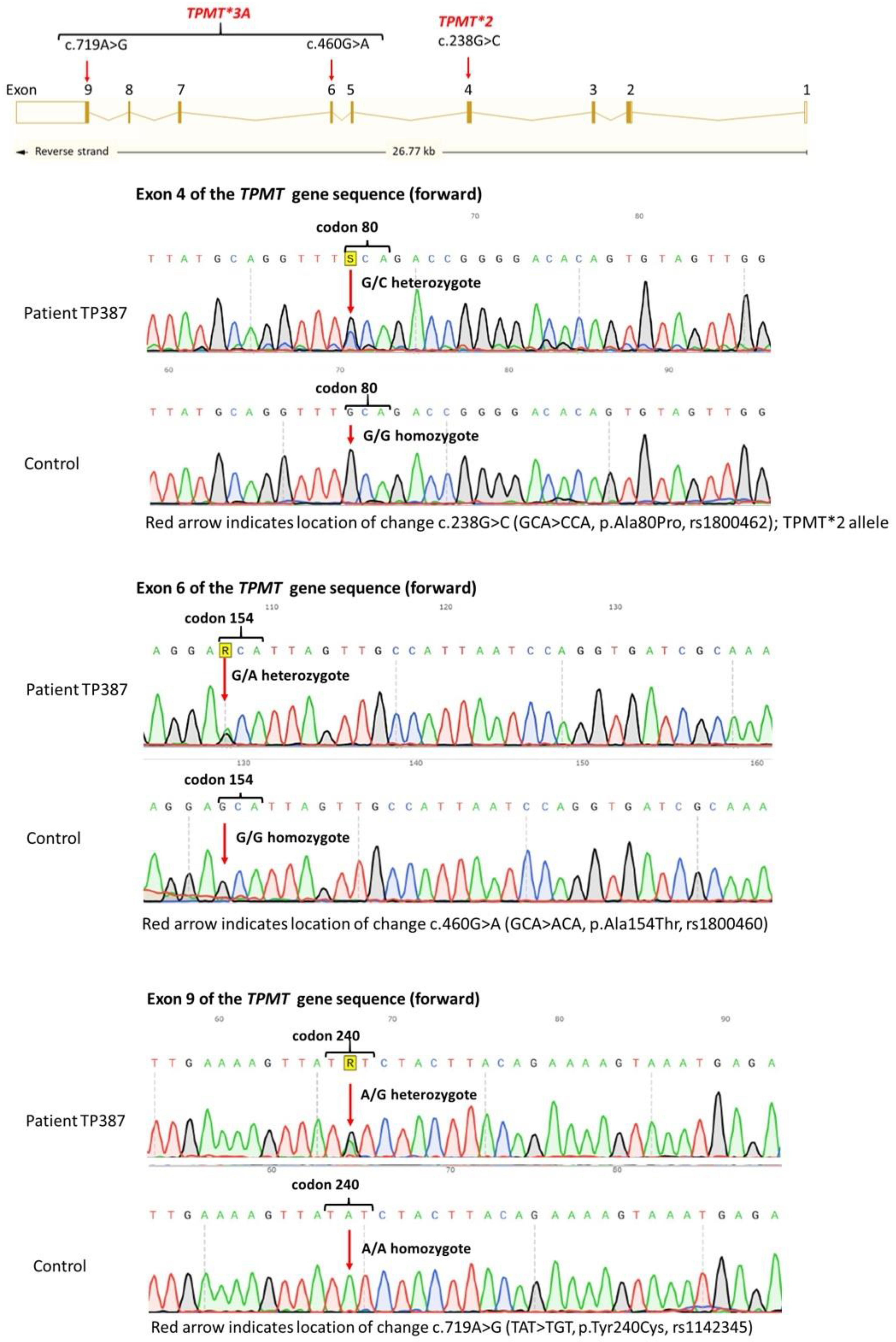
4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ARD | adverse drug reaction |
| 5-ASA | mesalazine |
| AZA | azathioprine |
| DNA-TG | DNA-incorporated thioguanine |
| IBD | inflammatory bowel diseases |
| 6-MP | 6-mercaptopurine |
| NUDT15 | nucleoside diphosphate-linked moiety X-type motif 15 |
| TPMT | thiopurine methyltransferase |
References
- Costantino, G.; Furfaro, F.; Belvedere, A.; Alibrandi, A.; Fries, W. Thiopurine treatment in inflammatory bowel disease: Response predictors, safety, and withdrawal in follow-up. J. Crohns Colitis 2012, 6, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gao, X.; Ding, L.; Liu, H.; Wang, X.; Chen, B.; Bi, H.; Xiao, Y.; Cheng, P.; Zhao, L.; et al. Prospective Evaluation of Pharmacogenomics and Metabolite Measurements upon Azathioprine Therapy in Inflammatory Bowel Disease: An Observational Study. Medicine 2016, 95, e3326. [Google Scholar]
- Otterness, D.; Szumlanski, C.; Lennard, L.; Klemetsdal, B.; Aarbakke, J.; Park-Hah, J.O.; Iven, H.; Schmiegelow, K.; Branum, E.; O’Brien, J.; et al. Human thiopurine methyltransferase pharmacogenetics: Gene sequence polymorphisms. Clin. Pharmacol. Ther. 1997, 62, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Weinshilboum, R.; Sladek, S. Mercaptopurine pharmacogenetics: Monogenic inheritance of erythrocyte thiopurine methyltransferase. Am. J. Hum. Genet. 1980, 32, 651–662. [Google Scholar]
- Black, A.; McLeod, H.; Capell, H.; Powrie, R.; Matowe, L.; Pritchard, S.; Collie-Duguid, E.S.; Reid, D.M. Thiopurine methyltransferase genotype predicts therapy-limiting severe toxicity from azathioprine. Ann. Intern. Med. 1998, 129, 716–718. [Google Scholar] [CrossRef]
- Lennard, L.; Van Loon, J.; Weinshilboum, R. Pharmacogenetics of acute azathioprine toxicity: Relationship to thiopurine methyltransferase genetic polymorphism. Clin. Pharmacol. Ther. 1989, 46, 149–154. [Google Scholar] [CrossRef]
- Kakuta, Y.; Kawai, Y.; Okamoto, D.; Takagawa, T.; Ikeya, K.; Sakuraba, H.; Nishida, A.; Nakagawa, S.; Miura, M.; Toyonaga, T.; et al. NUDT15 codon 139 is the best pharmacogenetic marker for predicting thiopurine-induced severe adverse events in Japanese patients with inflammatory bowel disease: A multicenter study. J. Gastroenterol. 2018, 53, 1065–1078. [Google Scholar] [CrossRef]
- Relling, M.; Schwab, M.; Whirl-Carrillo, M.; Suarez-Kurtz, G.; Pui, C.; Stein, C.; Moyer, A.M.; Evans, W.E.; Klein, T.E.; Antillon-Klussmann, F.G.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Thiopurine Dosing Based on TPMT and NUDT15 Genotypes: 2018 Update. Clin. Pharmacol. Ther. 2019, 105, 1095–1105. [Google Scholar] [CrossRef]
- Morikubo, H.; Kobayashi, T.; Ozaki, R.; Okabayashi, S.; Kuronuma, S.; Takeuchi, O.; Shiba, T.; Kiyohara, H.; Matsubayashi, M.; Sagami, S.; et al. Differential effects of mesalazine formulations on thiopurine metabolism through thiopurine S-methyltransferase inhibition. J. Gastroenterol. Hepatol. 2021, 36, 2116–2124. [Google Scholar] [CrossRef]
- Szumlanski, C.L.; Weinshilboum, R.M. Sulphasalazine inhibition of thiopurine methyltransferase: Possible mechanism for interaction with 6-mercaptopurine and azathioprine. Br. J. Clin. Pharmacol. 1995, 39, 456–459. [Google Scholar] [CrossRef]
- Skrzypczak-Zielinska, M.; Borun, P.; Milanowska, K.; Jakubowska-Burek, L.; Zakerska, O.; Dobrowolska-Zachwieja, A.; Pławski, A.; Froster, U.G.; Szalata, M.; Słomski, R. High-resolution melting analysis of the TPMT gene: A study in the Polish population. Genet. Test. Mol. Biomark. 2013, 17, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Schäffeler, E.; Marx, C.; Fischer, C.; Lang, T.; Behrens, C.; Gregor, M.; Eichelbaum, M.; Zanger, U.M.; Kaskas, A.B. Azathioprine therapy and adverse drug reactions in patients with inflammatory bowel disease: Impact of thiopurine S-methyltransferase polymorphism. Pharmacogenetics 2002, 12, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Barnhoorn, M.C.; Storm, B.N.; de Boer, N.K.; van der Voorn, M. Azathioprine-induced Myelotoxicity After Switching Mesalazine Compound. Inflamm. Bowel Dis. 2021, 27, e114–e115. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, H.; Agarwal, A.; Lim, M. A case of azathioprine-induced aplastic anemia. Int. J. Lab. Hematol. 2022, 44, 1015–1016. [Google Scholar] [CrossRef]
- Yeter, K.; Afkhami, M.; Brynes, R.; Quismorio, F., Jr. Aplastic anemia secondary to azathioprine in systemic lupus erythematosus: Report of a case with normal thiopurine S-methyltransferase enzyme activity and review of the literature. Lupus 2013, 22, 1526–1528. [Google Scholar] [CrossRef]
- Zarca, K.; Chansavang, A.; Loriot, M.A.; Durand-Zaleski, I.; Pallet, N. Cost-effectiveness analysis of pretreatment screening for NUDT15 defective alleles. Pharmacogenet Genom. 2020, 30, 175–183. [Google Scholar] [CrossRef]
- Eder, P.; Łodyga, M.; Łykowska-Szuber, L.; Bartnik, W.; Durlik, M.; Gonciarz, M.; Kłopocka, M.; Linke, K.; Małecka-Panas, E.; Radwan, P.; et al. [Guidelines for the management of ulcerative colitis. Recommendations of the Working Group of the Polish National Consultant in Gastroenterology and the Polish Society of Gastroenterology]. Prz. Gastroenterol. 2013, 8, 1–20. [Google Scholar] [CrossRef]
- Eder, P.; Łodyga, M.; Gawron-Kiszka, M.; Dobrowolska, A.; Gonciarz, M.; Hartleb, M.; Kłopocka, M.; Małecka-Wojciesko, E.; Radwan, P.; Reguła, J.; et al. Guidelines for the management of ulcerative colitis. Recommendations of the Polish Society of Gastroenterology and the Polish National Consultant in Gastroenterology. Prz. Gastroenterol. 2023, 18, 1–42. [Google Scholar] [CrossRef]
- Bernstein, C.N.; Eliakim, A.; Fedail, S.; Fried, M.; Gearry, R.; Goh, K.L.; Hamid, S.; Khan, A.G.; Khalif, I.; Ng, S.C.; et al. World Gastroenterology Organisation Global Guidelines Inflammatory Bowel Disease: Update August 2015. J. Clin. Gastroenterol. 2016, 50, 803–818. [Google Scholar] [CrossRef]
- Bayoumy, A.B.; Ansari, A.R.; Mulder, C.J.J.; Schmiegelow, K.; Florin, T.; De Boer, N.K.H. Innovating Thiopurine Therapeutic Drug Monitoring: A Systematic Review and Meta-Analysis on DNA-Thioguanine Nucleotides (DNA-TG) as an Inclusive Biomarker in Thiopurine Therapy. Clin. Pharmacokinet. 2024, 63, 1089–1109. [Google Scholar] [CrossRef]
- Singh, S.; Loftus, E.V., Jr.; Limketkai, B.N.; Haydek, J.P.; Agrawal, M.; Scott, F.I.; Ananthakrishnan, A.N. AGA Living Clinical Practice Guideline on Pharmacological Management of Moderate-to-Severe Ulcerative Colitis. Gastroenterology 2024, 167, 1307–1343. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Primer Sequence (5′→3′) | Chromosome Coordinates GRCh38/hg38 | Amplicon (bp) | |
|---|---|---|---|
| Exon 1 | F: CAAAGCACAACTGTAAGCGACT R: GAAAGACCCAGCTAGCAAAGAC | chr13:48037494 + 48038124 | 631 |
| Exon 2 | F: CGGCCTTCCAAAAGATTACA R: TGATCTAATCACCTCCCAAGG | chr13:48040685 + 48041309 | 625 |
| Exon 3 | F: AAGCAAATGCAAAGCATCAC R: GGCTGAAAGAGTGGGGGATA | chr13:48045510 + 48045959 | 450 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wroński, K.; Holecki, M.T.; Boguszewska, N.; Skrzypczak-Zielińska, M.; Chudek, J.T. Bone Marrow Aplasia and Neutropenic Fever Following Azathioprine Dose Escalation in a TPMT-Deficient Patient with Crohn’s Disease and Psoriatic Arthritis—A CARE–Compliant Case. Clin. Pract. 2025, 15, 114. https://doi.org/10.3390/clinpract15060114
Wroński K, Holecki MT, Boguszewska N, Skrzypczak-Zielińska M, Chudek JT. Bone Marrow Aplasia and Neutropenic Fever Following Azathioprine Dose Escalation in a TPMT-Deficient Patient with Crohn’s Disease and Psoriatic Arthritis—A CARE–Compliant Case. Clinics and Practice. 2025; 15(6):114. https://doi.org/10.3390/clinpract15060114
Chicago/Turabian StyleWroński, Krzysztof, Michał Tadeusz Holecki, Natalia Boguszewska, Marzena Skrzypczak-Zielińska, and Jerzy Tadeusz Chudek. 2025. "Bone Marrow Aplasia and Neutropenic Fever Following Azathioprine Dose Escalation in a TPMT-Deficient Patient with Crohn’s Disease and Psoriatic Arthritis—A CARE–Compliant Case" Clinics and Practice 15, no. 6: 114. https://doi.org/10.3390/clinpract15060114
APA StyleWroński, K., Holecki, M. T., Boguszewska, N., Skrzypczak-Zielińska, M., & Chudek, J. T. (2025). Bone Marrow Aplasia and Neutropenic Fever Following Azathioprine Dose Escalation in a TPMT-Deficient Patient with Crohn’s Disease and Psoriatic Arthritis—A CARE–Compliant Case. Clinics and Practice, 15(6), 114. https://doi.org/10.3390/clinpract15060114