
Bilateral Pheochromocytoma with Germline MAX Variant without Family History


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
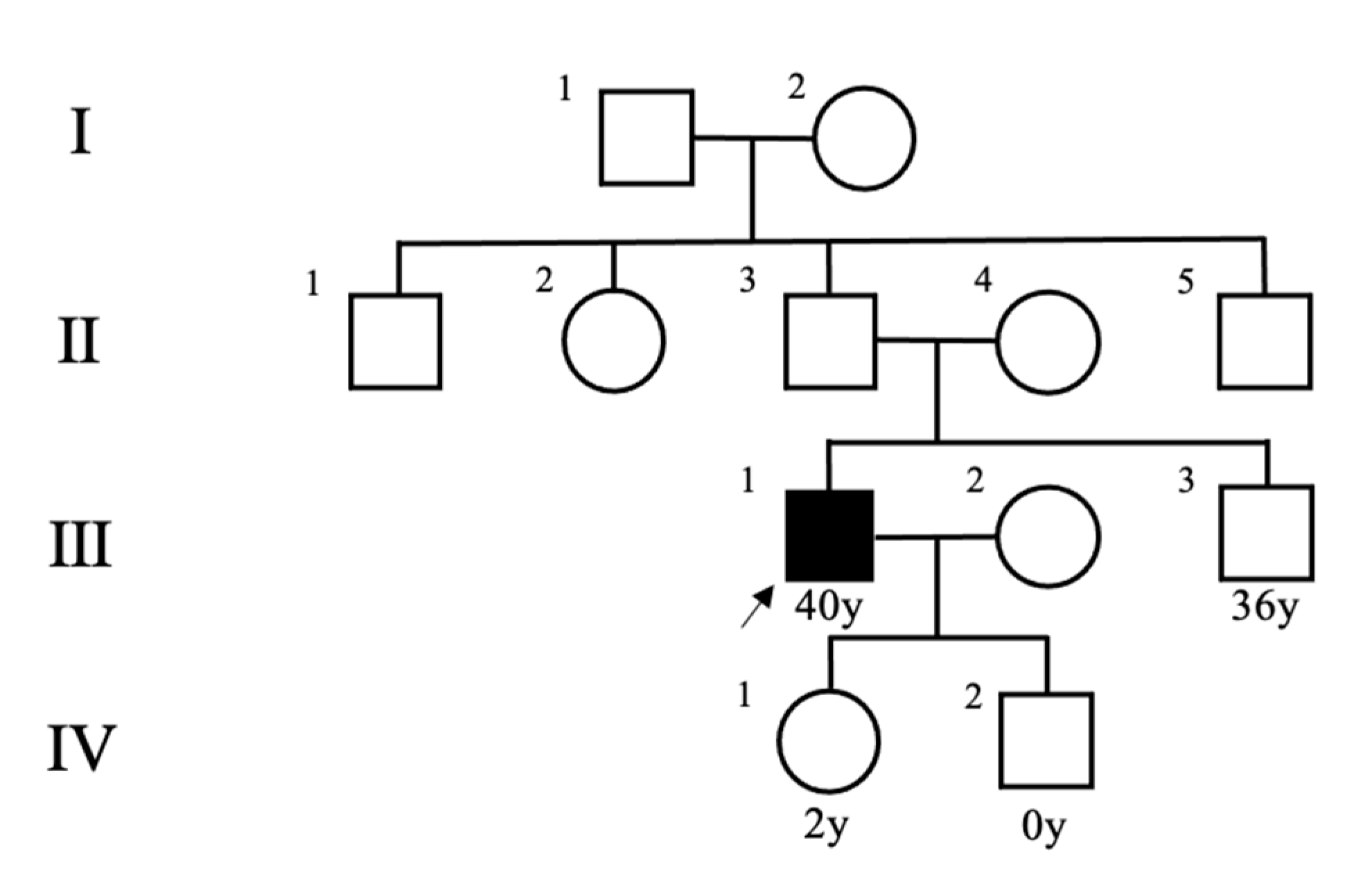
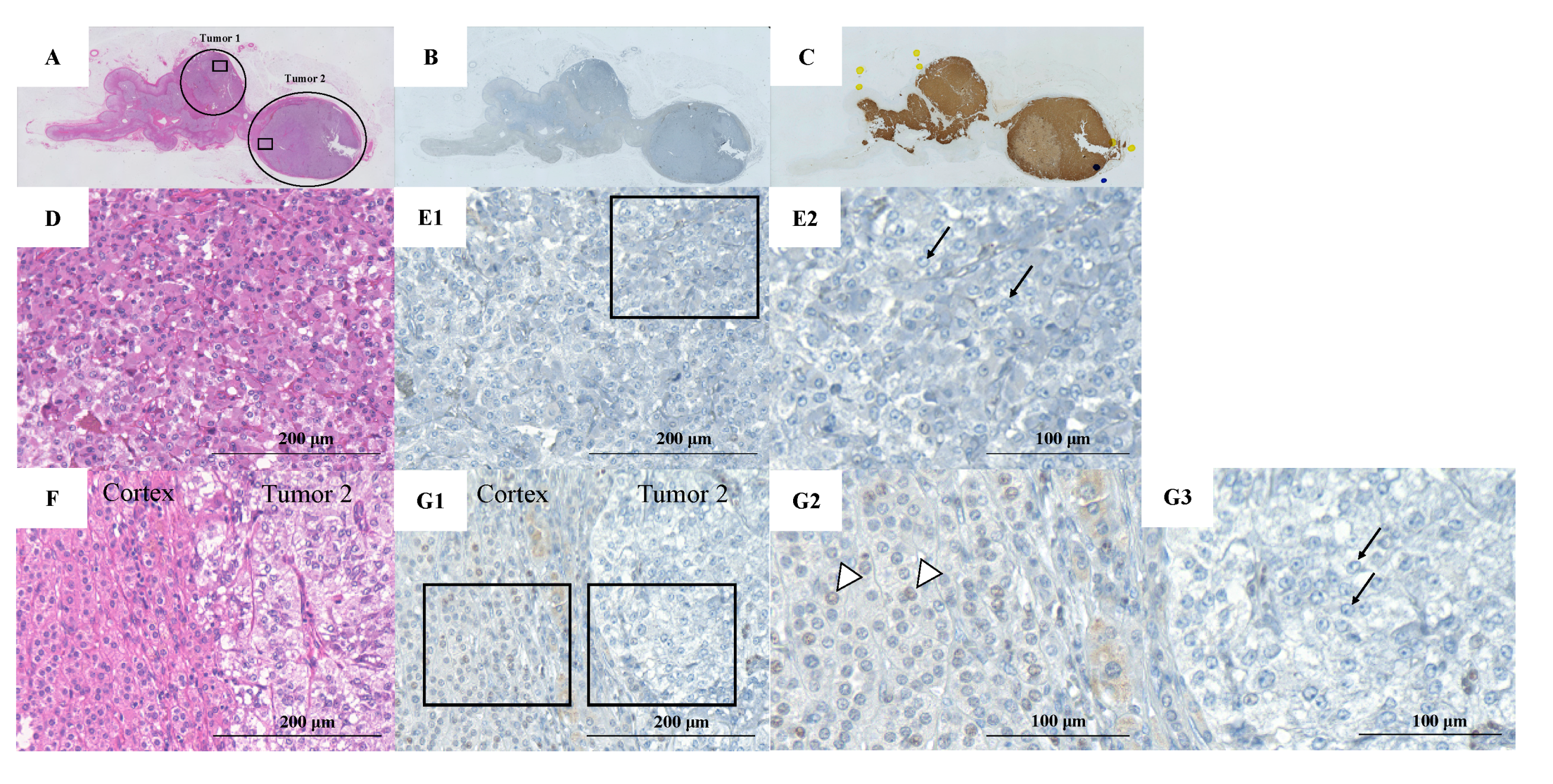
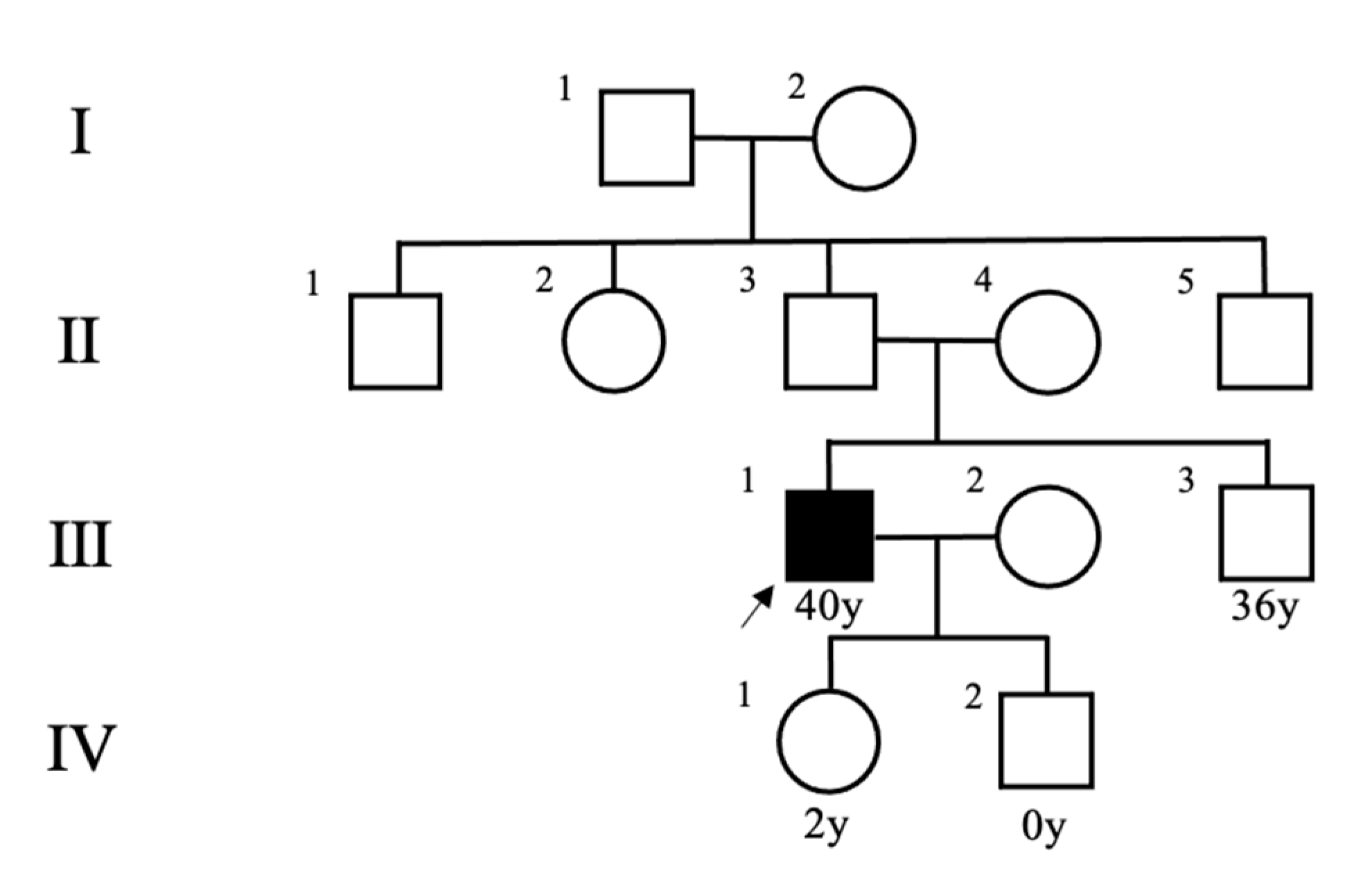
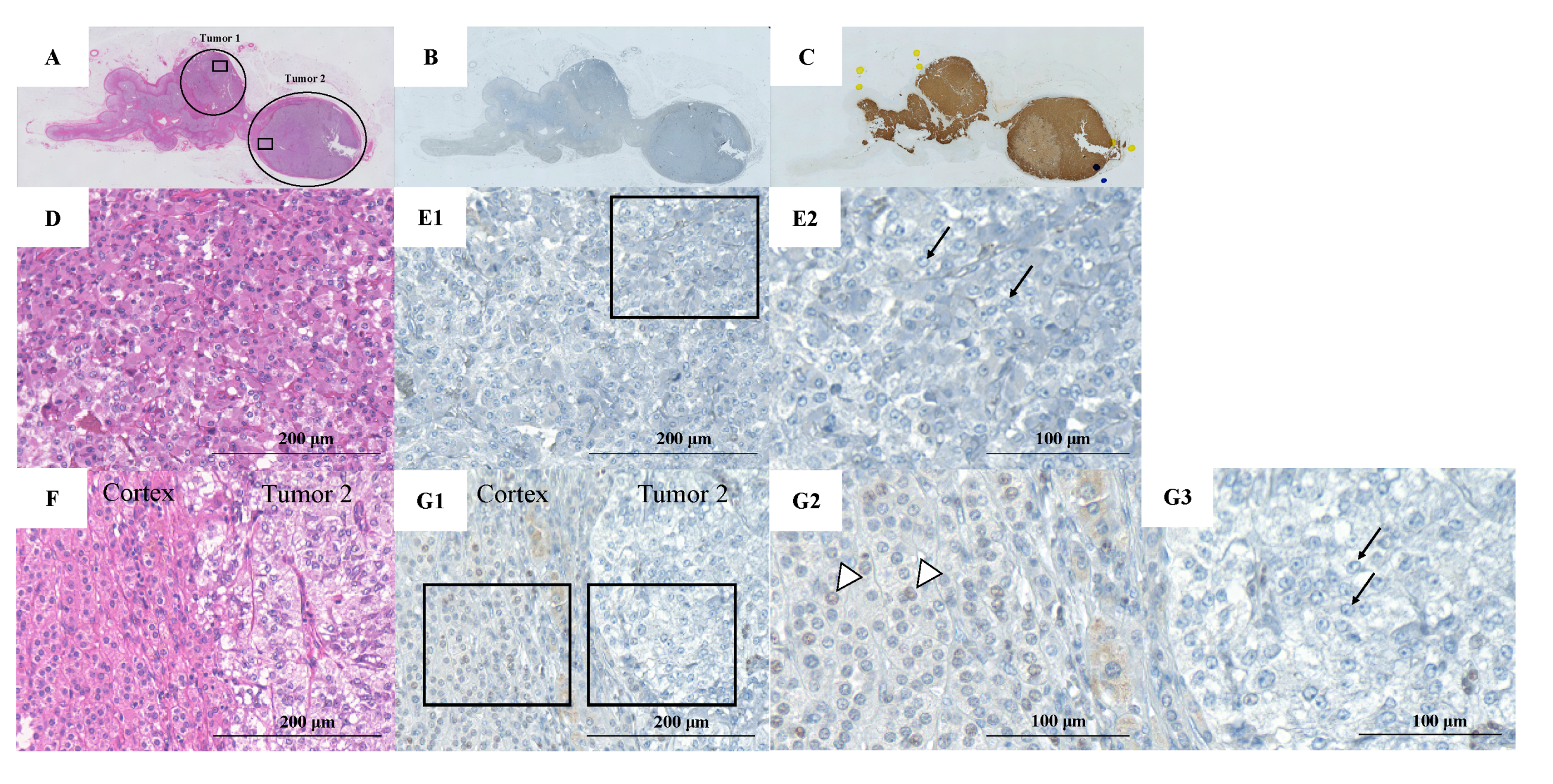
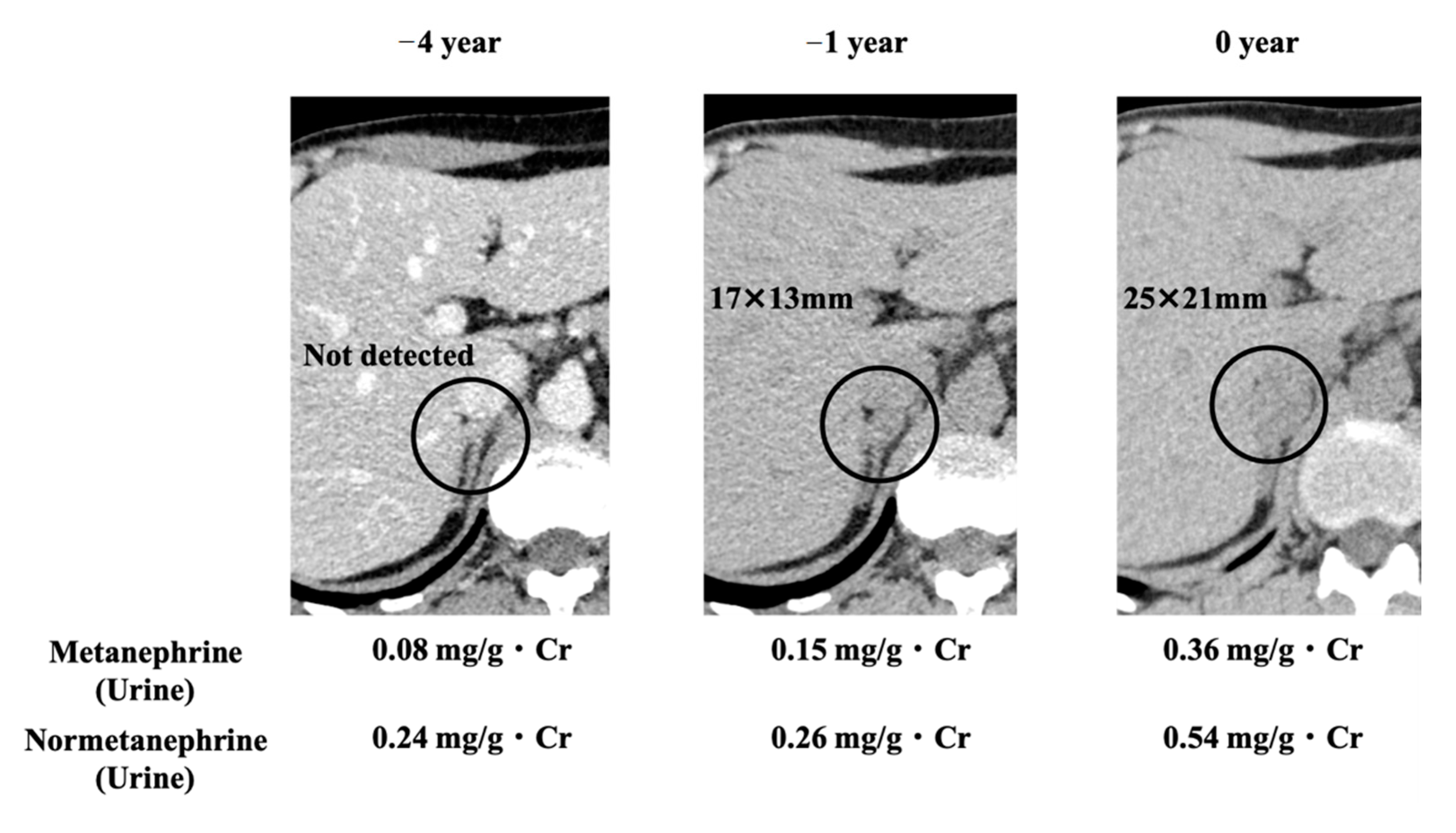
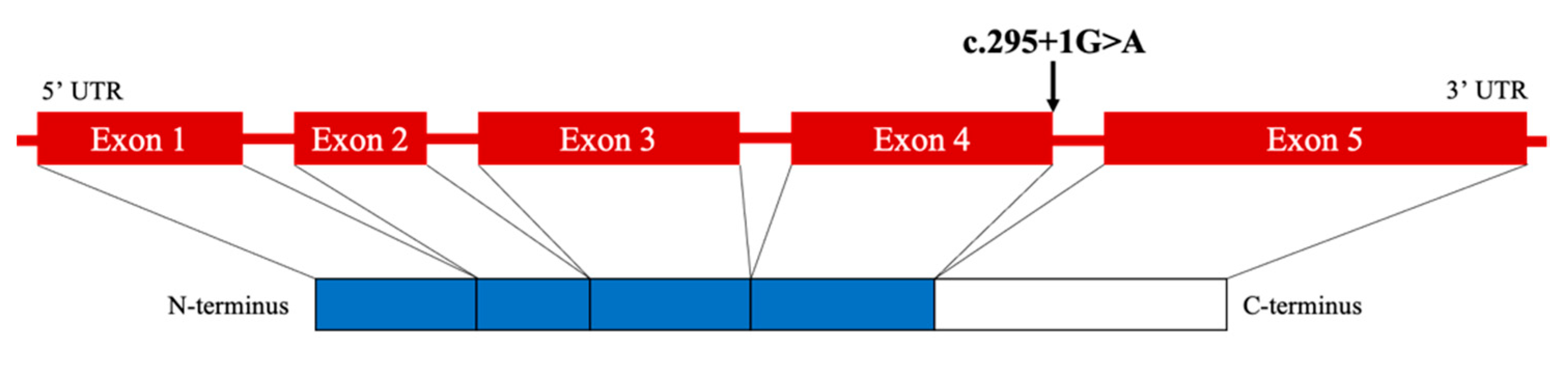
2. Case Presentation
3. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Pacak, K.; Taïeb, D. Pheochromocytoma (PHEO) and Paraganglioma (PGL). Cancers 2019, 11, 1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, E.R.; Eng, C. The pressure rises: Update on the genetics of phaeochromocytoma. Hum. Mol. Genet. 2002, 11, 2347–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannelli, M.; Castellano, M.; Schiavi, F.; Filetti, S.; Giacchè, M.; Mori, L.; Pignataro, V.; Bernini, G.; Giachè, V.; Bacca, A.; et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 1541–1547. [Google Scholar] [CrossRef] [Green Version]
- Cascón, A.; Pita, G.; Burnichon, N.; Landa, I.; López-Jiménez, E.; Monerto-Conde, C.; Leskelä, S.; Leandor-García, L.J.; Letón, R.; Rodriguez-Antona, C.; et al. Genetics of pheochromocytoma and paraganglioma in Spanish patients. J. Clin. Endocrinol. Metab. 2009, 94, 1701–1705. [Google Scholar] [CrossRef] [Green Version]
- Comino-Méndez, I.; Gracia-Aznárez, F.J.; Schiavi, F.; Landa, I.; Leandro-García, L.J.; Letón, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Amar, L.; Bertherat, J.; Baudin, E.; Ajzenberg, C.; Bressac-de Paillerets, B.; Chabre, O.; Chamontin, B.; Delemer, B.; Giraud, S.; Murat, A.; et al. Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. 2005, 23, 8812–8818. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Rohmer, V.; Amar, L.; Herman, P.; Leboulleux, S.; Darrouzet, V.; Niccoli, P.; Gaillard, D.; Chabrier, G.; Chabolle, F.; et al. PGL.NET network. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.; Whitworth, J.; Rattenberry, E.; Vialard, L.; Kilby, G.; Kumar, A.V.; Izatt, L.; Lalloo, F.; Brennan, P.; Cook, J.; et al. Evaluation of SDHB, SDHD and VHL gene susceptibility testing in the assessment of individuals with non-syndromic phaeochromocytoma, paraganglioma and head and neck paraganglioma. Clin. Endocrinol. 2013, 78, 898–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlic, Z.; Rybicki, L.; Peczkowska, M.; Golcher, H.; Kann, P.H.; Brauckhoff, M.; Müssig, K.; Muresan, M.; Schäffler, A.; Reisch, N.; et al. European-American Pheochromocytoma Study Group. Clinical Predictors and Algorithm for the Genetic Diagnosis of Pheochromocytoma Patients. Clin. Cancer. Res. 2009, 15, 6378–6385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korpershoek, E.; Favier, J.; Gaal, J.; Burnichon, N.; van Gessel, B.; Oudijk, L.; Badoual, C.; Gadessaud, N.; Venisse, A.; Bayley, J.; et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J. Clin. Endocrinol. Metab. 2011, 96, E1472–E1476. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Borson-Chazot, F.; Boutry-Kryza, N.; Wion, N.; Schillo, F.; Peix, J.; Brunaud, L.; Finat, A.; Calender, A.; Giraud, S. Screening of mutations in genes that predispose to hereditary paragangliomas and pheochromocytomas. Horm. Metab. Res. 2012, 44, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F. Endocrine Society. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Brito, J.P.; Asi, N.; Bancos, I.; Gionfriddo, M.R.; Zeballos-Palacios, C.L.; Leppin, A.L.; Undavalli, C.; Wang, Z.; Domecq, J.P.; Prustsky, G.; et al. Testing for germline mutations in sporadic pheochromocytoma/paraganglioma: A systematic review. Clin. Endocrinol. 2015, 82, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Welander, J.; Söderkvist, P.; Gimm, O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr. Relat. Cancer. 2011, 18, R253–R276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnichon, N.; Cascón, A.; Schiavi, F.; Morales, N.P.; Comino-Méndez, I.; Abermil, N.; Inglada-Pérez, L.; de Cubas, A.A.; Amar, L.; Barontini, M.; et al. MAX Mutations Cause Hereditary and Sporadic Pheochromocytoma and Paraganglioma. Clin. Cancer. Res. 2012, 18, 2828–2837. [Google Scholar] [CrossRef] [Green Version]
- Bausch, B.; Schiavi, F.; Ni, Y.; Welander, J.; Patocs, A.; Ngeow, J.; Wellner, U.; Malinoc, A.; Taschin, E.; Barbon, G.; et al. European-American-Asian Pheochromocytoma-Paraganglioma Registry Study Group. Clinical Characterization of the Pheochromocytoma and Paraganglioma Susceptibility Genes SDHA, TMEM127, MAX, and SDHAF2 for Gene-Informed Prevention. JAMA. Oncol. 2017, 3, 1204–1212. [Google Scholar] [CrossRef] [Green Version]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell. Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Diolaiti, D.; McFerrin, L.; Carroll, P.A.; Eisenman, R.N. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim. Biophys. Acta. 2015, 1849, 484–500. [Google Scholar] [CrossRef] [Green Version]
- Shibata, M.; Inaishi, T.; Miyajima, N.; Adachi, Y.; Takano, Y.; Nakanishi, K.; Takeuchi, D.; Noda, S.; Aita, Y.; Takekoshi, K.; et al. Synchronous bilateral pheochromocytomas and paraganglioma with novel germline mutation in MAX: A case report. Surg. Case. Rep. 2017, 3, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korpershoek, E.; Koffy, D.; Eussen, B.H.; Oudijk, L.; Papathomas, T.G.; van Nederveen, F.H.; Belt, E.J.; Franssen, G.J.; Restuccia, D.F.; Krol, N.M.; et al. Complex MAX Rearrangement in a Family with Malignant Pheochromocytoma, Renal Oncocytoma, and Erythrocytosis. J. Clin. Endocrinol. Metab. 2016, 101, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Roszko, K.L.; Blouch, E.; Blake, M.; Powers, J.F.; Tischler, A.S.; Hodin, R.; Sadow, P.; Lawson, E.A. Case Report of a Prolactinoma in a Patient with a Novel MAX Mutation and Bilateral Pheochromocytomas. J. Endocr. Soc. 2017, 1, 1401–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanet, P.; Guerin, C.; Pedini, P.; Essamet, W.; Castinetti, F.; Sebag, F.; Roche, P.; Cascon, A.; Tischler, A.S.; Pacak, K.; et al. Pathological and Genetic Characterization of Bilateral Adrenomedullary Hyperplasia in a Patient with Germline MAX Mutation. Endocr. Pathol. 2017, 28, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.F.; Castermans, E.; Oudijk, L.; Guitelman, M.A.; Beckers, P.; Potorac, I.; Neggers, S.J.; Sacre, N.; van der Lely, A.; Bours, V.; et al. Pheochromocytomas and pituitary adenomas in three patients with MAX exon deletions. Endocr. Relat. Cancer. 2018, 25, L37–L42. [Google Scholar] [CrossRef] [Green Version]
- Kobza, A.O.; Dizon, S.; Arnaout, A. Case Report of Bilateral Pheochromocytomas due to a Novel Max Mutation in a Patient Known to have a Pituitary Prolactinoma. AACE Clin. Case. Rep. 2018, 4, e453–e456. [Google Scholar] [CrossRef] [Green Version]
- Pozza, C.; Sesti, F.; Di Dato, C.; Sbardella, E.; Pofi, R.; Schiavi, F.; Bonifacio, V.; Isidori, A.M.; Faggiano, A.; Lenzi, A.; et al. A Novel MAX Gene Mutation Variant in a Patient with Multiple and "Composite" Neuroendocrine-Neuroblastic Tumors. Front. Endocrinol. 2020, 11, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Li, Z.; Ma, X.; Cui, Y.; Chen, S.; Tong, A. A Novel Phenotype of Germline Pathogenic Variants in MAX: Concurrence of Pheochromocytoma and Ganglioneuroma in a Chinese Family and Literature Review. Front. Endocrinol. 2020, 11, 558–563. [Google Scholar] [CrossRef]
- Choi, H.; Kim, K.J.; Hong, N.; Shin, S.; Choi, J.R.; Kang, S.W.; Lee, S.T.; Rhee, Y. Genetic Analysis and Clinical Characteristics of Hereditary Pheochromocytoma and Paraganglioma Syndrome in Korean Population. Endocrinol. Metab. 2020, 35, 858–872. [Google Scholar] [CrossRef]
- Duarte, D.B.; Ferreira, L.; Santos, A.P.; Costa, C.; Lima, J.; Santos, C.; Afonso, M.; Teixeira, M.R.; Carvalho, R.; Cardoso, M.H. Case Report: Pheochromocytoma and Synchronous Neuroblastoma in a Family with Hereditary Pheochromocytoma Associated with a MAX Deleterious Variant. Front. Endocrinol. 2021, 12, 609263–609271. [Google Scholar] [CrossRef]
- Seabrook, A.J.; Harris, J.E.; Velosa, S.B.; Kim, E.; McInerney-Leo, A.M.; Dwight, T.; Hockings, J.I.; Hockings, N.G.; Kirk, J.; Leo, P.J.; et al. Multiple Endocrine Tumors Associated with Germline MAX Mutations: Multiple Endocrine Neoplasia Type 5? J. Clin. Endocrinol. Metab. 2021, 106, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Mamedova, E.; Vasilyev, E.; Petrov, V.; Buryakina, S.; Tiulpakov, A.; Belaya, Z. Familial Acromegaly and Bilateral Asynchronous Pheochromocytomas in a Female Patient with a MAX Mutation: A Case Report. Front. Endocrinol. 2021, 12, 683492–683497. [Google Scholar] [CrossRef]
- Lam-Chung, C.E.; Rodríguez, L.L.; Vázquez, J.A.; Chávarri-Guerra, Y.; Arízaga-Ramírez, R.; Antonio, O.F.; González, J.D.; López-Hernández, M.A.; Weitzel, J.N.; Castillo, D.; et al. A Novel, Likely Pathogenic MAX Germline Variant in a Patient with Unilateral Pheochromocytoma. J. Endocr. Soc. 2021, 5, 1–6. [Google Scholar] [CrossRef]
- Petignot, S.; Daly, A.F.; Castermans, E.; Korpershoek, E.; Scagnol, I.; Beckers, P.; Dideberg, V.; Rohmer, V.; Bours, V.; Beckers, A. Pancreatic Neuroendocrine Neoplasm Associated with a Familial MAX Deletion. Horm. Metab. Res. 2020, 52, 784–787. [Google Scholar] [CrossRef]
- Muth, A.; Crona, J.; Gimm, O.; Elmgren, A.; Filipsson, K.; Askmalm, M.S.; Sandstedt, J.; Tengvar, M.; Tham, E. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J. Intern. Med. 2019, 285, 187–204. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hedge, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Jhawar, S.; Arakawa, Y.; Kumar, S.; Varghese, D.; Kim, Y.S.; Roper, N.; Elloumi, F.; Pommier, Y.; Pacak, K.; Del Rivero, J. New Insights on the genetics of pheochromocytoma and paraganglioma and its clinical implications. Cancers 2022, 14, 594–608. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hata, S.; Asano, M.; Tominaga, H.; Hamaguchi, M.; Hongo, F.; Usui, T.; Konishi, E.; Fukui, M. Bilateral Pheochromocytoma with Germline MAX Variant without Family History. Clin. Pract. 2022, 12, 299-305. https://doi.org/10.3390/clinpract12030035
Hata S, Asano M, Tominaga H, Hamaguchi M, Hongo F, Usui T, Konishi E, Fukui M. Bilateral Pheochromocytoma with Germline MAX Variant without Family History. Clinics and Practice. 2022; 12(3):299-305. https://doi.org/10.3390/clinpract12030035
Chicago/Turabian StyleHata, Shinnosuke, Mai Asano, Hiroyuki Tominaga, Masahide Hamaguchi, Fumiya Hongo, Takeshi Usui, Eiichi Konishi, and Michiaki Fukui. 2022. "Bilateral Pheochromocytoma with Germline MAX Variant without Family History" Clinics and Practice 12, no. 3: 299-305. https://doi.org/10.3390/clinpract12030035
APA StyleHata, S., Asano, M., Tominaga, H., Hamaguchi, M., Hongo, F., Usui, T., Konishi, E., & Fukui, M. (2022). Bilateral Pheochromocytoma with Germline MAX Variant without Family History. Clinics and Practice, 12(3), 299-305. https://doi.org/10.3390/clinpract12030035