Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) is closely associated with obesity and other cardiometabolic risk factors. MASLD has rapidly become the most common cause of liver disease worldwide, currently affecting 38% of the global population. Excess weight causes chronic inflammation and the activation of different pathways involved in liver damage. MASLD can progress from simple steatosis to steatohepatitis, giving way to its inflammatory component, metabolic dysfunction-associated steatohepatitis (MASH), previously recognized as non-alcoholic steatosis hepatitis (NASH). Chronic hepatitis C virus (HCV) infection remains a significant challenge to liver health as it triggers hepatic inflammation, metabolic disruption, and hepatic steatosis. The convergence of MASLD and chronic HCV infection can significantly alter the course of liver disease and accelerate the progression to severe liver damage. Currently, HCV treatment has a high cure rate. However, in patients who achieve a sustained virological response after treatment with direct-acting antivirals, weight gain, and excessive calorie intake may contribute to increased liver steatosis and a higher risk of liver disease progression. Therefore, the effective clinical and nutritional management of HCV patients, both before and after viral eradication, is crucial to reducing the risk of death from hepatocellular carcinoma. Understanding the complex interactions between MASLD and HCV infection is crucial for managing these patients appropriately. Herein, host and viral mechanisms inducing liver damage during the coexistence of MASLD and HCV infection are described, and their therapeutic and dietary management are discussed.
1. Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD) has become the most common chronic liver disease worldwide because of the obesity epidemic. According to recent estimations, MASLD affects nearly one-third of the global population [1]. MASLD commonly progresses from simple steatosis to metabolic dysfunction-associated steatohepatitis (MASH) due to inflammatory components [2]. Several factors, including environmental, metabolic, immune, genetic, and epigenetic factors, can affect the progression of MASLD to severe forms of the disease, such as liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) [3]. Hepatitis C (HCV) is a hepatotropic virus that disrupts hepatic metabolism, causing progressive liver damage [4]. Currently, chronic hepatitis C ranks among the leading causes of liver transplantation, and it is estimated that 242,000 people worldwide die each year from complications related to hepatitis C [5]. HCV itself can generate hepatic steatosis through mechanisms dependent on viral genotype. Both MASLD and chronic hepatitis C can independently contribute to liver-related complications, and their coexistence may have additive effects on liver health, especially by accelerating liver damage. In this sense, patients with untreated HCV had a prevalence of MASLD of 55%, spanning from 40 to 86% based on the regionality of the metabolic syndrome and HCV genotype [6]. Furthermore, the occurrence of liver steatosis before and after treatment with direct-acting antiviral agents (DAAs) is related to a lack of an improvement in liver fibrosis and an increased risk of liver cancer [7]. Therefore, the adequate clinical and nutritional management of these patients is essential for preventing deaths due to HCC.
A synergistic interaction between the metabolic components associated with HCV infection and MASLD that may accelerate the development of liver damage and HCC has been documented [8]. Thus, the early diagnosis of MASLD in HCV-infected patients would allow for prompt clinical, therapeutic, and lifestyle interventions to prevent further disease severity. This review examines the mechanisms contributing to liver injury during the convergence of metabolic and viral-induced steatosis, and the role of therapeutic and dietary management is discussed.
2. Effect of Chronic Inflammation in MASLD and HCV Infection
Obesity plays the most crucial role in the onset and progression of simple steatosis and MASH. When the capacity of adipose tissue to store fat is surpassed, hepatocytes begin storing excess lipids, primarily triglycerides. This ectopic fat storage can result in simple steatosis and inflammation. For its part, HCV can induce hepatic inflammation; thus, an intricate hepatic proinflammatory environment prevails during the coexistence of MASLD and chronic HCV infection (Figure 1). Hepatic inflammation is a complex process that protects hepatocytes from injury, favors liver repair, and establishes homeostasis [9]. However, prolonged inflammation leads to hepatocyte death, liver damage, and a decline in liver function [10,11]. Also, chronic inflammation contributes to metabolic disorders and progression from hepatic steatosis to MASH, fibrosis, cirrhosis, and HCC [10].

Figure 1.
Mechanisms linking MASLD to liver damage during HCV infection. Hypertrophic adipocytes during obesity produce adipokines and other factors that promote intra- and extrahepatic low-grade inflammation. In conjunction with viral proteins, low-grade inflammation accelerates liver damage progression during the coexistence of MASLD+ HCV. The genes involved in MASLD development during HCV infection are highlighted in blue. The arrows with a regular tip represent induction, while the arrows with a blunt tip represent inhibition. The dotted arrow indicates an impaired mechanism effect.
2.1. Adipose-Derived Adipokines Involved in the Onset of Steatosis-Related Liver Damage
Adipose tissue primarily stores fat but can also exert endocrine functions, producing numerous adipokines to regulate metabolic and inflammatory processes [12]. During the progression of excess weight, the adipocyte composition changes by favoring a forward influx of immune cells and overproduction of adipokines [13]. As discussed below, the altered expression pattern of these cytokines is associated with increased liver damage.
2.1.1. Leptin
Leptin is a peptide hormone produced and secreted by adipose tissue. It acts in the hypothalamus to influence food intake, energy expenditure, and fat storage, helping to maintain overall energy homeostasis [14]. Although produced and secreted from adipocytes, leptin acts upon its receptor, LEPR. Dysregulation of leptin production or receptor sensitivity can contribute to metabolic and weight-related disorders [15]. Obese individuals become hyperleptinemic and leptin-resistant due to increased adipogenesis [16]. Circulating leptin in obesity enhances adipocyte and systemic inflammation, up-regulating monocyte chemoattractant protein-1 (MCP-1), also known as CCL2.
Consequently, the infiltration of proinflammatory blood monocyte-derived macrophages and the production of TNF-α, IL-6, IL-12, and IL-1β increases [17]. High leptin levels are found in MASLD patients’ liver biopsies [18]. Leptin has shown a potential dual action in MASLD in vitro models. First, leptin was reported as a protective factor for MASLD, safeguarding hepatocyte cells from steatosis and lipotoxicity by preventing the up-regulation of lipogenesis and increasing fatty acid oxidation [19]. In contrast, leptin plays a unique role in developing hepatic fibrosis by activating hepatic stellar cells (HSC) via PPARgamma inhibition and proinflammatory responses (Figure 1) [20,21]. Elevated leptin levels have been observed in patients infected with HCV genotype 1, correlating with an exacerbation of liver steatosis [22,23]. Thus, leptin may play a distinct role in hepatic health depending on the etiology and stage of liver damage.
2.1.2. Adiponectin
Adiponectin is an adipocyte-specific factor contributing to insulin sensitivity, anti-inflammatory responses, and various metabolic processes, including glucose regulation and fatty acid oxidation [24]. Adiponectin mainly binds to its receptor AdipoR2. AdipoR2 activates 5-AMPK and PPAR-α pathways involved in fatty acid oxidation and inhibition of inflammation [25]. Adiponectin also downregulates the hepatic expression of CD36, thereby decreasing the influx of free fatty acids (FFAs) into the liver. Besides metabolic regulation, adiponectin has antifibrotic action in the liver via downregulating AOX-1, TGF-β, and connective tissue growth factor expression [26]. Adiponectin also has anti-inflammatory action in the liver by suppressing TNF-α and other proinflammatory cytokines and inducing anti-inflammatory cytokines, such as IL-10 [27]. Low adiponectin levels have been reported in obese subjects and patients with hepatic steatosis or MASH [28]. Recently, a significant association between elevated serum adiponectin levels and advanced Child-class liver cirrhosis in patients with HCV infection has been reported. ADIPOR1 mRNA were reduced in chronic HCV-infected patients, but not ADIPOR2 levels, suggesting a pattern of adiponectin resistance [29,30]. The beneficial effect of adiponectin is diminished in both MASLD and HCV patients. In the case of chronic hepatitis C, it is necessary to investigate the mechanisms that induce the downregulation of ADIPOR1 expression.
2.1.3. Tumor Necrosis Factor-Alpha (TNFα)
TNFα is a pleiotropic cytokine involved in diverse processes such as cell proliferation, metabolic activation, and inflammatory response [31]. The ligand of TNFα, the ligand-bound TNR receptor 1 (TNFR1), induces the production of reactive oxygen species (ROS) via the NOX1, thus coordinating the proinflammatory response (Figure 1) [32]. It has been reported that TNFR1 favors the recruitment of caspase-8, which initiates an apoptotic cascade in the liver [33]. TNFα expression increases insulin resistance (IR), IL-6, IL-1, and early MASH in diet-induced non-obese MASLD mice [34]. High levels of TNFα have been found in cirrhotic HCV patients as well as in MASH patients [35,36]. Thus, both etiologies may potentiate liver damage via TNFα activity.
2.1.4. Interleukin-6 (IL-6)
IL-6 is a pleiotropic cytokine expressed in different tissues. However, white adipose tissue is responsible for up to 35% of human IL-6 production [37]. IL-6 expression is highly correlated with obesity, contributing to a low grade of inflammation in this condition. IL-6 has differential functions according to tissue type. In the liver, it induces the acute phase response and infection defense, but aberrant activation of the IL-6 pathway can trigger hepatocyte apoptosis and mediate immune liver damage [38]. Also, IL-6 impairs insulin action through the STAT3-SOCS-3 pathway (Figure 1) [39,40]. High levels of IL-6 are reported in MASLD, MASH, and chronic HCV patients. However, the highest levels are found in MASLD patients, rather than in HCV patients. IL-6 activation results in the excessive stimulation of the IL-6/STAT3 signaling pathway in HCC cells. This process can increase the expression of the tissue inhibitor of metalloproteinases-1 (TIMP-1), driving the conversion of normal liver fibroblasts into carcinoma-associated fibroblasts (CAFs), thereby contributing to liver carcinogenesis [41]. Hence, IL-6 could be the key to rapidly conducting a dual MASLD + HCV state toward HCC development.
3. Immunometabolic Dysregulation Enhances Liver Damage in MASLD and Chronic HCV
Low-grade inflammation in obesity is characterized by metabolic dysfunction that increases the risk for cardiovascular disease, cancer, and other life-threatening conditions, including liver disease [42,43]. The lipotoxic microenvironment in MASLD increases hepatic oxidative stress, leading to T-cell recruitment, persistent proinflammatory response, and subsequent fibrosis and MASH development [43]. On the other hand, HCV is a metabolic regulator virus that, by itself, can modulate liver metabolism with hepatic insulin resistance and subsequent oxidative damage. Thus, the overlap of these two etiologies can exacerbate liver damage (Figure 1).
Insulin Resistance and Oxidative Stress
During weight gain, decreased glucose uptake by insulin-resistant skeletal muscle leads to compensatory hyperinsulinemia. Proinflammatory cytokines such as TNFα and IL-6 produced in hypertrophic adipocytes cause IR as they can stimulate the signaling pathways of the c-Jun amino-terminal kinase (JNK) and the IκB kinase-β (IKK-β)/nuclear factor-κB (NFκB). Once activated, JNK and NFκB phosphorylate the serine kinase insulin receptor substrate-1 (IRS1) and insulin receptor substrate-2 (IRS-2) that block insulin signaling, resulting in the incidence of IR [44]. In parallel, HCV can trigger hepatic and extrahepatic IR via core-protein-promoting IRS-1 degradation in viral genotype 3, while genotype 1 activates the mTOR signaling, reducing IRS1/2 signaling (Figure 1) [45,46]. The presence of IR has been associated with liver fibrosis in chronic HCV and MASLD patients [47]. Hyperinsulinemia and increased levels of insulin growth factors have been shown to promote cell proliferation in chronic inflammatory conditions, such as MASH and chronic HCV infection [48].
On the other hand, ROS are regular products of the mitochondrial respiratory chain. They constitute a highly reactive species produced in membranes and cytosolic and reticular components and are involved in TNFR1 activation [49]. ROS participates in intracellular liver damage, mitochondrial dysfunction, and cell death. Direct measurements in liver tissue from chronic HCV patients revealed an increase in ROS concentrations by two to five orders of magnitude [50]. One study demonstrated that HCV can potentially cause ROS production via the core, E1, E2, NS4B, and NS5A, the core protein’s most potent regulator [51]. They increased oxidative stress in vivo in a large cohort of subjects with MASLD. High oxidative stress is a well-established cause of liver injury due to indiscriminate oxidative biomolecular damage and dysregulated redox signaling [52]. Thus, HCV patients who have excess weight are expected to accelerate the progression of liver damage.
4. Genetic Variants Linked to the Development of MASLD in Hepatitis C Virus Infection
Susceptibility toward MASLD development in chronic HCV patients depends on the host’s genetic background. Genome-wide association studies revealed that many single variants confer susceptibility to steatosis by inducing hepatic fat accumulation [53]. One of the most widely described variants associated with MASLD is the non-synonymous isoleucine-to-methionine substitution at position 148 in the PNPLA3 gene (rs738409). This single nucleotide polymorphism (SNP) leads to the reduced enzyme activity of triglyceride lipase and reduced retinyl ester hydrolysis, resulting in hepatic triglyceride accumulation and hepatic stellate cell activation and fibrogenesis [54]. Chronic HCV patients who carry the GG genotype have a 4.33-fold increased risk of developing hepatic steatosis and a 2.99-fold increased risk of severe fibrosis compared to carriers of other genotypes [55]. Also, the PNPLA3 rs738409 and the HSD17B13 rs72613567TA variants have been associated with more severe liver disease, from mild fibrosis to significant fibrosis, cirrhosis, and HCC in chronic HCV infection [56]. Higher liver triglyceride content is also found in patients with the missense mutation E167K variant (rs58542926) in the TM6SF2 gene, which decreases hepatic lipid secretion [57]. The TM6SF2 E167K variant independently predicts steatosis in chronic HCV patients [58]. MTP is a protein involved in the hepatic lipid release by transferring lipids from the endoplasmic reticulum to the nascent apolipoprotein B and very low-density lipoprotein. The MTP rs1800591 variant confers a 6.72-fold increased risk of hepatic steatosis in chronic HCV genotype 3 patients [59]. These genetic variants are found in critical enzymes involved in lipid metabolism and can promote liver steatosis, resulting in the onset of liver damage (Figure 1).
5. Effect of HCV Genotypes on MASLD
HCV is a genetically heterogeneous virus with one to eight genotypes and over 50 subgenotypes [60]. Each genotype exerts a distinct influence on metabolism during infection, and specific genotypes may favor liver steatosis on their own or in the presence of obesity (Table 1). The development of steatosis in patients with chronic HCV infection ranges between 40–86%, with an average of 55% in all genotypes. The prevalence of liver steatosis in chronic HCV patients is higher than in the general adult population (55% compared to 20–30%) [6]. Genotype 3, also called the “steatogenic genotype”, is associated with a hepatic steatosis prevalence of up to 86% [61]. Patients infected with genotype 3 show more frequent and more severe steatosis with accelerated progression to liver damage and HCC than other genotypes, even without the presence of IR, obesity, or other metabolic risk factors related to MASLD. Genotype 3-induced liver steatosis is proportional to viral load and is resolved after successful viral treatment, indicating a direct cytopathic effect. MASLD related to genotype 3 involves the core protein inhibiting MTP, decreasing hepatic lipid export and triglyceride accumulation [62]. Genotype 3a activates p-Akt, increasing fatty acid synthesis via SREBP-1 and decreasing lipolysis by inhibiting PPARα [63,64].

Table 1.
Disease characteristics of liver steatosis in HCV genotype 3 and non-genotype 3.
On the other hand, genotypes 1, 2, and 4 promote steatosis primarily associated with pre-existing host metabolic risk factors, such as IR and visceral obesity [65]. The activation of proinflammatory pathways in obesity and IR is related to the emergence of steatosis in these patients. Also, increased free radical levels (MDA > 250 nmol/dL) have been significantly correlated with liver fibrosis in patients with an HCV genotype 1 [66]. However, no relationship has been found between genotype 1-viral load and the achievement of a sustained virologic response (SVR) or the extent of liver steatosis [67].
6. Treatment Considerations in Patients with MASLD and HCV Infection
The goal of HCV treatment is to reduce the occurrence of end-stage liver disease and its complications. Treatment success is assessed by SVR, which is defined as undetectable HCV RNA in the blood several months after treatment. Initially, HCV was treated with interferon (IFN)-based regimens. Nonetheless, the presence of steatosis and metabolic syndrome was recognized as a negative factor in antiviral IFN-based therapy [68,69]. Efforts to eliminate HCV have activated an ongoing global campaign to diminish the incidence and mortality caused by this virus [70].
Today, pharmacological treatment with DAAs against HCV has reached a high cure rate among all HCV genotypes [71]. However, some questions remain to be solved because post-treated patients and those who may be unaware of their conditions are at risk of HCV-induced HCC. (Figure 2).
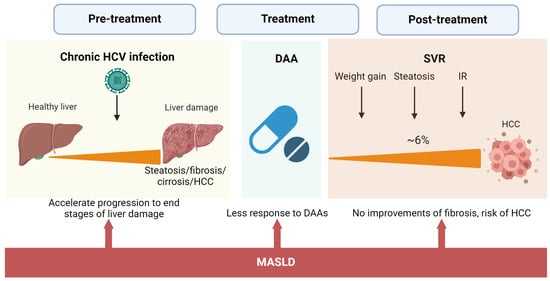
Figure 2.
Impact of MASLD on the natural history of HCV infection and after DAA treatment. The concurrence of MASLD and chronic HCV affects the natural history of HCV infection, accelerating the occurrence of advanced stages of liver damage. High BMI during and after treatment is associated with less SVR and no improvements in fibrosis grade. DAA effects on liver steatosis and metabolism significantly increase the risk of HCC even after SVR.
These patients with metabolic-associated steatotic conditions need medical and nutritional intervention. Eradication of HCV by DAAs is associated with weight gain, liver steatosis, and no improvement in IR [7,72,73]. A recent study of 1280 elderly patients with HCV eradication by DAAs and no history of HCC demonstrated that 25.8% of the patients developed MASLD at 24 weeks of SVR. In turn, MASLD at 24 weeks of SVR conferred a 3.04-fold increased risk of developing HCC. In the end, 6.7% of the patients developed HCC [74]. Thus, patients with dual etiology of liver damage are at higher risk for advanced hepatic fibrosis and HCC even after HCV eradication.
Similarly, having obesity before treatment with DAAs is negatively associated with improvement of fibrosis at 1-year follow-up [75]. Although DAAs have reached a high cure rate, 5% of the patients treated will not respond to therapy. A retrospective study involving 10,655 patients treated with DAAs to determine predictive factors associated with nonresponse to treatment found that an elevated pretreatment body mass index (BMI) was associated with nonresponse to DAAs [76]. Hence, managing MASLD before and after DAA treatment is vital to prevent the progression of the disease and prevent death by HCC.
The treatment of MASLD primarily focuses on modifying risk factors through therapies such as insulin sensitizers, antioxidants, weight reduction, physical activity, and diet [77]. Although there is little evidence regarding weight management before DAA treatment, a study reported that weight loss of >0.5 BMI before IFN + ribavirin therapy was associated with higher SVR [78]. Metformin is a primary insulin sensitizer in clinical practice. An in vitro study demonstrated that metformin inhibits HCV replication, activating the type I IFN antiviral signaling pathway via activation of AMPK and decreasing core protein expression [79]. Metformin also reduces the risk of HCC incidence after SVR among those with diabetes and chronic HCV [80]. When supplemented with metformin, statins lower serum cholesterol, reducing the risk for HCC in chronic patients who failed antiviral therapy [81]. Vitamin E is an antioxidant agent that is beneficial for treating non-diabetic MASLD patients [82]. Vitamin E supplementation of 400 IU twice daily for 12 weeks decreased serum alanine aminotransferase levels in patients infected with HCV genotype 3 [83]. Recently, resmetirom, a liver-targeted selective thyroid hormone receptor-β (THR-β) agonist, became the first FDA-approved drug for treating non-cirrhotic MASH patients. Resmetirom has been shown to improve cholesterol and triglyceride levels and reduce liver fat in MASH patients. However, its potential effects on HCV patients remain unexplored [84,85]. This evidence implies that MASLD management when HCV coexists may benefit both etiologies of liver damage. More studies are needed to investigate the effect of MASLD therapy in patients with both conditions.
7. Dietary Considerations in Patients with MASLD and Chronic HCV Infection
Management strategies for MASLD that focus on lifestyle modifications are essential for liver-diseased patients. Dietary interventions can improve inflammation, oxidative stress, IR, and BMI and reduce liver damage due to MASLD [86,87,88]. Furthermore, it has been documented that dietary interventions are a valuable resource that should be tailored by region based on genetic and environmental (cultural) differences among populations [89]. For example, epidemiological studies performed on US Hispanics of Mexican descent showed higher rates of obesity, diabetes, and MASLD [90], as well as higher rates of genetic susceptibility compared to other ethnic groups [1,91]. Studies in Mexican subpopulations have reported anthropometric and biochemical alterations in young obese patients with MASLD who consumed a hepatopathogenic diet [92]. However, it has also been documented that adherence to a traditional Mexican or genome-based diet in this population, i.e., consumption of a diet integrating Mexican staple foods such as maize products, legumes, pumpkin, zucchini, prickly pears, chia seeds, amaranth, tomato, squash, and chili with anti-inflammatory, antioxidant, anti-fibrogenic, and insulin-sensitizing properties, could decrease the risk of MASLD-related conditions [93,94].
Additionally, several foods and their components have shown beneficial effects for MASLD, such as preventing inflammation, decreasing cardiometabolic risk factors, and reducing liver fibrosis [95]. The availability and quantity of each micronutrient may depend on each population’s diet and food culture. As shown in Table 2, dietary recommendations for MASLD patients based on nutrients or functional components with beneficial effects on liver steatosis can significantly help with the clinical management of the disease.
Table 2.
Food source and concentration of nutrients with anti-HCV and MASLD activities.
The HCV lifecycle is closely related to hepatic lipid metabolism. Lipid droplets are involved in the HCV replication, assembly, and release stages. While saturated fatty acids (SFA) are required for successful HCV replication, polyunsaturated fatty acids (PUFAs) inhibit in vitro HCV replication [114,115]. A study revealed that an SFA-rich diet in HCV core protein transgenic mice increases liver steatosis, lipogenesis, inflammation, and the presence of liver tumors [115]. This study suggests that excessive intake of SFA-rich foods should be avoided in HCV-infected patients to prevent liver cancer. However, this finding might apply to MASLD patients or MASLD + HCV patients as the study reproduces steatosis-derived liver tumorigenesis without significant fibrosis. Another in vitro study demonstrated that arachidonic (AA), docosahexaenoic (DHA), and eicosapentaenoic acids (EPA) inhibit HCV replication by suppressing the expression of genes involved in lipogenesis [96,97]. However, the exact mechanism underlying these effects is unclear.
Similarly, several micronutrients have demonstrated anti-HCV replication in vitro (Figure 3, Table 2), but their effect in vivo is scarcely known [116]. BMI and dietary patterns of patients with an active HCV infection can influence the disease outcome [117,118]. A recent study of chronic HCV-infected patients found that patients with adherence to a fish-rich dietary pattern consisting mainly of fish, seafood, vegetable oils, and PUFAs ≥ 4.9% had lower viral load levels [118].
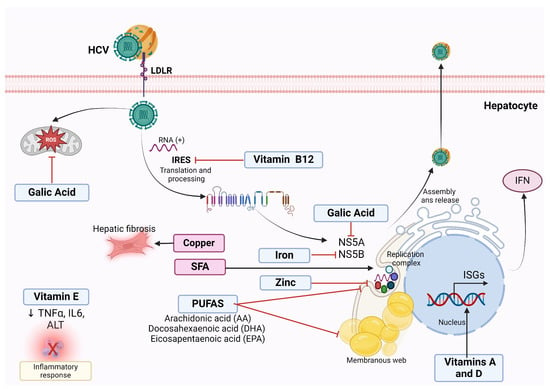
Figure 3.
Antiviral effect of nutrients against HCV infection. Polyunsaturated fatty acids (PUFAs) inhibit the formation of the membranous web necessary for successful HCV replication in vitro, thus reducing viral RNA replication and virus production. Gallic acid downregulates the expression of NS5A HCV protein required for HCV replication and decreases the ROS derivates from HCV infection in vitro. Vitamin B12 inhibits HCV internal ribosome entry site (IRES), essential for HCV translation, thus limiting HCV persistence in vitro. Vitamin E reduces TNFα and IL-6, suggesting an anti-inflammatory effect. Vitamins A and D induce the transcription of type-1 IFNs, enhancing the effect of IFN on HCV. Iron Inhibits NS5B polymerase activity in vitro. Zinc reduces HCV replication in vitro. On the contrary, saturated fatty acids (SFA) are necessary for HCV replication. Hepatic copper increases hepatic fibrosis and correlates positively with type IV collagen.
In this sense, PUFAs can decrease inflammation, ameliorate insulin sensitivity and liver steatosis, and counteract core protein effects [97]. DHA, EPA, and AA supplementation may help prevent or treat MASLD associated with HCV infection. Because of their absence of adverse effects, these treatments might be suitable for adults and children. Dietary intervention with PUFAs in MALSD + HCV patients could benefit both etiologies of liver damage. Furthermore, clinical practice guidelines must consider nutritional recommendations for patients with HCV infection to offer standard recommendations to the public.
8. Conclusions
There are numerous similarities between fat accumulation in HCV and MASLD. Besides fatty liver accumulation, HCV resembles MASLD in terms of IR, oxidative stress, metabolic dysfunction, and chronic inflammation. Both MASLD and HCV can independently trigger liver damage. When these two etiologies of liver damage converge, intricated mechanisms involving low-grade inflammation, IR, oxidative stress, and metabolic disturbances, as well as viral proteins, enhance the progression of liver damage. Future perspectives for managing MASLD in HCV patients should emphasize the development of clinical practice guidelines that integrate general and region-specific nutritional recommendations [119]. These guidelines are crucial for providing standardized advice tailored to diverse populations, particularly in regions where achieving HCV eradication remains challenging. Addressing metabolic disturbances must also be a priority, especially in patients with liver steatosis, but also in those without fatty liver before DAA treatment. Mandatory clinical follow-up should be implemented for all HCV patients after viral eradication, considering the risk of liver steatosis and HCC. A comprehensive approach combining clinical, nutritional, and metabolic management is essential for improving long-term outcomes in patients with MASLD and HCV chronic infection. HCC incidence will be an important health issue in HCV patients after virus eradication.
Author Contributions
Concept and design: K.G.-A., A.P. and L.A.T.-R.; Acquisition: K.G.-A. and S.R.; Analysis of data: C.O.-G. and L.L.-M.; Drafting of the manuscript: K.G.-A., L.A.T.-R. and S.R.; Critical review of the manuscript for important intellectual content: K.G.-A., L.A.T.-R., C.O.-G., L.L.-M., S.R. and A.P.; Supervision: K.G.-A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Consejo Nacional de Humanidades, Ciencias y Tecnologias (CONAHCYT), grant number CF-2023-I-1040 to K.G.-A.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Ekstedt, M.; Wong, G.L.; Hagstrom, H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J. Hepatol. 2023, 79, 842–852. [Google Scholar] [CrossRef]
- Pafili, K.; Roden, M. Nonalcoholic fatty liver disease (NAFLD) from pathogenesis to treatment concepts in humans. Mol. Metab. 2021, 50, 101122. [Google Scholar] [CrossRef]
- Hajarizadeh, B.; Grebely, J.; Dore, G.J. Epidemiology and natural history of HCV infection. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 553–562. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Hepatitis C. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 30 October 2024).
- Modaresi Esfeh, J.; Ansari-Gilani, K. Steatosis and hepatitis C. Gastroenterol. Rep. 2016, 4, 24–29. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soliman, H.; Ziada, D.; Hamisa, M.; Badawi, R.; Hawash, N.; Salama, M.; Abd-Elsalam, S. The Effect of HCV Eradication after Direct-Acting Antiviral Agents on Hepatic Steatosis: A Prospective Observational Study. Endocr. Metab. Immune Disord. Drug Targets 2022, 22, 100–107. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Gambardella, M.; Andreana, A.; Tripodi, M.F.; Utili, R.; Ruggiero, G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology 2001, 33, 1358–1364. [Google Scholar] [CrossRef]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Battineni, G.; Sagaro, G.G.; Chintalapudi, N.; Amenta, F.; Tomassoni, D.; Tayebati, S.K. Impact of Obesity-Induced Inflammation on Cardiovascular Diseases (CVD). Int. J. Mol. Sci. 2021, 22, 4798. [Google Scholar] [CrossRef]
- Rodriguez, A.; Ezquerro, S.; Mendez-Gimenez, L.; Becerril, S.; Fruhbeck, G. Revisiting the adipocyte: A model for integration of cytokine signaling in the regulation of energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E691–E714. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Abd Alkhaleq, H.; Kornowski, R.; Waldman, M.; Zemel, R.; Lev, D.L.; Shainberg, A.; Miskin, R.; Hochhauser, E. Leptin modulates gene expression in the heart, cardiomyocytes and the adipose tissue thus mitigating LPS-induced damage. Exp. Cell Res. 2021, 404, 112647. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Gruzdeva, O.; Borodkina, D.; Uchasova, E.; Dyleva, Y.; Barbarash, O. Leptin resistance: Underlying mechanisms and diagnosis. Diabetes Metab. Syndr. Obes. 2019, 12, 191–198. [Google Scholar] [CrossRef]
- Tazawa, R.; Uchida, K.; Fujimaki, H.; Miyagi, M.; Inoue, G.; Sekiguchi, H.; Murata, K.; Takata, K.; Kawakubo, A.; Takaso, M. Elevated leptin levels induce inflammation through IL-6 in skeletal muscle of aged female rats. BMC Musculoskelet. Disord. 2019, 20, 199. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Vallejo, L.; Chrysafi, P.; Kouvari, M.; Guatibonza-Garcia, V.; Mylonakis, S.C.; Katsarou, A.; Verrastro, O.; Markakis, G.; Eslam, M.; Papatheodoridis, G.; et al. Circulating hormones in biopsy-proven steatotic liver disease and steatohepatitis: A Multicenter Observational Study. Metabolism 2023, 148, 155694. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Wang, M.Y.; Kakuma, T.; Wang, Z.W.; Babcock, E.; McCorkle, K.; Higa, M.; Zhou, Y.T.; Unger, R.H. Liporegulation in diet-induced obesity. The antisteatotic role of hyperleptinemia. J. Biol. Chem. 2001, 276, 5629–5635. [Google Scholar] [CrossRef]
- Zhou, Y.; Jia, X.; Qin, J.; Lu, C.; Zhu, H.; Li, X.; Han, X.; Sun, X. Leptin inhibits PPARgamma gene expression in hepatic stellate cells in the mouse model of liver damage. Mol. Cell Endocrinol. 2010, 323, 193–200. [Google Scholar] [CrossRef]
- Becerril, S.; Rodriguez, A.; Catalan, V.; Ramirez, B.; Unamuno, X.; Gomez-Ambrosi, J.; Fruhbeck, G. iNOS Gene Ablation Prevents Liver Fibrosis in Leptin-Deficient ob/ob Mice. Genes 2019, 10, 184. [Google Scholar] [CrossRef]
- Pavlidis, C.; Panoutsopoulos, G.I.; Tiniakos, D.; Koutsounas, S.; Vlachogiannakos, J.; Zouboulis-Vafiadis, I. Serum leptin and ghrelin in chronic hepatitis C patients with steatosis. World J. Gastroenterol. 2011, 17, 5097–5104. [Google Scholar] [CrossRef] [PubMed]
- Baranova, A.; Jarrar, M.H.; Stepanova, M.; Johnson, A.; Rafiq, N.; Gramlich, T.; Chandhoke, V.; Younossi, Z.M. Association of serum adipocytokines with hepatic steatosis and fibrosis in patients with chronic hepatitis C. Digestion 2011, 83, 32–40. [Google Scholar] [CrossRef]
- Fang, H.; Judd, R.L. Adiponectin Regulation and Function. Compr. Physiol. 2018, 8, 1031–1063. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Udomsinprasert, W.; Honsawek, S.; Poovorawan, Y. Adiponectin as a novel biomarker for liver fibrosis. World J. Hepatol. 2018, 10, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Tsiaousi, E. The role of adiponectin in the pathogenesis and treatment of non-alcoholic fatty liver disease. Diabetes Obes. Metab. 2010, 12, 365–383. [Google Scholar] [CrossRef]
- Buechler, C.; Wanninger, J.; Neumeier, M. Adiponectin, a key adipokine in obesity related liver diseases. World J. Gastroenterol. 2011, 17, 2801–2811. [Google Scholar] [CrossRef]
- El-Daly, U.M.; Saber, M.M.; Abdellateif, M.S.; Nassar, H.R.; Namour, A.E.; Ismail, Y.M.; Zekri, A.N. The Possible Role of Adipokines in HCV Associated Hepatocellular Carcinoma. Asian Pac. J. Cancer Prev. 2020, 21, 599–609. [Google Scholar] [CrossRef]
- Corbetta, S.; Redaelli, A.; Pozzi, M.; Bovo, G.; Ratti, L.; Redaelli, E.; Pellegrini, C.; Beck-Peccoz, P.; Spada, A. Fibrosis is associated with adiponectin resistance in chronic hepatitis C virus infection. Eur. J. Clin. Investig. 2011, 41, 898–905. [Google Scholar] [CrossRef]
- Caldwell, A.B.; Cheng, Z.; Vargas, J.D.; Birnbaum, H.A.; Hoffmann, A. Network dynamics determine the autocrine and paracrine signaling functions of TNF. Genes Dev. 2014, 28, 2120–2133. [Google Scholar] [CrossRef]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. 2016, 11, 421–449. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.; Jung, F.; Baumann, A.; Brandt, A.; Staltner, R.; Sanchez, V.; Bergheim, I. TNFalpha is a key trigger of inflammation in diet-induced non-obese MASLD in mice. Redox Biol. 2023, 66, 102870. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; Cohen, L.B. Inflammation and Liver Cell Death in Patients with Hepatitis C Viral Infection. Curr. Issues Mol. Biol. 2021, 43, 2022–2035. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, Y.; Liu, J. Tumor necrosis factor-alpha signaling in nonalcoholic steatohepatitis and targeted therapies. J. Genet. Genom. 2022, 49, 269–278. [Google Scholar] [CrossRef]
- Mohamed-Ali, V.; Goodrick, S.; Rawesh, A.; Katz, D.R.; Miles, J.M.; Yudkin, J.S.; Klein, S.; Coppack, S.W. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J. Clin. Endocrinol. Metab. 1997, 82, 4196–4200. [Google Scholar] [CrossRef]
- Zhang, J.X.; Li, N.; Xu, Q.Y.; Yang, Y.; Xie, H.B.; Shen, T.; Zhu, Q.X. Kupffer cell depletion attenuates IL-6/STAT3 mediates hepatocyte apoptosis in immunological liver injury of trichloroethylene sensitized mice. Int. Immunopharmacol. 2020, 88, 106897. [Google Scholar] [CrossRef]
- Kim, J.H.; Bachmann, R.A.; Chen, J. Interleukin-6 and insulin resistance. Vitam. Horm. 2009, 80, 613–633. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef]
- Zheng, X.; Xu, M.; Yao, B.; Wang, C.; Jia, Y.; Liu, Q. IL-6/STAT3 axis initiated CAFs via up-regulating TIMP-1 which was attenuated by acetylation of STAT3 induced by PCAF in HCC microenvironment. Cell Signal. 2016, 28, 1314–1324. [Google Scholar] [CrossRef]
- Tian, X.; Chen, S.; Wang, P.; Xu, Q.; Zhang, Y.; Luo, Y.; Wu, S.; Wang, A. Insulin resistance mediates obesity-related risk of cardiovascular disease: A prospective cohort study. Cardiovasc. Diabetol. 2022, 21, 289. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306 e1220. [Google Scholar] [CrossRef]
- Gasmi, A.; Noor, S.; Menzel, A.; Dosa, A.; Pivina, L.; Bjorklund, G. Obesity and Insulin Resistance: Associations with Chronic Inflammation, Genetic and Epigenetic Factors. Curr. Med. Chem. 2021, 28, 800–826. [Google Scholar] [CrossRef] [PubMed]
- Pazienza, V.; Clement, S.; Pugnale, P.; Conzelman, S.; Foti, M.; Mangia, A.; Negro, F. The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology 2007, 45, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Labra, R.; Silva, L.; Subiabre, M.; Araos, J.; Salsoso, R.; Fuenzalida, B.; Saez, T.; Toledo, F.; Gonzalez, M.; Quezada, C.; et al. Akt/mTOR Role in Human Foetoplacental Vascular Insulin Resistance in Diseases of Pregnancy. J. Diabetes Res. 2017, 2017, 5947859. [Google Scholar] [CrossRef]
- Patel, S.; Jinjuvadia, R.; Patel, R.; Liangpunsakul, S. Insulin Resistance is Associated With Significant Liver Fibrosis in Chronic Hepatitis C Patients: A Systemic Review and Meta-Analysis. J. Clin. Gastroenterol. 2016, 50, 80–84. [Google Scholar] [CrossRef]
- Kumar, S.; Duan, Q.; Wu, R.; Harris, E.N.; Su, Q. Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv. Drug Deliv. Rev. 2021, 176, 113869. [Google Scholar] [CrossRef]
- Yuan, D.; Huang, S.; Berger, E.; Liu, L.; Gross, N.; Heinzmann, F.; Ringelhan, M.; Connor, T.O.; Stadler, M.; Meister, M.; et al. Kupffer Cell-Derived Tnf Triggers Cholangiocellular Tumorigenesis through JNK due to Chronic Mitochondrial Dysfunction and ROS. Cancer Cell 2017, 31, 771–789.e6. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and oxidative stress in the liver. Viruses 2013, 5, 439–469. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS ONE 2011, 6, e24957. [Google Scholar] [CrossRef]
- Del Ben, M.; Polimeni, L.; Carnevale, R.; Bartimoccia, S.; Nocella, C.; Baratta, F.; Loffredo, L.; Pignatelli, P.; Violi, F.; Angelico, F. NOX2-generated oxidative stress is associated with severity of ultrasound liver steatosis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2014, 14, 81. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Darlay, R.; Cockell, S.; Meroni, M.; Govaere, O.; Tiniakos, D.; Burt, A.D.; Bedossa, P.; Palmer, J.; Liu, Y.L.; et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort (☆). J. Hepatol. 2020, 73, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Banini, B.A.; Kumar, D.P.; Cazanave, S.; Seneshaw, M.; Mirshahi, F.; Santhekadur, P.K.; Wang, L.; Guan, H.P.; Oseini, A.M.; Alonso, C.; et al. Identification of a Metabolic, Transcriptomic, and Molecular Signature of Patatin-Like Phospholipase Domain Containing 3-Mediated Acceleration of Steatohepatitis. Hepatology 2021, 73, 1290–1306. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.H.; Xiang, M.Q.; Li, Q.L.; Shi, H.T.; Guo, J.J. PNPLA3 rs738409 Polymorphism Associated with Hepatic Steatosis and Advanced Fibrosis in Patients with Chronic Hepatitis C Virus: A Meta-Analysis. Gut Liver 2016, 10, 456–463. [Google Scholar] [CrossRef]
- De Benedittis, C.; Bellan, M.; Crevola, M.; Boin, E.; Barbaglia, M.N.; Mallela, V.R.; Ravanini, P.; Ceriani, E.; Fangazio, S.; Sainaghi, P.P.; et al. Interplay of PNPLA3 and HSD17B13 Variants in Modulating the Risk of Hepatocellular Carcinoma among Hepatitis C Patients. Gastroenterol. Res. Pract. 2020, 2020, 4216451. [Google Scholar] [CrossRef]
- Boren, J.; Adiels, M.; Bjornson, E.; Matikainen, N.; Soderlund, S.; Ramo, J.; Stahlman, M.; Ripatti, P.; Ripatti, S.; Palotie, A.; et al. Effects of TM6SF2 E167K on hepatic lipid and very low-density lipoprotein metabolism in humans. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Coppola, N.; Rosa, Z.; Cirillo, G.; Stanzione, M.; Macera, M.; Boemio, A.; Grandone, A.; Pisaturo, M.; Marrone, A.; Adinolfi, L.E.; et al. TM6SF2 E167K variant is associated with severe steatosis in chronic hepatitis C, regardless of PNPLA3 polymorphism. Liver Int. 2015, 35, 1959–1963. [Google Scholar] [CrossRef]
- Magri, M.C.; Manchiero, C.; Prata, T.V.G.; Nunes, A.; Oliveira Junior, J.S.; Dantas, B.P.; Tengan, F.M. The influence of gene-chronic hepatitis C virus infection on hepatic fibrosis and steatosis. Diagn. Microbiol. Infect. Dis. 2020, 97, 115025. [Google Scholar] [CrossRef]
- Guntipalli, P.; Pakala, R.; Kumari Gara, S.; Ahmed, F.; Bhatnagar, A.; Endaya Coronel, M.K.; Razzack, A.A.; Solimando, A.G.; Thompson, A.; Andrews, K.; et al. Worldwide prevalence, genotype distribution and management of hepatitis C. Acta Gastroenterol. Belg. 2021, 84, 637–656. [Google Scholar] [CrossRef]
- Sheridan, D.A.; Shawa, I.T.; Thomas, E.L.; Felmlee, D.J.; Bridge, S.H.; Neely, D.; Cobbold, J.F.; Holmes, E.; Bassendine, M.F.; Taylor-Robinson, S.D. Infection with the hepatitis C virus causes viral genotype-specific differences in cholesterol metabolism and hepatic steatosis. Sci. Rep. 2022, 12, 5562. [Google Scholar] [CrossRef]
- Mirandola, S.; Realdon, S.; Iqbal, J.; Gerotto, M.; Dal Pero, F.; Bortoletto, G.; Marcolongo, M.; Vario, A.; Datz, C.; Hussain, M.M.; et al. Liver microsomal triglyceride transfer protein is involved in hepatitis C liver steatosis. Gastroenterology 2006, 130, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Hourioux, C.; Patient, R.; Morin, A.; Blanchard, E.; Moreau, A.; Trassard, S.; Giraudeau, B.; Roingeard, P. The genotype 3-specific hepatitis C virus core protein residue phenylalanine 164 increases steatosis in an in vitro cellular model. Gut 2007, 56, 1302–1308. [Google Scholar] [CrossRef]
- Patra, T.; Sasaki, R.; Meyer, K.; Ray, R.B.; Ray, R. Transforming Growth Factor beta Acts as a Regulatory Molecule for Lipogenic Pathways among Hepatitis C Virus Genotype-Specific Infections. J. Virol. 2019, 93, e00811-19. [Google Scholar] [CrossRef]
- Hezode, C.; Roudot-Thoraval, F.; Zafrani, E.S.; Dhumeaux, D.; Pawlotsky, J.M. Different mechanisms of steatosis in hepatitis C virus genotypes 1 and 3 infections. J. Viral. Hepat. 2004, 11, 455–458. [Google Scholar] [CrossRef]
- Fierbinteanu-Braticevici, C.; Mohora, M.; Tribus, L.; Petrisor, A.; Cretoiu, S.M.; Cretoiu, D.; Usvat, R.; Ionita, L. Hepatocyte steatosis in patients infected with genotype 1 hepatitis C virus. Rom. J. Morphol. Embryol. 2010, 51, 235–242. [Google Scholar] [PubMed]
- Jonsson, J.R.; Barrie, H.D.; O’Rourke, P.; Clouston, A.D.; Powell, E.E. Obesity and steatosis influence serum and hepatic inflammatory markers in chronic hepatitis C. Hepatology 2008, 48, 80–87. [Google Scholar] [CrossRef]
- Aziz, H.; Gill, U.; Raza, A.; Gill, M.L. Metabolic syndrome is associated with poor treatment response to antiviral therapy in chronic hepatitis C genotype 3 patients. Eur. J. Gastroenterol. Hepatol. 2014, 26, 538–543. [Google Scholar] [CrossRef]
- Shah, S.R.; Patel, K.; Marcellin, P.; Foster, G.R.; Manns, M.; Kottilil, S.; Healey, L.; Pulkstenis, E.; Subramanian, G.M.; McHutchison, J.G.; et al. Steatosis is an independent predictor of relapse following rapid virologic response in patients with HCV genotype 3. Clin. Gastroenterol. Hepatol. 2011, 9, 688–693. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Combating Hepatitis B and C to Reach Elimination by 2030. Available online: https://www.who.int/publications/i/item/combating-hepatitis-b-and-c-to-reach-elimination-by-2030 (accessed on 30 September 2024).
- Di Marco, L.; La Mantia, C.; Di Marco, V. Hepatitis C: Standard of Treatment and What to Do for Global Elimination. Viruses 2022, 14, 505. [Google Scholar] [CrossRef]
- Strauhs-Nitsch, L.; Campiolo, M.F.; Morsoletto, D.B.G.; Pissaia Junior, A.; Ivantes, C.A.P. Curing Hepatitis C with the New Direct Acting Anti-virals Did Not Improve Insulin Resistance after One Year. Arq. Gastroenterol. 2020, 57, 267–271. [Google Scholar] [CrossRef]
- Schlevogt, B.; Boeker, K.H.W.; Mauss, S.; Klinker, H.; Heyne, R.; Link, R.; Simon, K.G.; Sarrazin, C.; Serfert, Y.; Manns, M.P.; et al. Weight Gain after Interferon-Free Treatment of Chronic Hepatitis C-Results from the German Hepatitis C-Registry (DHC-R). Biomedicines 2021, 9, 1495. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Amano, K.; Ide, T.; Isoda, H.; Honma, Y.; Morita, Y.; Yano, Y.; Nakamura, H.; Itano, S.; Miyajima, I.; et al. Metabolic management after sustained virologic response in elderly patients with hepatitis C virus: A multicenter study. Hepatol. Res. 2024, 54, 326–335. [Google Scholar] [CrossRef] [PubMed]
- McPhail, J.; Sims, O.T.; Guo, Y.; Wooten, D.; Herndon, J.S.; Massoud, O.I. Fibrosis improvement in patients with HCV treated with direct-acting antivirals. Eur. J. Gastroenterol. Hepatol. 2021, 33, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Shousha, H.I.; Saad, Y.; Saleh, D.A.; Dabes, H.; Alserafy, M.; ElShazly, Y.; Said, M. Simple predictors of nonresponse to direct-acting antivirals in chronic hepatitis C patients. Eur. J. Gastroenterol. Hepatol. 2020, 32, 1017–1022. [Google Scholar] [CrossRef]
- Raza, S.; Rajak, S.; Upadhyay, A.; Tewari, A.; Anthony Sinha, R. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front. Biosci. (Landmark Ed.) 2021, 26, 206–237. [Google Scholar] [CrossRef]
- Prabhukhot, R.; Browne, V.; Alwakeel, H.; Matarese, L.; Kandil, H. Weight Loss Prior to Hepatitis C Therapy Improves Virological Response Rates at 1 and 3 Months: 1126. Off. J. Am. Coll. Gastroenterol. ACG 2011, 106, S420. [Google Scholar] [CrossRef]
- Tsai, W.L.; Chang, T.H.; Sun, W.C.; Chan, H.H.; Wu, C.C.; Hsu, P.I.; Cheng, J.S.; Yu, M.L. Metformin activates type I interferon signaling against HCV via activation of adenosine monophosphate-activated protein kinase. Oncotarget 2017, 8, 91928–91937. [Google Scholar] [CrossRef]
- Tsai, P.C.; Kuo, H.T.; Hung, C.H.; Tseng, K.C.; Lai, H.C.; Peng, C.Y.; Wang, J.H.; Chen, J.J.; Lee, P.L.; Chien, R.N.; et al. Metformin reduces hepatocellular carcinoma incidence after successful antiviral therapy in patients with diabetes and chronic hepatitis C in Taiwan. J. Hepatol. 2023, 78, 281–292. [Google Scholar] [CrossRef]
- Tsai, P.C.; Huang, C.F.; Yeh, M.L.; Hsieh, M.H.; Kuo, H.T.; Hung, C.H.; Tseng, K.C.; Lai, H.C.; Peng, C.Y.; Wang, J.H.; et al. Metformin and statins reduce hepatocellular carcinoma risk in chronic hepatitis C patients with failed antiviral therapy. Clin. Mol. Hepatol. 2024, 30, 468–486. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Bunchorntavakul, C.; Wootthananont, T.; Atsawarungruangkit, A. Effects of vitamin E on chronic hepatitis C genotype 3: A randomized, double-blind, placebo-controlled study. J. Med. Assoc. Thai 2014, 97 (Suppl. S11), S31–S40. [Google Scholar] [PubMed]
- U.S. Food and Drug Administration. Novel Drug Approvals for 2024. Available online: https://www.fda.gov/drugs/novel-drug-approvals-fda/novel-drug-approvals-2024 (accessed on 16 November 2024).
- Rinella, M.E.; Neuschwander-Tetri, B.A.; Siddiqui, M.S.; Abdelmalek, M.F.; Caldwell, S.; Barb, D.; Kleiner, D.E.; Loomba, R. AASLD Practice Guidance on the clinical assessment and management of nonalcoholic fatty liver disease. Hepatology 2023, 77, 1797–1835. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jin, P.; Liu, Y.; Zhang, Z.; Wu, X.; Weng, M.; Cao, S.; Wang, Y.; Zeng, C.; Yang, R.; et al. A comprehensive approach to lifestyle intervention based on a calorie-restricted diet ameliorates liver fat in overweight/obese patients with NAFLD: A multicenter randomized controlled trial in China. Nutr. J. 2024, 23, 64. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Liu, H.; Li, C. Dietary Regulation of Oxidative Stress in Chronic Metabolic Diseases. Foods 2021, 10, 1854. [Google Scholar] [CrossRef]
- Yang, Z.; Song, S.; Li, L.; Yuan, Z.; Li, Y. Association between the composite dietary antioxidant index and metabolic dysfunction-associated steatotic liver disease in adults: A cross-sectional study from NHANES 2017-2020. Sci. Rep. 2024, 14, 13801. [Google Scholar] [CrossRef]
- Roman, S.; Ojeda-Granados, C.; Ramos-Lopez, O.; Panduro, A. Genome-based nutrition: An intervention strategy for the prevention and treatment of obesity and nonalcoholic steatohepatitis. World J. Gastroenterol. 2015, 21, 3449–3461. [Google Scholar] [CrossRef]
- Kwan, S.Y.; Sabotta, C.M.; Cruz, L.R.; Wong, M.C.; Ajami, N.J.; McCormick, J.B.; Fisher-Hoch, S.P.; Beretta, L. Gut phageome in Mexican Americans: A population at high risk for metabolic dysfunction-associated steatotic liver disease and diabetes. mSystems 2024, 9, e0043424. [Google Scholar] [CrossRef]
- Torres-Reyes, L.A.; Gonzalez-Aldaco, K.; Panduro, A.; Jose-Abrego, A.; Roman, S. Whole-Exome Sequencing identified Olfactory Receptor genes as a key contributor to extreme obesity with progression to nonalcoholic steatohepatitis in Mexican patients: Olfactory receptor genes in obese NASH patients. Ann. Hepatol. 2022, 27, 100767. [Google Scholar] [CrossRef]
- Sepulveda-Villegas, M.; Roman, S.; Rivera-Iniguez, I.; Ojeda-Granados, C.; Gonzalez-Aldaco, K.; Torres-Reyes, L.A.; Jose-Abrego, A.; Panduro, A. High prevalence of nonalcoholic steatohepatitis and abnormal liver stiffness in a young and obese Mexican population. PLoS ONE 2019, 14, e0208926. [Google Scholar] [CrossRef]
- Lopez-Pentecost, M.; Tamez, M.; Mattei, J.; Jacobs, E.T.; Thomson, C.A.; Garcia, D.O. Adherence to a Traditional Mexican Diet Is Associated with Lower Hepatic Steatosis in US-Born Hispanics of Mexican Descent with Overweight or Obesity. Nutrients 2023, 15, 4997. [Google Scholar] [CrossRef]
- Ojeda-Granados, C.; Panduro, A.; Rivera-Iniguez, I.; Sepulveda-Villegas, M.; Roman, S. A Regionalized Genome-Based Mexican Diet Improves Anthropometric and Metabolic Parameters in Subjects at Risk for Obesity-Related Chronic Diseases. Nutrients 2020, 12, 645. [Google Scholar] [CrossRef] [PubMed]
- Kosmalski, M.; Frankowski, R.; Deska, K.; Rozycka-Kosmalska, M.; Pietras, T. Exploring the Impact of Nutrition on Non-Alcoholic Fatty Liver Disease Management: Unveiling the Roles of Various Foods, Food Components, and Compounds. Nutrients 2023, 15, 2838. [Google Scholar] [CrossRef]
- Leu, G.Z.; Lin, T.Y.; Hsu, J.T. Anti-HCV activities of selective polyunsaturated fatty acids. Biochem. Biophys. Res. Commun. 2004, 318, 275–280. [Google Scholar] [CrossRef]
- Miyoshi, H.; Moriya, K.; Tsutsumi, T.; Shinzawa, S.; Fujie, H.; Shintani, Y.; Fujinaga, H.; Goto, K.; Todoroki, T.; Suzuki, T.; et al. Pathogenesis of lipid metabolism disorder in hepatitis C: Polyunsaturated fatty acids counteract lipid alterations induced by the core protein. J. Hepatol. 2011, 54, 432–438. [Google Scholar] [CrossRef]
- Govea-Salas, M.; Rivas-Estilla, A.M.; Rodriguez-Herrera, R.; Lozano-Sepulveda, S.A.; Aguilar-Gonzalez, C.N.; Zugasti-Cruz, A.; Salas-Villalobos, T.B.; Morlett-Chavez, J.A. Gallic acid decreases hepatitis C virus expression through its antioxidant capacity. Exp. Ther. Med. 2016, 11, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, S.; Fukuda, R.; Ishimura, N.; Rumi, M.A.; Kazumori, H.; Uchida, Y.; Kadowaki, Y.; Ishihara, S.; Kinoshita, Y. 9-cis retinoic acid enhances the antiviral effect of interferon on hepatitis C virus replication through increased expression of type I interferon receptor. J. Lab. Clin. Med. 2003, 141, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Murayama, A.; Kato, T. Inhibition of hepatitis C virus by vitamin D. Vitam. Horm. 2021, 117, 227–238. [Google Scholar] [CrossRef]
- Li, D.; Lott, W.B.; Martyn, J.; Haqshenas, G.; Gowans, E.J. Differential effects on the hepatitis C virus (HCV) internal ribosome entry site by vitamin B12 and the HCV core protein. J. Virol. 2004, 78, 12075–12081. [Google Scholar] [CrossRef]
- Fillebeen, C.; Rivas-Estilla, A.M.; Bisaillon, M.; Ponka, P.; Muckenthaler, M.; Hentze, M.W.; Koromilas, A.E.; Pantopoulos, K. Iron inactivates the RNA polymerase NS5B and suppresses subgenomic replication of hepatitis C Virus. J. Biol. Chem. 2005, 280, 9049–9057. [Google Scholar] [CrossRef]
- Yuasa, K.; Naganuma, A.; Sato, K.; Ikeda, M.; Kato, N.; Takagi, H.; Mori, M. Zinc is a negative regulator of hepatitis C virus RNA replication. Liver Int. 2006, 26, 1111–1118. [Google Scholar] [CrossRef]
- Rafiei, H.; Omidian, K.; Bandy, B. Dietary Polyphenols Protect Against Oleic Acid-Induced Steatosis in an in Vitro Model of NAFLD by Modulating Lipid Metabolism and Improving Mitochondrial Function. Nutrients 2019, 11, 541. [Google Scholar] [CrossRef]
- Hefer, M.; Petrovic, A.; Roguljic, L.K.; Kolaric, T.O.; Kizivat, T.; Wu, C.H.; Tabll, A.A.; Smolic, R.; Vcev, A.; Smolic, M. Green Tea Polyphenol (-)-Epicatechin Pretreatment Mitigates Hepatic Steatosis in an In Vitro MASLD Model. Curr. Issues Mol. Biol. 2024, 46, 8981–8994. [Google Scholar] [CrossRef] [PubMed]
- Chee, N.M.; Sinnanaidu, R.P.; Chan, W.K. Vitamin E Improves Serum Markers and Histology in Adults with Metabolic Dysfunction-Associated Steatotic Liver Disease: Systematic Review and Meta-Analysis. J. Gastroenterol. Hepatol. 2024. early view. [Google Scholar] [CrossRef] [PubMed]
- Šmíd, V.; Dvořák, K.; Šedivý, P.; Kosek, V.; Leníček, M.; Dezortová, M.; Hajšlová, J.; Hájek, M.; Vítek, L.; Bechyňská, K.; et al. Effect of Omega-3 Polyunsaturated Fatty Acids on Lipid Metabolism in Patients With Metabolic Syndrome and NAFLD. Hepatol. Commun. 2022, 6, 1336–1349. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimpour-Koujan, S.; Sohrabpour, A.A.; Giovannucci, E.; Vatannejad, A.; Esmaillzadeh, A. Effects of vitamin D supplementation on liver fibrogenic factors, vitamin D receptor and liver fibrogenic microRNAs in metabolic dysfunction-associated steatotic liver disease (MASLD) patients: An exploratory randomized clinical trial. Nutr. J. 2024, 23, 24. [Google Scholar] [CrossRef]
- Lee, E.S.; Kwon, M.H.; Kim, H.M.; Woo, H.B.; Ahn, C.M.; Chung, C.H. Curcumin analog CUR5-8 ameliorates nonalcoholic fatty liver disease in mice with high-fat diet-induced obesity. Metabolism 2020, 103, 154015. [Google Scholar] [CrossRef]
- Ma, K.; Sheng, W.; Song, X.; Song, J.; Li, Y.; Huang, W.; Liu, Y. Chlorogenic Acid from Burdock Roots Ameliorates Oleic Acid-Induced Steatosis in HepG2 Cells through AMPK/ACC/CPT-1 Pathway. Molecules 2023, 28, 7257. [Google Scholar] [CrossRef]
- Sánchez-Tapia, M.; Tobón-Cornejo, S.; Noriega, L.G.; Vázquez-Manjarrez, N.; Coutiño-Hernández, D.; Granados-Portillo, O.; Román-Calleja, B.M.; Ruíz-Margáin, A.; Macías-Rodríguez, R.U.; Tovar, A.R.; et al. Hepatic Steatosis Can Be Partly Generated by the Gut Microbiota-Mitochondria Axis via 2-Oleoyl Glycerol and Reversed by a Combination of Soy Protein, Chia Oil, Curcumin and Nopal. Nutrients 2024, 16, 2594. [Google Scholar] [CrossRef]
- U.S. Department of Agriculture Database. Agricultural Research Service. FoodData Central. Available online: https://fdc.nal.usda.gov/ (accessed on 30 October 2024).
- Rothwell, J.A.; Perez-Jimenez, J.; Neveu, V.; Medina-Remon, A.; M’Hiri, N.; Garcia-Lobato, P.; Manach, C.; Knox, C.; Eisner, R.; Wishart, D.S.; et al. Phenol-Explorer 3.0: A major update of the Phenol-Explorer database to incorporate data on the effects of food processing on polyphenol content. Database 2013, 2013, bat070. [Google Scholar] [CrossRef]
- Kapadia, S.B.; Chisari, F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 2005, 102, 2561–2566. [Google Scholar] [CrossRef]
- Diao, P.; Wang, X.; Jia, F.; Kimura, T.; Hu, X.; Shirotori, S.; Nakamura, I.; Sato, Y.; Nakayama, J.; Moriya, K.; et al. A saturated fatty acid-rich diet enhances hepatic lipogenesis and tumorigenesis in HCV core gene transgenic mice. J. Nutr. Biochem. 2020, 85, 108460. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Read, S.A.; Shackel, N.A.; Hebbard, L.; George, J.; Ahlenstiel, G. The Role of Micronutrients in the Infection and Subsequent Response to Hepatitis C Virus. Cells 2019, 8, 603. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Aldaco, K.; Roman, S.; Torres-Valadez, R.; Ojeda-Granados, C.; Torres-Reyes, L.A.; Panduro, A. Hepatitis C virus clearance and less liver damage in patients with high cholesterol, low-density lipoprotein cholesterol and APOE epsilon4 allele. World J. Gastroenterol. 2019, 25, 5826–5837. [Google Scholar] [CrossRef] [PubMed]
- Ojeda-Granados, C.; Panduro, A.; Gonzalez-Aldaco, K.; Rivera-Iniguez, I.; Campos-Medina, L.; Roman, S. Adherence to a Fish-Rich Dietary Pattern Is Associated with Chronic Hepatitis C Patients Showing Low Viral Load: Implications for Nutritional Management. Nutrients 2021, 13, 3337. [Google Scholar] [CrossRef]
- Panduro, A.; Roman, S. Advancements in genomic medicine and the need for updated regional clinical practice guidelines in the field of hepatology. Ann. Hepatol. 2020, 19, 1–2. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).