
The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family




Abstract
1. Xenobiotics Disposition and Excretion
2. Historical Aspects of Cytochrome P450s Research
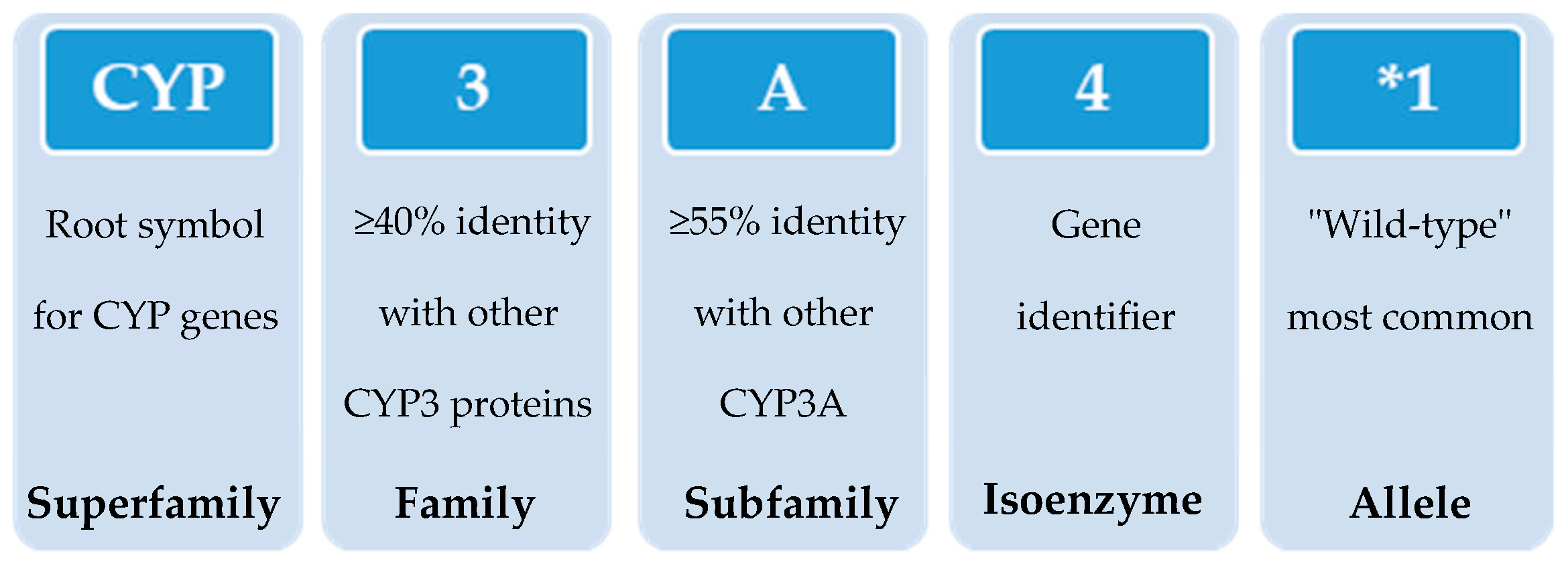
3. Classification and Nomenclature of Human Cytochrome P450s
4. Location and Function of Cytochrome P450s in Mammals
5. The Central Role of Cytochrome P450s as Xenobiotic-Metabolizing Enzymes
6. Genetic Determinants in Cytochrome P450s Expression and Activity
7. Nongenetic Factors Influencing Cytochrome P450s Expression and Activity
8. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Croom, E. Metabolism of Xenobiotics of Human Environments. In Progress in Molecular Biology and Translational Science; Ernest, H., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 112, pp. 31–88. ISBN 978-0-12-415813-9. [Google Scholar]
- Juchau, M.R.; Chen, H. Developmental Enzymology. In Handbook of Developmental Neurotoxicology; William, S., Jr., Louis, W.C., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; pp. 321–337. ISBN 978-0-12-648860-9. [Google Scholar]
- Evans, T.J. Chapter 2—Toxicokinetics and Toxicodynamics. In Small Animal Toxicology, 3rd ed.; Peterson, M.E., Talcott, P.A., Eds.; W.B. Saunders: Saint Louis, MO, USA, 2013; pp. 13–19. ISBN 978-1-4557-0717-1. [Google Scholar]
- Johnson, C.H.; Patterson, A.D.; Idle, J.R.; Gonzalez, F.J. Xenobiotic Metabolomics: Major Impact on the Metabolome. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Stanley, L.A. Drug Metabolism. In Pharmacognosy; Simone, B., Rupika, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 527–545. ISBN 978-0-12-802104-0. [Google Scholar]
- Rendic, S.; Guengerich, F.P. Survey of Human Oxidoreductases and Cytochrome P450 Enzymes Involved in the Metabolism of Xenobiotic and Natural Chemicals. Chem. Res. Toxicol. 2015, 28, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.; Ioannides, C.; Parke, D.V. Cytochromes P450 and Species Differences in Xenobiotic Metabolism and Activation of Carcinogen. Environ. Health Perspect. 1998, 106, 633–641. [Google Scholar] [CrossRef]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef]
- Bachmann, K. Drug Metabolism. In Pharmacology; Miles, H., William, M., Kenneth, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 131–173. ISBN 978-0-12-369521-5. [Google Scholar]
- Tillement, J.-P.; Tremblay, D. Clinical Pharmacokinetic Criteria for Drug Research. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 11–30. ISBN 978-0-08-045044-5. [Google Scholar]
- Murray, M.; Zhou, F. Trafficking and Other Regulatory Mechanisms for Organic Anion Transporting Polypeptides and Organic Anion Transporters That Modulate Cellular Drug and Xenobiotic Influx and That Are Dysregulated in Disease. Br. J. Pharmacol. 2017, 174, 1908–1924. [Google Scholar] [CrossRef]
- Janov, P.; Iller, M. Phase II Drug Metabolism. In Topics on Drug Metabolism; Paxton, J., Ed.; InTech: London, UK, 2012; pp. 35–60. ISBN 978-953-51-0099-7. [Google Scholar]
- Guengerich, F.P. Cytochrome P450 and Chemical Toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Strolin Benedetti, M.; Tipton, K.F.; Whomsley, R. Amine Oxidases and Monooxygenases in the in Vivo Metabolism of Xenobiotic Amines in Humans: Has the Involvement of Amine Oxidases Been Neglected? Amine Oxidases Monooxygenases Amine Metab. Hum. Fundam. Clin. Pharmacol. 2007, 21, 467–480. [Google Scholar] [CrossRef]
- Gan, J.; Ma, S.; Zhang, D. Non-Cytochrome P450-Mediated Bioactivation and Its Toxicological Relevance. Drug Metab. Rev. 2016, 48, 473–501. [Google Scholar] [CrossRef] [PubMed]
- Furge, L.L.; Guengerich, F.P. Cytochrome P450 Enzymes in Drug Metabolism and Chemical Toxicology: An Introduction. Biochem. Mol. Biol. Educ. 2006, 34, 66–74. [Google Scholar] [CrossRef]
- Bernhardt, R. Cytochromes P450 as Versatile Biocatalysts. J. Biotechnol. 2006, 124, 128–145. [Google Scholar] [CrossRef]
- Grillo, M.P. Bioactivation by Phase-II-Enzyme-Catalyzed Conjugation of Xenobiotics. In Encyclopedia of Drug Metabolism and Interactions; Lyubimov, A.V., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; ISBN 978-0-470-92192-0. [Google Scholar] [CrossRef]
- Iyanagi, T. Molecular Mechanism of Phase I and Phase II Drug-Metabolizing Enzymes: Implications for Detoxification. In International Review of Cytology; Elsevier: Amsterdam, The Netherlands, 2007; Volume 260, pp. 35–112. ISBN 978-0-12-374114-1. [Google Scholar]
- Roy, U.; Barber, P.; Tse-Dinh, Y.-C.; Batrakova, E.V.; Mondal, D.; Nair, M. Role of MRP Transporters in Regulating Antimicrobial Drug Inefficacy and Oxidative Stress-Induced Pathogenesis during HIV-1 and TB Infections. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Döring, B.; Petzinger, E. Phase 0 and Phase III Transport in Various Organs: Combined Concept of Phases in Xenobiotic Transport and Metabolism. Drug Metab. Rev. 2014, 46, 261–282. [Google Scholar] [CrossRef]
- Petzinger, E.; Geyer, J. Drug Transporters in Pharmacokinetics. Naunyn. Schmiedebergs Arch. Pharmacol. 2006, 372, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K. Cytochrome P450 and Anticancer Drugs. Curr. Drug Metab. 2006, 7, 23–37. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Human Cytochrome P450 Enzymes. In Cytochrome P450: Structure, Mechanism, and Biochemistry; Ortiz de Montellano, P.R., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 523–785. ISBN 978-3-319-12108-6. [Google Scholar]
- Frederiks, C.N.; Lam, S.W.; Guchelaar, H.J.; Boven, E. Genetic Polymorphisms and Paclitaxel- or Docetaxel-Induced Toxicities: A Systematic Review. Cancer Treat. Rev. 2015, 41, 935–950. [Google Scholar] [CrossRef]
- Stingl, J.C.; Brockmöller, J.; Viviani, R. Genetic Variability of Drug-Metabolizing Enzymes: The Dual Impact on Psychiatric Therapy and Regulation of Brain Function. Mol. Psychiatry 2013, 18, 273–287. [Google Scholar] [CrossRef]
- Annalora, A.J.; Marcus, C.B.; Iversen, P.L. Alternative Splicing in the Cytochrome P450 Superfamily Expands Protein Diversity to Augment Gene Function and Redirect Human Drug Metabolism. Drug Metab. Dispos. 2017, 45, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Nelson, D.R.; Feyereisen, R. Evolution of the Cytochrome P450 Genes. Xenobiotica Fate Foreign Compd. Biol. Syst. 1989, 19, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Omura, T. Future Perception in P450 Research. J. Inorg. Biochem. 2018, 186, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Substrate Oxidation by Cytochrome P450 Enzymes. In Cytochrome P450; Ortiz de Montellano, P.R., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 111–176. ISBN 978-3-319-12107-9. [Google Scholar]
- Estabrook, R.W. A Passion for P450s (Rememberances of the Early History of Research on Cytochrome P450). Drug Metab. Dispos. Biol. Fate Chem. 2003, 31, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Mueller, G.C.; Miller, J.A. The Metabolism of 4-Dimethylaminoazobenzene by Rat Liver Homogenates. J. Biol. Chem. 1948, 176, 535–544. [Google Scholar] [CrossRef]
- Klingenberg, M. Pigments of Rat Liver Microsomes. Arch. Biochem. Biophys. 1958, 75, 376–386. [Google Scholar] [CrossRef]
- Garfinkel, D. Studies on Pig Liver Microsomes. I. Enzymic and Pigment Composition of Different Microsomal Fractions. Arch. Biochem. Biophys. 1958, 77, 493–509. [Google Scholar] [CrossRef]
- Omura, T.; Sato, R. A New Cytochrome in Liver Microsomes. J. Biol. Chem. 1962, 237, 1375–1376. [Google Scholar] [CrossRef]
- Conney, A.H. Pharmacological Implications of Microsomal Enzyme Induction. Pharmacol. Rev. 1967, 19, 317–366. [Google Scholar]
- Hildebrandt, A.; Remmer, H.; Estabrook, R.W. Cytochrome P-450 of Liver Microsomes—One Pigment or Many. Biochem. Biophys. Res. Commun. 1968, 30, 607–612. [Google Scholar] [CrossRef]
- Gillette, J.R.; Davis, D.C.; Sasame, H.A. Cytochrome P-450 and Its Role in Drug Metabolism. Annu. Rev. Pharmacol. 1972, 12, 57–84. [Google Scholar] [CrossRef]
- Wada, O.; Yano, Y. Adaptive Responses of the Liver to Foreign Compounds, with Special Reference to Microsomal Drug-Matabolizing Enzymes. Rev. Environ. Health 1974, 1, 261–282. [Google Scholar] [PubMed]
- Parke, D.V. Induction of the Drug-Metabolizing Enzymes. Basic Life Sci. 1975, 6, 207–271. [Google Scholar] [CrossRef] [PubMed]
- Estabrook, R.W.; Cooper, D.Y.; Rosenthal, O. The Light Reversible Carbon Monoxide Inhibition of the Steroid C21-Hydroxylase System of the Adrenal Cortex. Biochem. Z. 1963, 338, 741–755. [Google Scholar]
- Cooper, D.Y.; Estabrook, R.W.; Rosenthal, O. The Stoichiometry of C21 Hydroxylation of Steroids by Adrenocortical Microsomes. J. Biol. Chem. 1963, 238, 1320–1323. [Google Scholar] [CrossRef]
- Lu, A.Y.; Coon, M.J. Role of Hemoprotein P-450 in Fatty Acid Omega-Hydroxylation in a Soluble Enzyme System from Liver Microsomes. J. Biol. Chem. 1968, 243, 1331–1332. [Google Scholar] [CrossRef]
- Imai, Y.; Sato, R. A Gel-Electrophoretically Homogeneous Preparation of Cytochrome P-450 from Liver Microsomes of Phenobarbital-Pretreated Rabbits. Biochem. Biophys. Res. Commun. 1974, 60, 8–14. [Google Scholar] [CrossRef]
- Haugen, D.A.; van der Hoeven, T.A.; Coon, M.J. Purified Liver Microsomal Cytochrome P-450. Separation and Characterization of Multiple Forms. J. Biol. Chem. 1975, 250, 3567–3570. [Google Scholar] [CrossRef]
- Alvares, A.P.; Schilling, G.; Levin, W.; Kuntzman, R. Studies on the Induction of CO-Binding Pigments in Liver Microsomes by Phenobarbital and 3-Methylcholanthrene. Biochem. Biophys. Res. Commun. 1967, 29, 521–526. [Google Scholar] [CrossRef]
- Sladek, N.E.; Mannering, G.J. Induction of Drug Metabolism. II. Qualitative Differences in the Microsomal N-Demethylating Systems Stimulated by Polycyclic Hydrocarbons and by Phenobarbital. Mol. Pharmacol. 1969, 5, 186–199. [Google Scholar] [PubMed]
- Schenkman, J.B.; Remmer, H.; Estabrook, R.W. Spectral Studies of Drug Interaction with Hepatic Microsomal Cytochrome. Mol. Pharmacol. 1967, 3, 113–123. [Google Scholar] [PubMed]
- Katagiri, M.; Ganguli, B.N.; Gunsalus, I.C. A Soluble Cytochrome P-450 Functional in Methylene Hydroxylation. J. Biol. Chem. 1968, 243, 3543–3546. [Google Scholar] [CrossRef]
- Groves, J.T.; McClusky, G.A. Aliphatic Hydroxylation by Highly Purified Liver Microsomal Cytochrome P-450. Evidence for a Carbon Radical Intermediate. Biochem. Biophys. Res. Commun. 1978, 81, 154–160. [Google Scholar] [CrossRef]
- Mizukami, Y.; Sogawa, K.; Suwa, Y.; Muramatsu, M.; Fujii-Kuriyama, Y. Gene Structure of a Phenobarbital-Inducible Cytochrome P-450 in Rat Liver. Proc. Natl. Acad. Sci. USA 1983, 80, 3958–3962. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Gonzalez, F.J. P450 Genes: Structure, Evolution, and Regulation. Annu. Rev. Biochem. 1987, 56, 945–993. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, M.; Ingelman-Sundberg, M.; Evans, W.E. Pharmacogenomics and Individualized Drug Therapy. Annu. Rev. Med. 2006, 57, 119–137. [Google Scholar] [CrossRef]
- Johansson, I.; Ingelman-Sundberg, M. Genetic Polymorphism and Toxicology—With Emphasis on Cytochrome P450. Toxicol. Sci. 2011, 120, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Solus, J.F.; Arietta, B.J.; Harris, J.R.; Sexton, D.P.; Steward, J.Q.; McMunn, C.; Ihrie, P.; Mehall, J.M.; Edwards, T.L.; Dawson, E.P. Genetic Variation in Eleven Phase I Drug Metabolism Genes in an Ethnically Diverse Population. Pharmacogenomics 2004, 5, 895–931. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Feng, S. Role of Metabolic Enzymes P450 (CYP) on Activating Procarcinogen and Their Polymorphisms on the Risk of Cancers. Curr. Drug Metab. 2015, 16, 850–863. [Google Scholar] [CrossRef] [PubMed]
- Stranger, B.E.; Forrest, M.S.; Dunning, M.; Ingle, C.E.; Beazley, C.; Thorne, N.; Redon, R.; Bird, C.P.; de Grassi, A.; Lee, C.; et al. Relative Impact of Nucleotide and Copy Number Variation on Gene Expression Phenotypes. Science 2007, 315, 848–853. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. The Human Genome Project and Novel Aspects of Cytochrome P450 Research. Toxicol. Appl. Pharmacol. 2005, 207, 52–56. [Google Scholar] [CrossRef]
- Nelson, D.R. The Cytochrome P450 Homepage. Hum. Genomics 2009, 4, 59–65. [Google Scholar] [CrossRef]
- Barnes, H.J.; Arlotto, M.P.; Waterman, M.R. Expression and Enzymatic Activity of Recombinant Cytochrome P450 17 Alpha-Hydroxylase in Escherichia Coli. Proc. Natl. Acad. Sci. USA 1991, 88, 5597–5601. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.R.; Coon, M.J.; Porter, T.D. Alcohol-Inducible Cytochrome P-450IIE1 Lacking the Hydrophobic NH2-Terminal Segment Retains Catalytic Activity and Is Membrane-Bound When Expressed in Escherichia Coli. J. Biol. Chem. 1991, 266, 7321–7324. [Google Scholar] [CrossRef]
- Li, Y.C.; Chiang, J.Y. The Expression of a Catalytically Active Cholesterol 7 Alpha-Hydroxylase Cytochrome P450 in Escherichia Coli. J. Biol. Chem. 1991, 266, 19186–19191. [Google Scholar] [CrossRef]
- Fisher, C.W.; Caudle, D.L.; Martin-Wixtrom, C.; Quattrochi, L.C.; Tukey, R.H.; Waterman, M.R.; Estabrook, R.W. High-Level Expression of Functional Human Cytochrome P450 1A2 in Escherichia Coli. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1992, 6, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Kranendonk, M.; Mesquita, P.; Laires, A.; Vermeulen, N.P.; Rueff, J. Expression of Human Cytochrome P450 1A2 in Escherichia Coli: A System for Biotransformation and Genotoxicity Studies of Chemical Carcinogens. Mutagenesis 1998, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Kranendonk, M.; Carreira, F.; Theisen, P.; Laires, A.; Fisher, C.W.; Rueff, J.; Estabrook, R.W.; Vermeulen, N.P. Escherichia Coli MTC, a Human NADPH P450 Reductase Competent Mutagenicity Tester Strain for the Expression of Human Cytochrome P450 Isoforms 1A1, 1A2, 2A6, 3A4, or 3A5: Catalytic Activities and Mutagenicity Studies. Mutat. Res. 1999, 441, 73–83. [Google Scholar] [CrossRef]
- Poulos, T.L.; Finzel, B.C.; Gunsalus, I.C.; Wagner, G.C.; Kraut, J. The 2.6-A Crystal Structure of Pseudomonas Putida Cytochrome P-450. J. Biol. Chem. 1985, 260, 16122–16130. [Google Scholar] [CrossRef]
- Poulos, T.L.; Finzel, B.C.; Howard, A.J. High-Resolution Crystal Structure of Cytochrome P450cam. J. Mol. Biol. 1987, 195, 687–700. [Google Scholar] [CrossRef]
- Ravichandran, K.G.; Boddupalli, S.S.; Hasermann, C.A.; Peterson, J.A.; Deisenhofer, J. Crystal Structure of Hemoprotein Domain of P450BM-3, a Prototype for Microsomal P450’s. Science 1993, 261, 731–736. [Google Scholar] [CrossRef]
- Williams, P.A.; Cosme, J.; Ward, A.; Angove, H.C.; Matak Vinković, D.; Jhoti, H. Crystal Structure of Human Cytochrome P450 2C9 with Bound Warfarin. Nature 2003, 424, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The Structure of Human Microsomal Cytochrome P450 3A4 Determined by X-Ray Crystallography to 2.05-A Resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef]
- Rowland, P.; Blaney, F.E.; Smyth, M.G.; Jones, J.J.; Leydon, V.R.; Oxbrow, A.K.; Lewis, C.J.; Tennant, M.G.; Modi, S.; Eggleston, D.S.; et al. Crystal Structure of Human Cytochrome P450 2D6. J. Biol. Chem. 2006, 281, 7614–7622. [Google Scholar] [CrossRef]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the Oxidation of Polycyclic Aromatic Hydrocarbons Exhibited by the Structure of Human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef] [PubMed]
- Duarte, M.P.; Palma, B.B.; Gilep, A.A.; Laires, A.; Oliveira, J.S.; Usanov, S.A.; Rueff, J.; Kranendonk, M. The Stimulatory Role of Human Cytochrome b5 in the Bioactivation Activities of Human CYP1A2, 2A6 and 2E1: A New Cell Expression System to Study Cytochrome P450-Mediated Biotransformation (a Corrigendum Report on Duarte et al. (2005) Mutagenesis 20, 93–100). Mutagenesis 2007, 22, 75–81. [Google Scholar] [CrossRef]
- Palma, B.B.; Silva, E.; Sousa, M.; Urban, P.; Rueff, J.; Kranendonk, M. Functional Characterization of Eight Human CYP1A2 Variants: The Role of Cytochrome b5. Pharmacogenet. Genomics 2013, 23, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Nakamura, M.; Komatsu, T.; Ohyama, K.; Hatanaka, N.; Asahi, S.; Shimada, N.; Guengerich, F.P.; Shimada, T.; Nakajima, M.; et al. Roles of NADPH-P450 Reductase and Apo- and Holo-Cytochrome b5 on Xenobiotic Oxidations Catalyzed by 12 Recombinant Human Cytochrome P450s Expressed in Membranes of Escherichia Coli. Protein Expr. Purif. 2002, 24, 329–337. [Google Scholar] [CrossRef]
- Yamaori, S.; Yamazaki, H.; Suzuki, A.; Yamada, A.; Tani, H.; Kamidate, T.; Fujita, K.; Kamataki, T. Effects of Cytochrome b(5) on Drug Oxidation Activities of Human Cytochrome P450 (CYP) 3As: Similarity of CYP3A5 with CYP3A4 but Not CYP3A7. Biochem. Pharmacol. 2003, 66, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Storbeck, K.-H.; Swart, A.C.; Fox, C.L.; Swart, P. Cytochrome b5 Modulates Multiple Reactions in Steroidogenesis by Diverse Mechanisms. J. Steroid Biochem. Mol. Biol. 2015, 151, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Hlavica, P. Mechanistic Basis of Electron Transfer to Cytochromes P450 by Natural Redox Partners and Artificial Donor Constructs. Adv. Exp. Med. Biol. 2015, 851, 247–297. [Google Scholar] [CrossRef] [PubMed]
- Waskell, L.; Kim, J.-J.P. Electron Transfer Partners of Cytochrome P450. In Cytochrome P450: Structure, Mechanism, and Biochemistry; Ortiz de Montellano, P.R., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 33–68. ISBN 978-3-319-12108-6. [Google Scholar]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human Cytochromes P450 in Health and Disease. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Adesnik, M.; Coon, M.J.; Estabrook, R.W.; Gonzalez, F.J.; Guengerich, F.P.; Gunsalus, I.C.; Johnson, E.F.; Kemper, B.; Levin, W. The P450 Gene Superfamily: Recommended Nomenclature. DNA 1987, 6, 1–11. [Google Scholar] [CrossRef]
- Maron, D.M.; Ames, B.N. Revised Methods for the Salmonella Mutagenicity Test. Mutat. Res. 1983, 113, 173–215. [Google Scholar] [CrossRef]
- Rueff, J.; Rodrigues, A.S.; Kranendonk, M. A Personally Guided Tour on Some of Our Data with the Ames Assay-A Tribute to Professor Bruce Ames. Mutat. Res. 2019, 846, 503094. [Google Scholar] [CrossRef]
- Hakura, A.; Shimada, H.; Nakajima, M.; Sui, H.; Kitamoto, S.; Suzuki, S.; Satoh, T. Salmonella/Human S9 Mutagenicity Test: A Collaborative Study with 58 Compounds. Mutagenesis 2005, 20, 217–228. [Google Scholar] [CrossRef]
- Baillie, T.A.; Rettie, A.E. Role of Biotransformation in Drug-Induced Toxicity: Influence of Intra- and Inter-Species Differences in Drug Metabolism. Drug Metab. Pharmacokinet. 2011, 26, 15–29. [Google Scholar] [CrossRef]
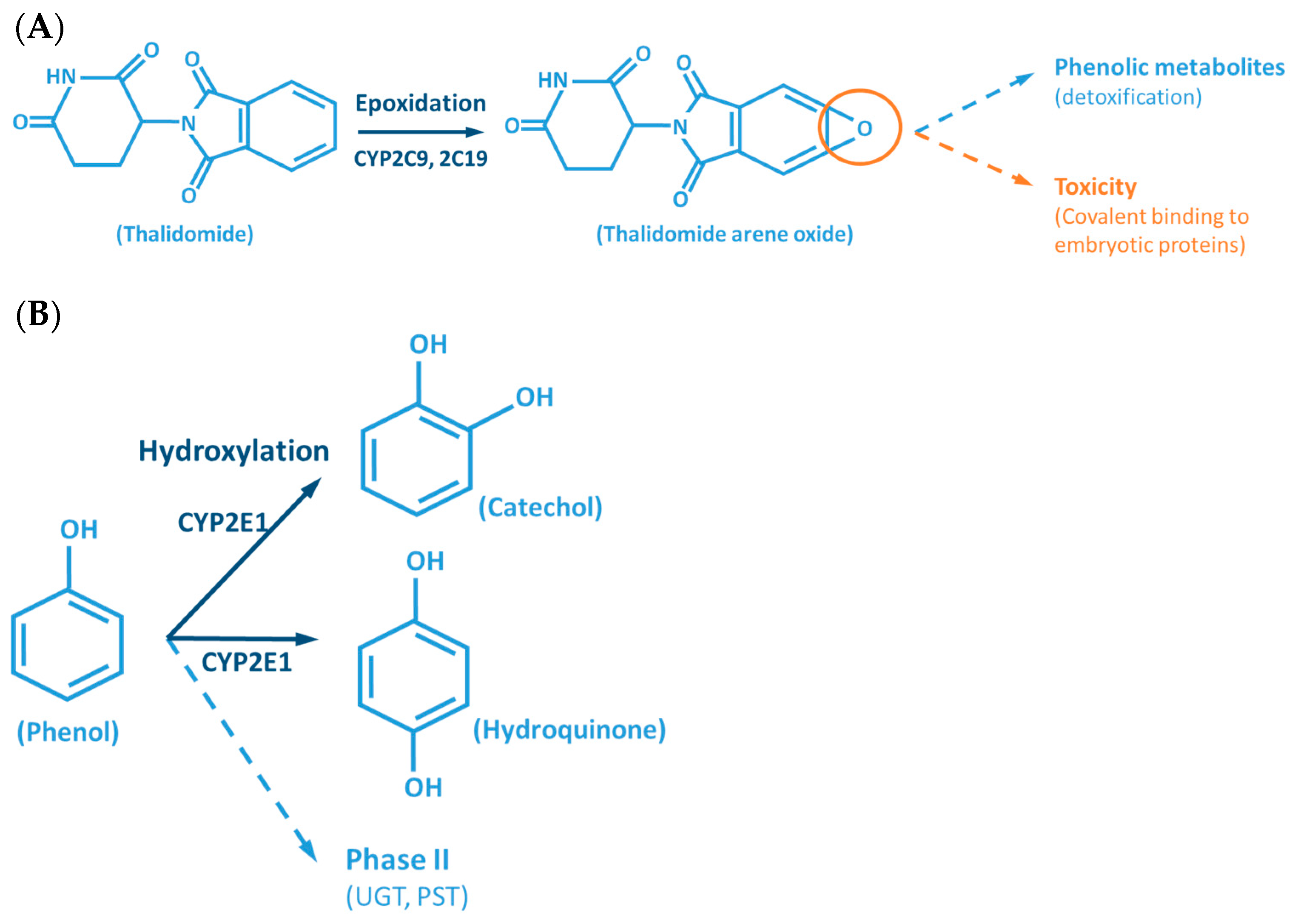
- Lu, J.; Helsby, N.; Palmer, B.D.; Tingle, M.; Baguley, B.C.; Kestell, P.; Ching, L.-M. Metabolism of Thalidomide in Liver Microsomes of Mice, Rabbits, and Humans. J. Pharmacol. Exp. Ther. 2004, 310, 571–577. [Google Scholar] [CrossRef]
- Gordon, G.B.; Spielberg, S.P.; Blake, D.A.; Balasubramanian, V. Thalidomide Teratogenesis: Evidence for a Toxic Arene Oxide Metabolite. Proc. Natl. Acad. Sci. USA 1981, 78, 2545–2548. [Google Scholar] [CrossRef] [PubMed]
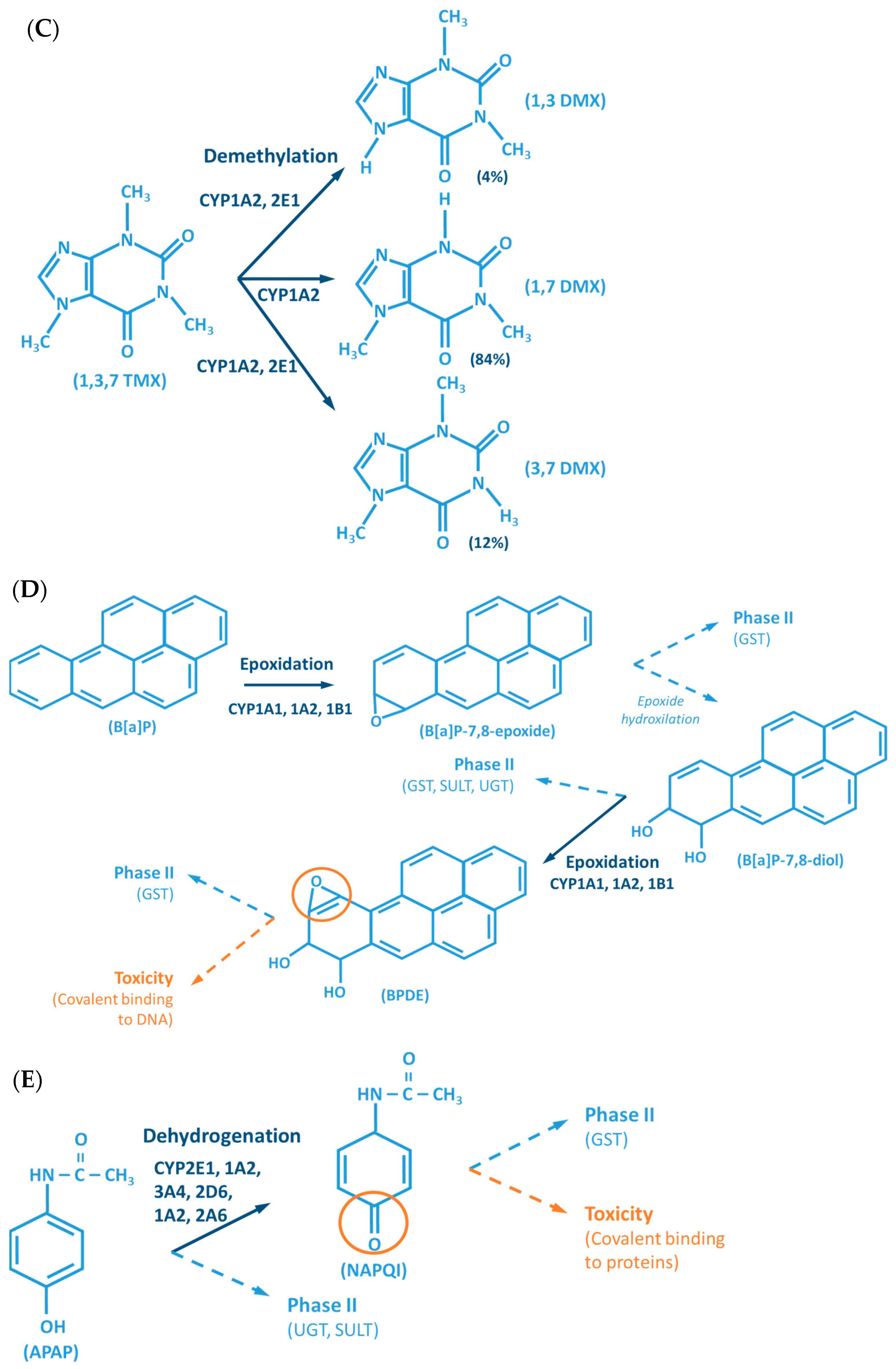
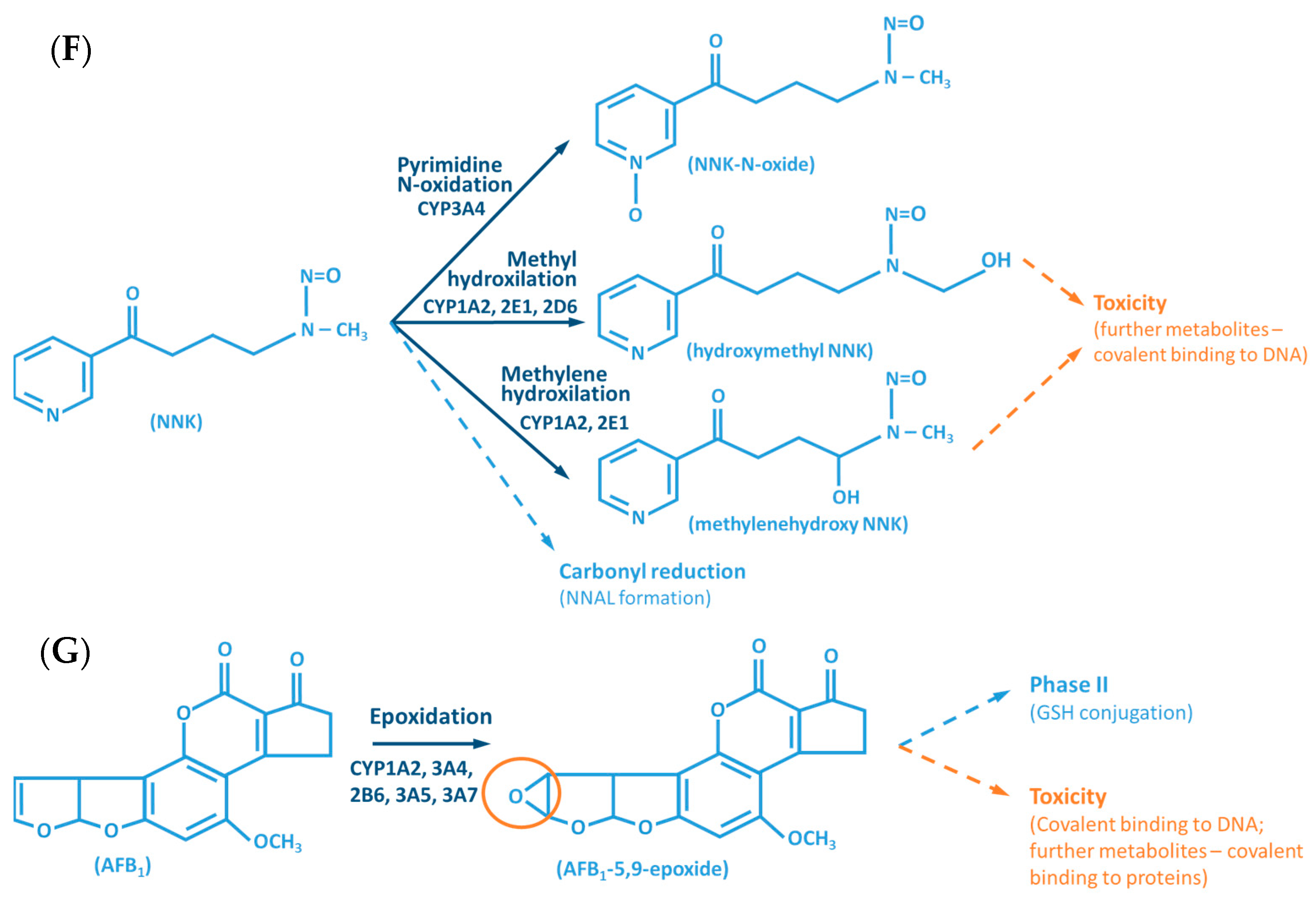
- Williams, D.E.; Buhler, D.R. Purified Form of Cytochrome P-450 from Rainbow Trout with High Activity toward Conversion of Aflatoxin B1 to Aflatoxin B1-2,3-Epoxide. Cancer Res. 1983, 43, 4752–4756. [Google Scholar]
- Eaton, D.L.; Gallagher, E.P. Mechanisms of Aflatoxin Carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 135–172. [Google Scholar] [CrossRef]
- Nelson, D.R. Cytochrome P450 Nomenclature. Methods Mol. Biol. Clifton N. J. 1998, 107, 15–24. [Google Scholar] [CrossRef]
- Nelson, D.R. Progress in Tracing the Evolutionary Paths of Cytochrome P450. Biochim. Biophys. Acta 2011, 1814, 14–18. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Waterman, M.R.; Egli, M. Recent Structural Insights into Cytochrome P450 Function. Trends Pharmacol. Sci. 2016, 37, 625–640. [Google Scholar] [CrossRef]
- Omura, T. Forty Years of Cytochrome P450. Biochem. Biophys. Res. Commun. 1999, 266, 690–698. [Google Scholar] [CrossRef]
- Collins, S.L.; Patterson, A.D. The Gut Microbiome: An Orchestrator of Xenobiotic Metabolism. Acta Pharm. Sin. B 2020, 10, 19–32. [Google Scholar] [CrossRef]
- Zimmermann, M.; Zimmermann-Kogadeeva, M.; Wegmann, R.; Goodman, A.L. Mapping Human Microbiome Drug Metabolism by Gut Bacteria and Their Genes. Nature 2019, 570, 462–467. [Google Scholar] [CrossRef]
- Selwyn, F.P.; Cheng, S.L.; Klaassen, C.D.; Cui, J.Y. Regulation of Hepatic Drug-Metabolizing Enzymes in Germ-Free Mice by Conventionalization and Probiotics. Drug Metab. Dispos. Biol. Fate Chem. 2016, 44, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Aleksunes, L.M.; Klaassen, C.D. Coordinated Regulation of Hepatic Phase I and II Drug-Metabolizing Genes and Transporters Using AhR-, CAR-, PXR-, PPARα-, and Nrf2-Null Mice. Drug Metab. Dispos. Biol. Fate Chem. 2012, 40, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F. On the Recognition of Mammalian Microsomal Cytochrome P450 Substrates and Their Characteristics: Towards the Prediction of Human P450 Substrate Specificity and Metabolism. Biochem. Pharmacol. 2000, 60, 293–306. [Google Scholar] [CrossRef]
- Urban, P.; Truan, G.; Pompon, D. Access Channels to the Buried Active Site Control Substrate Specificity in CYP1A P450 Enzymes. Biochim. Biophys. Acta 2015, 1850, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A. Interindividual Differences in Caffeine Metabolism and Factors Driving Caffeine Consumption. Pharmacol. Rev. 2018, 70, 384–411. [Google Scholar] [CrossRef]
- Song, M.-K.; Yoon, J.-S.; Song, M.; Choi, H.-S.; Shin, C.-Y.; Kim, Y.-J.; Ryu, W.-I.; Lee, H.-S.; Ryu, J.-C. Gene Expression Analysis Identifies DNA Damage-Related Markers of Benzo[a]Pyrene Exposure in HepG2 Human Hepatocytes. Toxicol. Environ. Health Sci. 2012, 4, 19–29. [Google Scholar] [CrossRef]
- Barnes, J.L.; Zubair, M.; John, K.; Poirier, M.C.; Martin, F.L. Carcinogens and DNA Damage. Biochem. Soc. Trans. 2018, 46, 1213–1224. [Google Scholar] [CrossRef]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Pathways of Acetaminophen Metabolism at the Therapeutic versus Toxic Doses. Pharmacogenet. Genomics 2015, 25, 416–426. [Google Scholar] [CrossRef]
- Wu, J.; Guan, X.; Dai, Z.; He, R.; Ding, X.; Yang, L.; Ge, G. Molecular Probes for Human Cytochrome P450 Enzymes: Recent Progress and Future Perspectives. Coord. Chem. Rev. 2021, 427, 213600. [Google Scholar] [CrossRef]
- Isin, E.M.; Guengerich, F.P. Complex Reactions Catalyzed by Cytochrome P450 Enzymes. Biochim. Biophys. Acta 2007, 1770, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Masters, B.S.S. The Journey from NADPH-Cytochrome P450 Oxidoreductase to Nitric Oxide Synthases. Biochem. Biophys. Res. Commun. 2005, 338, 507–519. [Google Scholar] [CrossRef]
- Hamdane, D.; Xia, C.; Im, S.-C.; Zhang, H.; Kim, J.-J.P.; Waskell, L. Structure and Function of an NADPH-Cytochrome P450 Oxidoreductase in an Open Conformation Capable of Reducing Cytochrome P450. J. Biol. Chem. 2009, 284, 11374–11384. [Google Scholar] [CrossRef] [PubMed]
- Aigrain, L.; Pompon, D.; Moréra, S.; Truan, G. Structure of the Open Conformation of a Functional Chimeric NADPH Cytochrome P450 Reductase. EMBO Rep. 2009, 10, 742–747. [Google Scholar] [CrossRef]
- Ellis, J.; Gutierrez, A.; Barsukov, I.L.; Huang, W.-C.; Grossmann, J.G.; Roberts, G.C.K. Domain Motion in Cytochrome P450 Reductase: Conformational Equilibria Revealed by NMR and Small-Angle x-Ray Scattering. J. Biol. Chem. 2009, 284, 36628–36637. [Google Scholar] [CrossRef] [PubMed]
- Campelo, D.; Lautier, T.; Urban, P.; Esteves, F.; Bozonnet, S.; Truan, G.; Kranendonk, M. The Hinge Segment of Human NADPH-Cytochrome P450 Reductase in Conformational Switching: The Critical Role of Ionic Strength. Front. Pharmacol. 2017, 8, 755. [Google Scholar] [CrossRef]
- Campelo, D.; Esteves, F.; Brito Palma, B.; Costa Gomes, B.; Rueff, J.; Lautier, T.; Urban, P.; Truan, G.; Kranendonk, M. Probing the Role of the Hinge Segment of Cytochrome P450 Oxidoreductase in the Interaction with Cytochrome P450. Int. J. Mol. Sci. 2018, 19, 3914. [Google Scholar] [CrossRef]
- Rendic, S.; Guengerich, F.P. Contributions of Human Enzymes in Carcinogen Metabolism. Chem. Res. Toxicol. 2012, 25, 1316–1383. [Google Scholar] [CrossRef]
- Jin, Y.; Zollinger, M.; Borell, H.; Zimmerlin, A.; Patten, C.J. CYP4F Enzymes Are Responsible for the Elimination of Fingolimod (FTY720), a Novel Treatment of Relapsing Multiple Sclerosis. Drug Metab. Dispos. Biol. Fate Chem. 2011, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Edson, K.Z.; Rettie, A.E. CYP4 Enzymes as Potential Drug Targets: Focus on Enzyme Multiplicity, Inducers and Inhibitors, and Therapeutic Modulation of 20-Hydroxyeicosatetraenoic Acid (20-HETE) Synthase and Fatty Acid ω-Hydroxylase Activities. Curr. Top. Med. Chem. 2013, 13, 1429–1440. [Google Scholar] [CrossRef]
- Seguin, R.P.; Herron, J.M.; Lopez, V.A.; Dempsey, J.L.; Xu, L. Metabolism of Benzalkonium Chlorides by Human Hepatic Cytochromes P450. Chem. Res. Toxicol. 2019, 32, 2466–2478. [Google Scholar] [CrossRef]
- Hsu, M.-H.; Savas, U.; Lasker, J.M.; Johnson, E.F. Genistein, Resveratrol, and 5-Aminoimidazole-4-Carboxamide-1-β-D-Ribofuranoside Induce Cytochrome P450 4F2 Expression through an AMP-Activated Protein Kinase-Dependent Pathway. J. Pharmacol. Exp. Ther. 2011, 337, 125–136. [Google Scholar] [CrossRef]
- Wahlang, B.; Falkner, K.C.; Cave, M.C.; Prough, R.A. Role of Cytochrome P450 Monooxygenase in Carcinogen and Chemotherapeutic Drug Metabolism. Adv. Pharmacol. San Diego Calif 2015, 74, 1–33. [Google Scholar] [CrossRef]
- Stepanov, I.; Upadhyaya, P.; Carmella, S.G.; Feuer, R.; Jensen, J.; Hatsukami, D.K.; Hecht, S.S. Extensive Metabolic Activation of the Tobacco-Specific Carcinogen 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-Butanone in Smokers. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1764–1773. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guengerich, F.P. Principles of Covalent Binding of Reactive Metabolites and Examples of Activation of Bis-Electrophiles by Conjugation. Arch. Biochem. Biophys. 2005, 433, 369–378. [Google Scholar] [CrossRef]
- Mizutani, T. PM Frequencies of Major CYPs in Asians and Caucasians. Drug Metab. Rev. 2003, 35, 99–106. [Google Scholar] [CrossRef]
- Distlerath, L.M.; Reilly, P.E.; Martin, M.V.; Davis, G.G.; Wilkinson, G.R.; Guengerich, F.P. Purification and Characterization of the Human Liver Cytochromes P-450 Involved in Debrisoquine 4-Hydroxylation and Phenacetin O-Deethylation, Two Prototypes for Genetic Polymorphism in Oxidative Drug Metabolism. J. Biol. Chem. 1985, 260, 9057–9067. [Google Scholar] [CrossRef]
- Chun, Y.-J.; Kim, D. Cancer Activation and Polymorphisms of Human Cytochrome P450 1B1. Toxicol. Res. 2016, 32, 89–93. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Sim, S.C.; Gomez, A.; Rodriguez-Antona, C. Influence of Cytochrome P450 Polymorphisms on Drug Therapies: Pharmacogenetic, Pharmacoepigenetic and Clinical Aspects. Pharmacol. Ther. 2007, 116, 496–526. [Google Scholar] [CrossRef]
- Burkhard, F.Z.; Parween, S.; Udhane, S.S.; Flück, C.E.; Pandey, A.V. P450 Oxidoreductase Deficiency: Analysis of Mutations and Polymorphisms. J. Steroid Biochem. Mol. Biol. 2017, 165, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Udhane, S.S.; Parween, S.; Kagawa, N.; Pandey, A.V. Altered CYP19A1 and CYP3A4 Activities Due to Mutations A115V, T142A, Q153R and P284L in the Human P450 Oxidoreductase. Front. Pharmacol. 2017, 8, 580. [Google Scholar] [CrossRef] [PubMed]
- Esteves, F.; Campelo, D.; Gomes, B.C.; Urban, P.; Bozonnet, S.; Lautier, T.; Rueff, J.; Truan, G.; Kranendonk, M. The Role of the FMN-Domain of Human Cytochrome P450 Oxidoreductase in Its Promiscuous Interactions With Structurally Diverse Redox Partners. Front. Pharmacol. 2020, 11, 299. [Google Scholar] [CrossRef]
- Esteves, F.; Urban, P.; Rueff, J.; Truan, G.; Kranendonk, M. Interaction Modes of Microsomal Cytochrome P450s with Its Reductase and the Role of Substrate Binding. Int. J. Mol. Sci. 2020, 21, 6669. [Google Scholar] [CrossRef]
- Morgan, E. Impact of Infectious and Inflammatory Disease on Cytochrome P450–Mediated Drug Metabolism and Pharmacokinetics. Clin. Pharmacol. Ther. 2009, 85, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.T. Regulation of Drug-Metabolizing Enzymes and Drug Metabolism by Inflammatory Responses. In Drug Metabolism in Diseases; Wen, X., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 21–58. ISBN 978-0-12-802949-7. [Google Scholar]
- Beedanagari, S.R.; Taylor, R.T.; Bui, P.; Wang, F.; Nickerson, D.W.; Hankinson, O. Role of Epigenetic Mechanisms in Differential Regulation of the Dioxin-Inducible Human CYP1A1 and CYP1B1 Genes. Mol. Pharmacol. 2010, 78, 608–616. [Google Scholar] [CrossRef]
- Pan, Y.-Z.; Gao, W.; Yu, A.-M. MicroRNAs Regulate CYP3A4 Expression via Direct and Indirect Targeting. Drug Metab. Dispos. Biol. Fate Chem. 2009, 37, 2112–2117. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, L.O.; Edenberg, H.J. Epigenetics of Gene Expression in Human Hepatoma Cells: Expression Profiling the Response to Inhibition of DNA Methylation and Histone Deacetylation. BMC Genomics 2006, 7, 181. [Google Scholar] [CrossRef]
- Anttila, S.; Hakkola, J.; Tuominen, P.; Elovaara, E.; Husgafvel-Pursiainen, K.; Karjalainen, A.; Hirvonen, A.; Nurminen, T. Methylation of Cytochrome P4501A1 Promoter in the Lung Is Associated with Tobacco Smoking. Cancer Res. 2003, 63, 8623–8628. [Google Scholar]
- Tsuchiya, Y.; Nakajima, M.; Takagi, S.; Taniya, T.; Yokoi, T. MicroRNA Regulates the Expression of Human Cytochrome P450 1B1. Cancer Res. 2006, 66, 9090–9098. [Google Scholar] [CrossRef] [PubMed]
- Mohri, T.; Nakajima, M.; Fukami, T.; Takamiya, M.; Aoki, Y.; Yokoi, T. Human CYP2E1 Is Regulated by MiR-378. Biochem. Pharmacol. 2010, 79, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Lamba, V.; Ghodke, Y.; Guan, W.; Tracy, T.S. MicroRNA-34a Is Associated with Expression of Key Hepatic Transcription Factors and Cytochromes P450. Biochem. Biophys. Res. Commun. 2014, 445, 404–411. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Gomez, A. The Past, Present and Future of Pharmacoepigenomics. Pharmacogenomics 2010, 11, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Correia, M.A.; Hollenberg, P.F. Inhibition of Cytochrome P450 Enzymes. In Cytochrome P450; Ortiz de Montellano, P.R., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 177–259. ISBN 978-3-319-12107-9. [Google Scholar]
- Gentry, K.A.; Anantharamaiah, G.M.; Ramamoorthy, A. Probing Protein-Protein and Protein-Substrate Interactions in the Dynamic Membrane-Associated Ternary Complex of Cytochromes P450, b5, and Reductase. Chem. Commun. Camb. Engl. 2019, 55, 13422–13425. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Jamali, F. The Effect of Infliximab on Hepatic Cytochrome P450 and Pharmacokinetics of Verapamil in Rats with Pre-Adjuvant Arthritis: A Drug-Disease and Drug-Drug Interaction. Basic Clin. Pharmacol. Toxicol. 2009, 105, 24–29. [Google Scholar] [CrossRef]
- Tompkins, L.M.; Wallace, A.D. Mechanisms of Cytochrome P450 Induction. J. Biochem. Mol. Toxicol. 2007, 21, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, O.; Turpeinen, M.; Hakkola, J.; Honkakoski, P.; Hukkanen, J.; Raunio, H. Inhibition and Induction of Human Cytochrome P450 Enzymes: Current Status. Arch. Toxicol. 2008, 82, 667–715. [Google Scholar] [CrossRef]
- Ishihara, Y.; Hamaguchi, A.; Sekine, M.; Hirakawa, A.; Shimamoto, N. Accumulation of Cytochrome P450 Induced by Proteasome Inhibition during Cardiac Ischemia. Arch. Biochem. Biophys. 2012, 527, 16–22. [Google Scholar] [CrossRef]
- Pondugula, S.R.; Dong, H.; Chen, T. Phosphorylation and Protein-Protein Interactions in PXR-Mediated CYP3A Repression. Expert Opin. Drug Metab. Toxicol. 2009, 5, 861–873. [Google Scholar] [CrossRef]
- Mahpour, A.; Mullen, A.C. Our Emerging Understanding of the Roles of Long Non-Coding RNAs in Normal Liver Function, Disease, and Malignancy. JHEP Rep. Innov. Hepatol. 2021, 3, 100177. [Google Scholar] [CrossRef] [PubMed]
- Congiu, M.; Mashford, M.L.; Slavin, J.L.; Desmond, P.V. Coordinate Regulation of Metabolic Enzymes and Transporters by Nuclear Transcription Factors in Human Liver Disease. J. Gastroenterol. Hepatol. 2009, 24, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, K.; Yoda, N.; Toriyabe, T.; Yamazoe, Y. Constitutive Androstane Receptor Transcriptionally Activates Human CYP1A1 and CYP1A2 Genes through a Common Regulatory Element in the 5’-Flanking Region. Biochem. Pharmacol. 2010, 79, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Nakajima, M.; Higashi, E.; Yoshida, R.; Nagata, K.; Yamazoe, Y.; Yokoi, T. Induction of Human CYP2A6 Is Mediated by the Pregnane X Receptor with Peroxisome Proliferator-Activated Receptor-Gamma Coactivator 1alpha. J. Pharmacol. Exp. Ther. 2006, 319, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, T.; Kawamoto, T.; Zelko, I.; Honkakoski, P.; Negishi, M. The Repressed Nuclear Receptor CAR Responds to Phenobarbital in Activating the Human CYP2B6 Gene. J. Biol. Chem. 1999, 274, 6043–6046. [Google Scholar] [CrossRef]
- Wang, H.; Faucette, S.; Sueyoshi, T.; Moore, R.; Ferguson, S.; Negishi, M.; LeCluyse, E.L. A Novel Distal Enhancer Module Regulated by Pregnane X Receptor/Constitutive Androstane Receptor Is Essential for the Maximal Induction of CYP2B6 Gene Expression. J. Biol. Chem. 2003, 278, 14146–14152. [Google Scholar] [CrossRef]
- Willson, T.M.; Kliewer, S.A. PXR, CAR and Drug Metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Ueda, R.; Iketaki, H.; Nagata, K.; Kimura, S.; Gonzalez, F.J.; Kusano, K.; Yoshimura, T.; Yamazoe, Y. A Common Regulatory Region Functions Bidirectionally in Transcriptional Activation of the Human CYP1A1 and CYP1A2 Genes. Mol. Pharmacol. 2006, 69, 1924–1930. [Google Scholar] [CrossRef] [PubMed]
- Jorge-Nebert, L.F.; Jiang, Z.; Chakraborty, R.; Watson, J.; Jin, L.; McGarvey, S.T.; Deka, R.; Nebert, D.W. Analysis of Human CYP1A1 and CYP1A2 Genes and Their Shared Bidirectional Promoter in Eight World Populations. Hum. Mutat. 2010, 31, 27–40. [Google Scholar] [CrossRef]
- Benowitz, N.L.; Lessov-Schlaggar, C.N.; Swan, G.E.; Jacob, P. Female Sex and Oral Contraceptive Use Accelerate Nicotine Metabolism. Clin. Pharmacol. Ther. 2006, 79, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Higashi, E.; Fukami, T.; Itoh, M.; Kyo, S.; Inoue, M.; Yokoi, T.; Nakajima, M. Human CYP2A6 Is Induced by Estrogen via Estrogen Receptor. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 1935–1941. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Goldstein, J.A. The Transcriptional Regulation of the Human CYP2C Genes. Curr. Drug Metab. 2009, 10, 567–578. [Google Scholar] [CrossRef]
- Helsby, N.A.; Burns, K.E. Molecular Mechanisms of Genetic Variation and Transcriptional Regulation of CYP2C19. Front. Genet. 2012, 3, 206. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Schaefer, O.; Kawakami, H.; Inoue, T.; Liehner, S.; Saito, A.; Ishiguro, N.; Kishimoto, W.; Ludwig-Schwellinger, E.; Ebner, T.; et al. Simultaneous Absolute Protein Quantification of Transporters, Cytochromes P450, and UDP-Glucuronosyltransferases as a Novel Approach for the Characterization of Individual Human Liver: Comparison with MRNA Levels and Activities. Drug Metab. Dispos. Biol. Fate Chem. 2012, 40, 83–92. [Google Scholar] [CrossRef]
- Jover, R.; Moya, M.; Gómez-Lechón, M.J. Transcriptional Regulation of Cytochrome P450 Genes by the Nuclear Receptor Hepatocyte Nuclear Factor 4-Alpha. Curr. Drug Metab. 2009, 10, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, K.; Saito, T.; Takahashi, Y.; Ozeki, T.; Kiyotani, K.; Fujieda, M.; Yamazaki, H.; Kunitoh, H.; Kamataki, T. Identification of a Novel Polymorphic Enhancer of the Human CYP3A4 Gene. Mol. Pharmacol. 2004, 65, 326–334. [Google Scholar] [CrossRef]
- Schröder, A.; Wollnik, J.; Wrzodek, C.; Dräger, A.; Bonin, M.; Burk, O.; Thomas, M.; Thasler, W.E.; Zanger, U.M.; Zell, A. Inferring Statin-Induced Gene Regulatory Relationships in Primary Human Hepatocytes. Bioinforma. Oxf. Engl. 2011, 27, 2473–2477. [Google Scholar] [CrossRef]
- Martínez-Jiménez, C.P.; Gómez-Lechón, M.J.; Castell, J.V.; Jover, R. Transcriptional Regulation of the Human Hepatic CYP3A4: Identification of a New Distal Enhancer Region Responsive to CCAAT/Enhancer-Binding Protein Beta Isoforms (Liver Activating Protein and Liver Inhibitory Protein). Mol. Pharmacol. 2005, 67, 2088–2101. [Google Scholar] [CrossRef]
- Rodríguez-Antona, C.; Bort, R.; Jover, R.; Tindberg, N.; Ingelman-Sundberg, M.; Gómez-Lechón, M.J.; Castell, J.V. Transcriptional Regulation of Human CYP3A4 Basal Expression by CCAAT Enhancer-Binding Protein Alpha and Hepatocyte Nuclear Factor-3 Gamma. Mol. Pharmacol. 2003, 63, 1180–1189. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein Sensors for Membrane Sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef]
- Santes-Palacios, R.; Marroquín-Pérez, A.L.; Hernández-Ojeda, S.L.; Camacho-Carranza, R.; Govezensky, T.; Espinosa-Aguirre, J.J. Human CYP1A1 Inhibition by Flavonoids. Toxicol. Vitro Int. J. Publ. Assoc. BIBRA 2020, 62, 104681. [Google Scholar] [CrossRef]
- Ronis, M.J.J. Effects of Soy Containing Diet and Isoflavones on Cytochrome P450 Enzyme Expression and Activity. Drug Metab. Rev. 2016, 48, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Elkahwaji, J.; Robin, M.A.; Berson, A.; Tinel, M.; Lettéron, P.; Labbe, G.; Beaune, P.; Elias, D.; Rougier, P.; Escudier, B.; et al. Decrease in Hepatic Cytochrome P450 after Interleukin-2 Immunotherapy. Biochem. Pharmacol. 1999, 57, 951–954. [Google Scholar] [CrossRef]
- Abdel-Razzak, Z.; Garlatti, M.; Aggerbeck, M.; Barouki, R. Determination of Interleukin-4-Responsive Region in the Human Cytochrome P450 2E1 Gene Promoter. Biochem. Pharmacol. 2004, 68, 1371–1381. [Google Scholar] [CrossRef]
- Gorski, J.C.; Hall, S.D.; Becker, P.; Affrime, M.B.; Cutler, D.L.; Haehner-Daniels, B. In Vivo Effects of Interleukin-10 on Human Cytochrome P450 Activity. Clin. Pharmacol. Ther. 2000, 67, 32–43. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Ukairo, O.; Khetani, S.R.; McVay, M.; Kanchagar, C.; Seghezzi, W.; Ayanoglu, G.; Irrechukwu, O.; Evers, R. Establishment of a Hepatocyte-Kupffer Cell Coculture Model for Assessment of Proinflammatory Cytokine Effects on Metabolizing Enzymes and Drug Transporters. Drug Metab. Dispos. Biol. Fate Chem. 2015, 43, 774–785. [Google Scholar] [CrossRef]
- Siewert, E.; Bort, R.; Kluge, R.; Heinrich, P.C.; Castell, J.; Jover, R. Hepatic Cytochrome P450 Down-Regulation during Aseptic Inflammation in the Mouse Is Interleukin 6 Dependent. Hepatol. Baltim. Md. 2000, 32, 49–55. [Google Scholar] [CrossRef]
- Shah, R.R.; Smith, R.L. Inflammation-Induced Phenoconversion of Polymorphic Drug Metabolizing Enzymes: Hypothesis with Implications for Personalized Medicine. Drug Metab. Dispos. 2015, 43, 400–410. [Google Scholar] [CrossRef]
- Charles, K.A.; Rivory, L.P.; Brown, S.L.; Liddle, C.; Clarke, S.J.; Robertson, G.R. Transcriptional Repression of Hepatic Cytochrome P450 3A4 Gene in the Presence of Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 7492–7497. [Google Scholar] [CrossRef] [PubMed]
- Renton, K.W.; Nicholson, T.E. Hepatic and Central Nervous System Cytochrome P450 Are Down-Regulated during Lipopolysaccharide-Evoked Localized Inflammation in Brain. J. Pharmacol. Exp. Ther. 2000, 294, 524–530. [Google Scholar] [PubMed]
- Abdulla, D.; Goralski, K.B.; Renton, K.W. The Regulation of Cytochrome P450 2E1 during LPS-Induced Inflammation in the Rat. Toxicol. Appl. Pharmacol. 2006, 216, 1–10. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
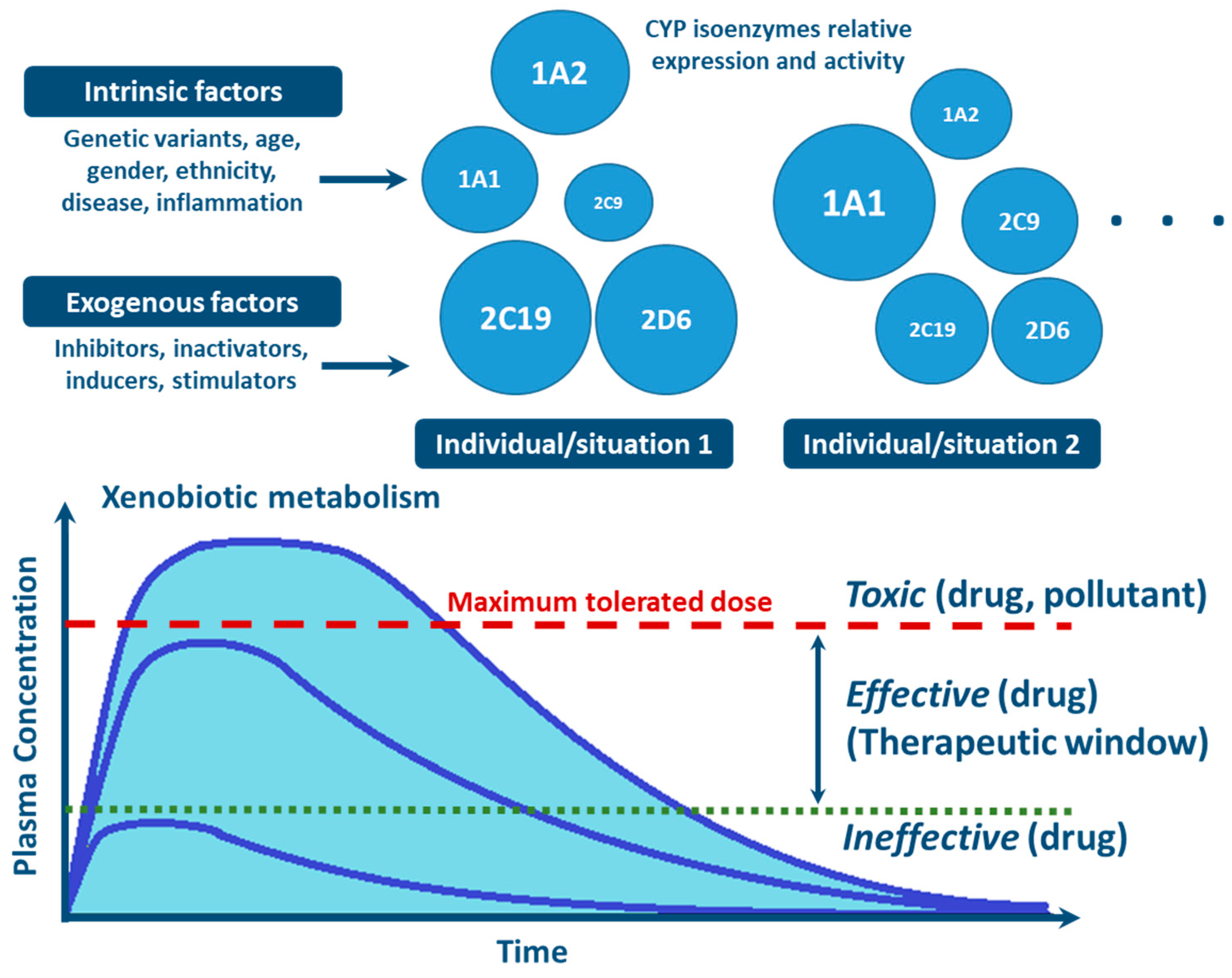{kind=link}
{kind=link}
| Classes of Compounds | CYP Isoenzymes |
|---|---|
| Sterols | 1B1, 7A1, 7B1, 8B1, 11A1, 11B1, 11B2, 17A1, 19A1, 21A2, 27A1, 39A1, 46A1, 51A1 |
| Xenobiotics | 1A1, 1A2, 2A6, 2A13, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 2F1, 3A4, 3A5, 3A7 |
| Fatty acids | 2J2, 2U1, 4A11, 4B1, 4F11, 4F12, 4F22, 4V2, 4X1, 4Z1 |
| Eicosanoids | 4F2, 4F3, 4F8, 5A1, 8A1 |
| Vitamins | 2R1, 24A1, 26A1, 26B1, 26C1, 27B1, 27C1 |
| Unknown | 2A7, 2S1, 2W1, 4A22, 20A1 |
| CYCYP Isoenzyme | Polymorphism Frequency | Functional Effects | Allelic Variants | Participation in the Metabolism of Xenobiotics |
|---|---|---|---|---|
| 1A1 | Relatively high | Rare | 13 |  |
| 1A2 | High | Rare | 21 | |
| 1B1 | Frequent missense mutations | Rare | 26 | |
| 2A6 | Higher in Orientals than in Caucasians | Significant | 45 | |
| 2B6 | High | Significant | 38 | |
| 2C8 | High | Significant | 14 | |
| 2C9 | Relatively rare in Caucasians | Very significant | 70 | |
| 2C19 | High | Very significant | 38 | |
| 2D6 | Very High | Highly significant | 145 | |
| 2E1 | Low | No significance | 7 | |
| 3A4 | Low | Low significance | 32 | |
| 3A5 | High | Significant | 9 | |
| Other (a) | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esteves, F.; Rueff, J.; Kranendonk, M. The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family. J. Xenobiot. 2021, 11, 94-114. https://doi.org/10.3390/jox11030007
Esteves F, Rueff J, Kranendonk M. The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family. Journal of Xenobiotics. 2021; 11(3):94-114. https://doi.org/10.3390/jox11030007
Chicago/Turabian StyleEsteves, Francisco, José Rueff, and Michel Kranendonk. 2021. "The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family" Journal of Xenobiotics 11, no. 3: 94-114. https://doi.org/10.3390/jox11030007
APA StyleEsteves, F., Rueff, J., & Kranendonk, M. (2021). The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family. Journal of Xenobiotics, 11(3), 94-114. https://doi.org/10.3390/jox11030007