Abstract
Prostate cancer is one of the most common cancers diagnosed in men in the United States and the second leading cause of cancer-related deaths worldwide. Since over 60% of prostate cancer cases occur in men over 65 years of age, and this population will increase steadily in the coming years, prostate cancer will be a major cancer-related burden in the foreseeable future. Accumulating data from more recent research suggest that the tumor microenvironment (TME) plays a previously unrecognized role in every stage of cancer development, including initiation, proliferation, and metastasis. Prostate cancer is not only diagnosed in the late stages of life, but also progresses relatively slowly. This makes prostate cancer an ideal model system for exploring the potential of natural products as cancer prevention and/or treatment reagents because they usually act relatively slowly compared to most synthetic drugs. Resveratrol (RSV) is a naturally occurring stilbenoid and possesses strong anti-cancer properties with few adverse effects. Accumulating data from both in vitro and in vivo experiments indicate that RSV can interfere with prostate cancer initiation and progression by targeting the TME. Therefore, this review is aimed to summarize the recent advancement in RSV-inhibited prostate cancer initiation, proliferation, and metastasis as well as the underlying molecular mechanisms, with particular emphasis on the effect of RSV on TME. This will not only better our understanding of prostate cancer TMEs, but also pave the way for the development of RSV as a potential reagent for prostate cancer prevention and/or therapy.
1. Introduction
For men, prostate cancer is the second most common cancer in the world, resulting in over 350,000 deaths in 2018 [1]. Most prostate cancers are adenocarcinoma, originating from epithelial cells in the peripheral zone of the prostate [2]. The survival rate for patients with prostate cancer confined to the primary site is nearly 100% [3]. If left unchecked, prostate cancer progresses and eventually metastasizes to form new cancers in other tissues throughout the body. The overwhelming majority of prostate cancer–related deaths occur due to metastasis, most often to the bone [4]. Since prostate cancer incidence increases exponentially for men over 65 years, and this population will increase dramatically in the coming years, prostate cancer is expected to be a big burden both socially and economically in the foreseeable future [1].
Because of the importance of the androgen/androgen receptor (AR) signaling pathway in prostate cancer initiation and progression [5], most basic and clinical research has been focused on the androgen/AR axis. Androgens such as testosterone and dihydrotestosterone bind AR in the cytoplasm and the ligand-bound AR is translocated to the nucleus to interact with AR responsive elements (ARE) and transcriptionally regulate the expression of AR target genes such as prostate-specific antigen (PSA) [6]. Dysregulation of the androgen/AR axis can lead to cancer cell proliferation, escaping from apoptosis, and metastasis [7,8]. Multiple lines of evidence from recent research indicate that, in addition to the androgen/AR axis, the tumor microenvironment (TME) plays an indispensable role in prostate cancer initiation and progression [9,10]. The TME is composed of fibroblasts, the extracellular matrix (ECM), immune cells, and other factors in the tumor cell’s surroundings [11]. There is constant communication between tumor cells and the TME [12]. Fibroblasts, especially cancer-associated fibroblasts (CAFs), play important roles in each of the developmental stages of different cancers, including prostate cancer [9,13]. In fact, AR signaling is not only involved but also plays an important role in TME [14,15], although the extent of and exact mechanisms involved in AR-mediated changes in TME are still being elucidated.
Compared to most other cancers, prostate cancer progression is usually much slower, so the most common approaches taken with early and low-risk prostate cancers are non-invasive active surveillance and watchful waiting [16]. Advanced prostate cancer is treated with radical prostatectomy, radiotherapy, chemotherapy, and androgen-deprivation therapy (ADT) through physical or chemical castration to repress androgen-induced AR signaling [17]. Although responsive to ADT at the beginning, most prostate cancers become ADT-resistant over time [18] due to the formation of constitutively active AR variants or the activation of AR by other factors [9]. For castration-resistant prostate cancer (CRPC) [19], treatment is usually the combination of traditional ADT and second-line antiandrogen drugs such as abiraterone acetate, an androgen synthesis inhibitor, or enzalutamide, to inhibit AR directly [20,21]. Accumulating data demonstrate that resveratrol (RSV), a stilbenoid produced in multiple plants, possesses strong anti-cancer properties [22,23]. By targeting both AR [24] and the TME [25,26], RSV can induce growth inhibition, cell cycle arrest, apoptosis, as well as inhibit metastasis of different cancer types, including prostate cancer [22,27]. More importantly, since RSV has few side effects at therapeutic concentrations, it can be used as a treatment regimen without fear of complications [28]. To pave the road for the development of RSV as a preventive and/or therapeutic reagent, either by itself or in combination with other drugs, our review focuses on the effects of RSV on the TME in prostate cancer initiation, proliferation, and metastasis.
2. RSV Inhibits Prostate Cancer Initiation by Targeting the TME
The exact point at which prostate tissue transitions from normal to cancerous as well as the underlying molecular mechanisms remain ambiguous. However, multiple factors, including the TME, play important roles in the initiation of tumors, including those in the prostate. Although different components of the TME have their roles in cancer development, the communication between the prostate stromal and epithelial cells plays an indispensable role in prostate cancer initiation. Results from research using a mouse model to study cancer initiation in submandibular glands indicate that the interaction between stroma and epithelium is essential because malignancy only occurred when stromal and epithelial cells grew together [29]. Following suit, Chung et al. demonstrated the essentiality of stromal cells in prostate cancer initiation [30]. Since transitional carcinoma cells from mice bladder can grow together with embryonic urogenital sinus mesenchyme (UGM), UGM was used to mimic the reactive human prostate stroma to induce prostatic neoplasms. These results indicate that crosstalk between the stromal and epithelial cells not only exists but also is imperative for the initiation of prostate neoplasia. However, since humans have a much higher proportion of smooth muscle and overall stromal cells than mice [31], this difference needs to be taken into consideration for any solid conclusions about the role of stroma in prostate cancer initiation. Therefore, similar studies in humans will be an important step for an accurate depiction of reactive stroma in prostate cancer initiation.
The development of prostate cancer is very similar to the development of benign prostate hyperplasia (BPH) because they share similarities at the molecular, cellular, genetic, hormonal, and inflammatory levels [32]. Therefore, studying the initiation of benign prostate hyperplasia may also provide insight into prostate cancer initiation. Hayward et al. performed a tissue recombination study by co-culturing benign prostate epithelial cells with stromal cells from either BPH or normal prostate tissues [33] and found that significant epithelial proliferation occurred only when co-cultured with BPH stromal cell lines, indicating that stromal cells have an important role in BPH development. It is important to note that although stromal cells can influence the development of malignancy [34], the communication between the stroma and epithelium is bidirectional. Epithelium-derived cancer cells are capable of influencing the stroma, specifically that of the smooth muscle. Co-culturing of 18-day embryonic rat UGM tissue with either normal epithelial cells or neoplastic prostate epithelial cells showed that the cells from the normal epithelia induce ductal structure formation in smooth muscle, while the cells from neoplastic prostate epithelia failed to do so [33]. These findings altogether indicate that the stromal cells play an important role in prostate cancer initiation, and malignant cells are capable of affecting stromal cell behavior. Given the importance of stroma–epithelium crosstalk in prostate cancer initiation, interrupting this communication could be a plausible strategy in the prevention of prostate cancer initiation [35].
Although multiple lines of evidence indicate that stroma–epithelium crosstalk is essential in the initiation of prostate cancer, the exact role of stromal cells in prostate cancer initiation is currently unknown. Based on the fact that the stromal cells in non-cancerous tissues are more stable than in cancerous tissues, it has been postulated that it is the cancer-associated fibroblasts (CAFs), not the normal stromal cells, that induce tumor initiation [35]. Cunah et al. engrafted BPH-1 cells with either normal stroma or CAFs to adult male mice and found that tumors formed only when the cells were co-engrafted with CAFs [36]. Therefore, it is hypothesized that the CAFs may secrete excess growth factors and/or cytokines to protect the tumor cells from apoptotic death and subsequently lead to tumor formation [37]. As depicted in Figure 1, CAFs secrete more transforming growth factor-beta (TGF-β) than the normal stroma [38]. By activating TGF-β Receptor II (TBRII), the TGF-β can enhance cell proliferation, differentiation, and extracellular matrix production. Given that the prostate epithelial cells are TGF-β Receptor II (TBRII)-positive [39], it is fully conceivable that TGF-β/TBRII can mediate the communication between CAFs and prostate epithelial cells (Figure 1).
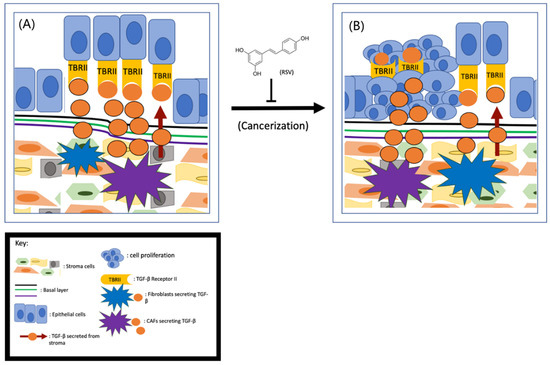
Figure 1.
RSV inhibits prostate cancer initiation (A) CAFs- secreted TGF-β interacts with its receptors TBRII on prostate cells drives tumor initiation (B) and RSV is capable of inhibiting this process.
Since the cancer-preventive effects of RSV were first reported in 1997 [22], both epidemiological and case-controlled studies have demonstrated that RSV and/or consumption of foods/drinks with high levels of RSV can reduce prostate cancer incidence [40]. These findings suggest that RSV could potentially inhibit prostate cancer initiation. Based on the role of CAFs in prostate cancer initiation, early research was focused on the effect of RSV on CAFs. To determine if RSV can inhibit CAF-induced prostate cancer initiation, Wen et al. examined the effect of RSV on TGF-β secretion and found that RSV is capable of downregulating TGF-β expression from CAFs [8]. Further research demonstrated that the downregulation of TGF-β is partially due to RSV’s repressive effect on the Wnt/Beta-catenin pathway [41]. Given that the Wnt pathway in prostate epithelial cells can be activated by CAFs, and RSV is capable of inhibiting TGF-β secretion from CAFs, RSV is likely to inhibit prostate cancer initiation by targeting the TGF-β/TBRII axis. Also, activation of the JAK2/pSTAT1 pathway activates mast cells, leading to inflammation and the transition of stroma to CAFs [42]. Since RSV can cause mast cells to reduce fibrosis by targeting the SCF and c-kit pathway [43], it is also likely that RSV inhibits prostate cancer initiation through repression of mast cell–mediated stromal cell transition to CAFs (Figure 1). However, to demonstrate that RSV can inhibit prostate cancer initiation unambiguously, it is important to conduct both in vitro and in vivo experiments to demonstrate RSV’s role in inhibiting prostate cell cancerization.
3. RSV Inhibits Prostate Cancer Cell Proliferation by Targeting the TME
Two of the most fundamental traits of cancer cells are their abilities to proliferate without regulation and evade cell death [44], and both of these traits can be mediated by the TME. As described in the previous section, crosstalk between the stroma and epithelium under tumorigenic conditions is a key driver in prostate cancer growth [44,45]. The normal fibroblasts acquire a modified phenotype (phenoconversion) and become perpetually activated CAFs [46,47]. The normal fibroblasts secrete only enough factors to maintain normal tissue function and have a relatively low proliferative potential [48]. But the increased secretion of growth factors and cytokines by the CAFs [49] exposes the tumor cells to an altered TME favoring cell growth and proliferation. Therefore, the transition of pre-carcinogenic stroma to CAFs is a critical mechanism for prostate cancer cell proliferation and cancer progression.
3.1. RSV Inhibits Prostate Cancer Proliferation by Interrupting Stroma–Tumor Cell Communication
It has been shown that RSV can affect the proliferation of prostate cancer cells both in vitro and in vivo. For example, treating androgen-responsive human prostate cancer cells (LNCaP) with different concentrations of RSV demonstrated that RSV can inhibit cancer cell proliferation in a concentration-dependent manner. One possible explanation is that RSV targets the AR- and ER-dependent signaling pathways because RSV is capable of counteracting both androgen- and estrogen-induced cell growth [50]. RSV also can inhibit prostate cancer cell proliferation in both transgenic rat adenocarcinoma of prostate (TRAMP) mouse and tumor xenograft models. Harper et al. showed that when transgenic mice were fed a diet containing RSV, cell proliferation in the dorsolateral prostate and ventral prostate decreased significantly [51]. The authors also observed that the transgenic mice had a marked increase (42–62%) of well-differentiated prostate tumors compared to the control animals, suggesting that RSV can halt tumor progression. Wang, et al. injected LNCaP cells into mice on an RSV-enriched diet and found that RSV can delay tumor growth. Of note, both Harper and Wang’s studies suggest that RSV targeted the hormone-based signaling pathways [50,51]. However, the RSV-mediated delay was only temporary, as the tumor volumes of the xenograft mice fed RSV eventually caught up with those of the controls. The underlying mechanisms remain unknown.
Increased levels of interleukin-6 (IL-6) have been linked to poor survival among prostate cancer patients, and it is known that CAFs secrete high levels of IL-6 into the TME. One of the signaling pathways activated by IL-6 is JAK/STAT3. Activation of this pathway in the cells leads to tumor cell proliferation and escape from apoptosis. Mechanistically, the IL-6-activated JAK leads to STAT3 dimerization and translocation to the nucleus to upregulate the expression of proliferative proteins like Bcl-x and Mcl-1 [52,53]. These anti-apoptotic proteins collectively (Figure 2) prevent cell death by reducing mitochondrial outer membrane permeability [54]. When LNCAP and 22RV1 cells were treated with IL-6, the STAT3 was phosphorylated and activated [55], accompanied by enhanced cell proliferation. Consistently, the activity of the IL-6/STAT3 signaling pathway is proportional to the growth rate of advanced prostate cancer [56]. Additionally, it has been found that in LNCAP cells the IL-6-activated JAK/STAT3 pathway also downregulates p53, a major cell cycle regulator [57,58]. Multiple lines of evidence indicate that the CAF-secreted IL-6 may also influence tumor growth by affecting AR activity. Hobisch et al. first observed that in prostate cancer cells IL-6 can activate AR in a dose-dependent manner [59]. Soon after, it was found that in LNCaP cells the IL-6-upregulated serine-threonine kinase Pim1 can phosphorylate (serine-213) and activate AR [60,61]. In addition, Kemskova et al. demonstrated that Pim1 is also regulated by STAT3. These findings altogether indicate that the IL-6/JAK/STAT3 pathway can enhance prostate cancer cell proliferation in an AR-dependent manner [62] (Figure 2). Therefore, monoclonal and immunotherapeutic treatments targeting the IL-6/JAK/STAT3 pathway are currently under vigorous investigation [55,63].
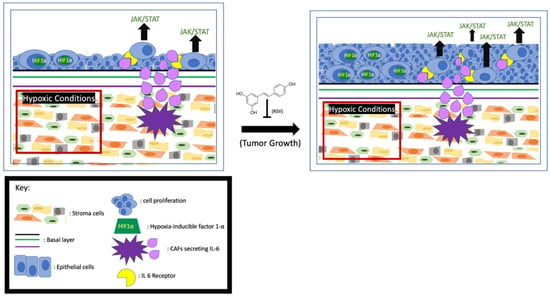
Figure 2.
RSV inhibits prostate cancer proliferation. Hypoxia-induced HIF1α and activation of JAK/STAT by the CAFs- secreted IL-6 enhance tumor growth, and RSV is capable of inhibiting cell proliferation.
It has been reported that RSV can exert a strong anti-proliferative and pro-apoptosis effect on the CAFs in both prostate and lung cancer cell lines [64]. Meanwhile, RSV blunts and even reverses phenoconversion from normal prostate fibroblast to myofibroblast. RSV also indirectly affects IL-6 signaling by inhibiting its downstream factor STAT3 to mitigate proliferative effects. For example, experimental results from mouse fibroblast (NIH3T3) cells and LNCaP cells showed the inhibitory effect of RSV on Src tyrosine kinase, an activator of STAT3, repressing the STAT3 signaling pathway [65,66] and subsequently reducing cell growth and increasing apoptosis (Figure 2). The authors posit that apoptosis is triggered by downregulating downstream factors like Bcl-x, Mcl-, and Cyclin D1 [67,68]. Furthermore, Lee et al. demonstrated that inhibiting IL-6 RSV can repress STAT3-upregulated AR and cell proliferation in prostate cancer cells [25].
3.2. RSV Inhibits Cancer Cell Proliferation by Affecting Hypoxic Conditions of the TME
It is well established that aerobic glycolysis, also known as the Warburg effect, occurs in cells of solid tumors. Hypoxia and high acidity are the two main characteristics of the Warburg effect, and both of them favor the proliferation of cells in solid cancer [69]. Numerous signaling pathways in prostate cancer cells respond to the hypoxic TME, and these pathways collectively lead to increased angiogenesis, survival, proliferation, and/or metastasis [70]. Hypoxia-inducible factor 1-α (HIF1α) plays a central role in cellular adaptation to hypoxic conditions. In hypoxia, the stabilized HIF1α transcriptionally regulates the pro-proliferative and anti-apoptotic genes [71]. Meanwhile, O2 is more frequently reduced to reactive oxygen species (ROS), such as superoxide (O2−) [72,73], which can further stabilize HIF1α [74,75]. Understanding the mechanisms by which tumor cells proliferate in response to the hypoxic TME may prove advantageous for targeted inhibition of cancer cell proliferation.
RSV appears to be both pro- and anti-oxidant, depending on the circumstances [76]. In non-cancer tissues, RSV serves as an antioxidant [77], and therefore RSV can exert a beneficial effect on a wide variety of issues, including neuronal [78], anti-inflammatory [79], and cardiac [80]. However, to cancer cells with low pH environments due to the Warburg Effect, RSV shows more pro-oxidant characteristics. Shamim et al. found that RSV can induce cancer cell death by inducing ROS accumulation, which subsequently leads to oxidative DNA damage and apoptosis [81,82,83]. Using the TRAMP model, Wang et al. demonstrated that RSV-enhanced cancer cell death is due to the upregulation of HIF1α, which enhances ROS concentration in the TME beyond the limit for survival [84]. Since RSV preferentially increases superoxide over other ROS species like hydrogen peroxide [85], Low et al. expounded on the pro-apoptotic effects of elevated superoxide levels in the mitochondria [86] and found that superoxide can activate caspases 9 and 3, and subsequently promote the release of cytochrome C and apoptosis (Figure 2). It is important to note that low concentration of RSV can serve as a pro-oxidant that favors cell survival, and pro-apoptotic effects occur only at relatively higher RSV concentrations to stimulate superoxide production. The bidirectional nature of RSV needs to be considered when RSV is used as a therapeutic reagent.
4. RSV Inhibits Prostate Cancer Metastasis by Targeting the TME
The migration of cells in solid cancers toward their surrounding tissues is the first step of metastasis. Migratory cancer cells undergo both molecular and cellular changes in response to their TME. These changes are collectively known as epithelial-mesenchymal transition (EMT), in which the epithelial cells lose their cell–cell adhesion and gain migratory and invasive properties. Generally, the stroma in non-cancer tissues is rather stable. However, both genetic and epigenetic changes have been found in CAFs [87]. Given that constant communication occurs between stromal cells and cancerous epithelial cells [13], it is conceivable that CAFs could affect the migratory properties of the cancer cells. Multiple lines of evidence indicate that the CAFs are capable of promoting EMT [30,87] and consequently enhance cancer cell migration and invasion [88,89]. Therefore, understanding stroma-mediated EMT is of importance not only in understanding the underlying molecular mechanism but also for identifying potential agents to prevent or even reverse metastasis [89].
4.1. Resveratrol Interrupts the Communication between Stromal and Cancer Cells
One of the effects that CAFs exerted on cancer cells to enhance EMT is to secret different factors into the TME, which are subsequently sensed by cancer cells [90]. Indeed, the level of hepatocyte growth factor (HGF) in the TME of metastatic prostate cancer is higher than that of non-metastatic cancers [90]. Glenn A. Gmyrek et al. [91] have experimentally demonstrated that HGF is capable of triggering pro-migratory phosphorylation of prostate cancer cells and enhancing migration and metastasis. The same group of researchers further demonstrated that the cultured stromal cells showed a myofibroblastic phenotype and speculated that the myofibroblastic subpopulation of prostate stromal cells could be the source of HGF in vivo [91]. Further analysis found that in prostate tissues the HGF is expressed in the stromal cells but not the epithelium, suggesting the HGF in the TME could be the result of paracrine secretion. Since CAFs secrete higher levels of HGF than normal stromal cells, the CAFs were postulated as the culprits driving cancer cell migration [26,91]. Later, it was experimentally demonstrated that stromal cells are capable of enhancing prostate cancer cell migration and that this effect is HGF-dependent [92]. Mechanistically, the CAF-derived HGF interacts with c-Met receptors on cancer cells to activate numerous signaling pathways, especially those related to EMT and metastasis [90] (Figure 3). Therefore, any strategies interrupting the HGF/c-Met interaction by either inhibiting the secretion of HGF or repressing the cancer cell responses to HGF could inhibit EMT and metastasis. To determine if RSV can repress stromal cell–mediated prostate cancer migration, Hsieh TC et al. co-cultured stromal and cancer cells in the presence or absence of RSV and found that RSV can indeed inhibit stromal cell–enhanced cancer cell migration. Since treatment of the co-culture with either RSV or antibodies specifically against HGF abolished stroma-enhanced migration to a similar degree, the authors proposed that RSV inhibits cancer cell migration by counteracting the stromal cell–derived HGF [92]. These findings altogether indicate that (1) stromal cell–secreted HGF plays important roles in prostate cancer migration and invasion, and (2) RSV inhibits prostate cancer migration at least partially by counteracting HGF (Figure 3).
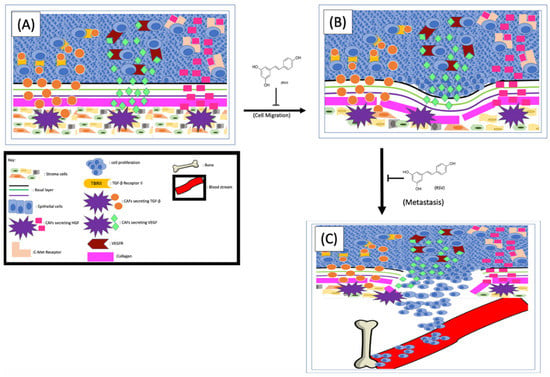
Figure 3.
RSV inhibits EMT and cancer cell metastasis. The CAFs-released TGF-β, VEGF and HGF (A) interact with their corresponding receptors on cancer cells and enhance EMT processes (B). The cancer cells metastasize from their primary site (B) to the bone (C). RSV is capable of inhibiting prostate cancer metastasis by repressing EMT and cancer cell migration/invasion.
It is well known that the ion channel transient receptor potential ankyrin 1 (TRPA1) on CAFs is one of the main control mechanisms for HGF secretion. To further understand the mechanism of RSV-inhibited cancer migration, both mouse [93] and rat [94] models were used, and the results demonstrated that RSV can block HGF secretion by inhibiting TRPA1. Additionally, Vancauwenberghe et al. [26] further demonstrated that by inhibiting the TRPA1, RSV is capable of blocking the secretion of both HGF and vascular endothelial growth factor (VEGF), an important factor involving the angiogenesis of tumor tissues. These findings altogether suggest that RSV could be potentially used as a reagent to inhibit prostate cancer metastasis (Figure 3). Of note, this same study [89] also noticed that RSV can exert an opposite effect on cells expressing a mutant TRPA1, which not only increased expression and secretion of HGF and VEGF, but also enhanced prostate cancer metastasis. Therefore, it is important to stratify prostate cancer patients based on their TRPA1 types and apply RSV only to those with wild-type TRPA1.
4.2. Resveratrol Inhibits Prostate Cancer Metastasis by Targeting the Extracellular Matrix
The extracellular matrix (ECM) is a network formed between cells composed of non-cellular components such as protein, glycosaminoglycan, and glycoconjugate. The ECM can provide both structural and biochemical support to the resident cells, and therefore the ECM can affect cellular behaviors. One of the largest components of the ECM is collagen, which is synthesized and secreted by fibroblasts located in the stroma [95]. Communication between collagen and cancer cells exists in every cancer development stage [96]. Additionally, cadherin plays important structural and signaling roles in cell–cell and cell–ECM interactions [97]. E-cadherin usually maintains the integrity of epithelial tissue, and N-cadherin mediates the contact between cells and the matrix. During the EMT process, N-cadherin is usually upregulated with concurrent downregulation of E-cadherin [98]. Combined dysregulations make the tumor cells more motile and favor metastasis, thereby allowing cancer cells to escape from the site of origin. Therefore, the ECM is an additional barrier to break before the cancer cells can escape from the TME.
Activation of the PI3K signaling pathway can lead to collagen production and fibrosis [99]. The transforming growth factor beta-1 (TGFβ-1), secreted from human prostate stromal cells, can reactivate the stroma by stimulating the phenotypic switch of fibroblasts to myofibroblasts, thus further increasing collagen secretion [95]. It has been shown that RSV can inhibit both the TGFβ and PI3K signaling pathways, thereby decreasing the production and secretion of collagen from the stroma. Since collagen is so prevalent in both benign and malignant tissue, it has been shown that, in organs with highly collagenous reactive stroma, the structural properties of collagen contribute to tumor invasion and metastasis [99]. In normal tissue, the collagen production and assembly are regulated by the counterbalance of matrix metalloproteinase (MMP), MMP inhibitors, and other enzymes. The collagen in TME affects tumor cells’ proliferation, differentiation, gene expression, migration, invasion, and metastasis. Since the MMPs in TME are more prevalent, they degrade and remodel the basement membrane, which favors tumor cell migration and metastasis [100]. The inhibitory effects of RSV on TGFβ and/or CXCL12-mediated myofibroblast phenoconversion of prostate fibroblasts in vitro [64] in a concentration-dependent manner have been reported. Since phenoconversion is necessary for increased collagen production [101], and increased collagen has been shown to contribute to tumor invasion and metastasis, it is conceivable that RSV might be able to inhibit prostate cell metastasis by preventing fibroblast to myofibroblast transition (Figure 3).
The stromal-cell–secreted MMPs play an indispensable role in cancer cell metastasis, with MMP-2 and MMP-9 being the best-studied MMPs in prostate cancer metastasis. In tissues with non-migrating cells, the MMPs are inhibited by tissue inhibitors of metalloproteinase (TIMPs). However, upregulated expressions of MMP-2 and MMP-9, accompanied by downregulated TIMPs, were found in high-grade tumors [102], suggesting that interrupting the delicate balance between MMPs and TIMPs could affect tumor cell migration and cancer metastasis. Once secreted, both MMP-2 and MMP-9 promote metastasis by either enhancing the degradation of barriers of the ECM, such as collagen and laminins, or affecting the balance between cell–cell and cell–matrix attachments [103]. Since the inhibitory effect of RSV on MMPs has been shown in many cancer types, and RSV is capable of inhibiting both MMP-2 and MMP-9 [102], the inhibitory effect of RSV on prostate cancer was examined in different models. The results from the animal models indicate that RSV and the tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) can act individually; the combination of them inhibits prostate cancer metastasis synergistically [104]. Also, it has been noted that androgen treatment of the LNCaP cells can morphologically alter the cells from spindle-like into fibroblast mesenchymal cell shape, and RSV is capable of inhibiting the androgen-induced morphological change [105]. Since this kind of morphological change usually occurs during the EMT process, it further implies that RSV may play a role in prostate cancer cell EMT. In addition, it appears that RSV is also capable of regulating the expression of cadherin. Loh et al. showed that RSV can restore the epithelial phenotype of the mesenchymal cells and inhibit the expression of EMT-related markers [106]. Consistently, our unpublished data also support the notion that RSV can inhibit EMT by up- and downregulating E-cadherin and N-cadherin, respectively, in prostate cancer cells.
4.3. Inhibitory Effects of RSV on Prostate Cancer Cell Bone Metastasis
Once escaped from their primary sites into the bloodstream, cancer cells can travel through the blood and resettle in different organs of the body, although prostate cancer cells preferentially metastasize to the bone. Therefore, metastatic bone cancer is the leading cause of death for patients with prostate cancer [4]. A better understanding of the communication between prostate cancer cells and the bone microenvironment is the first step in designing strategies to prevent bone metastasis of prostate cancer [107].
The CXCR4/CXCL12 axis plays essential roles in all stages of prostate cancer progression [108], including bone metastasis [109]. Interaction between CXCL12 and CXCR4 leads to the activation of multiple downstream pathways such as MAPK and JNK [110] and ultimately causes cytoskeletal rearrangements, actin polymerization, and integrin-dependent adhesion to endothelial cells [110]. On the one hand, higher levels of CXCL12 are expressed in bone marrow stromal cells [109]. On the other hand, most prostate cancer cells express elevated levels of CXCR4, which makes the CXCL12/CXCR4 axis more active in these cells. Indeed, CXCL12 can enhance the adhesion of prostate cancer cells with osteosarcoma cells [111]. In addition, Sun et al. compared the levels of CXCL12 in different tissues and found that, in both human and mouse models, the levels of CXCL12 are higher in the prostate cancer–preferred metastatic tissues than in those rarely targeted [112]. To further demonstrate how essential CXCR4 is to prostate cancer bone metastasis, the authors neutralized the receptor by injecting antibodies against CXCR4 and found that prostate-cancer-to-bone metastases were significantly inhibited [112].
Compared to the surrounding non-cancerous tissues, the vascular endothelial growth factor (VEGF) is upregulated in most solid cancers, including prostate cancer, and one of the major sources of VEGF in the TME is CAFs [113]. Consistently, overexpression of VEGF receptors (VEGFR) has been found in cancerous endothelial cells, which play an indispensable role in cancer cell migration and metastasis [114]. More recent results indicate that the interaction between the tumor and stromal cells can further induce HIF1α-mediated VEGF secretion from the CAFs [38,115], which could be part of the reason why elevated levels of VEGF are found in cancerous tissues but not the benign tissues [116]. One of the effects of VEGF is to support tumor neovascularization through the secretion of proteolytic enzymes to dismantle the ECM, which favors EMT [116]. The precise mechanism for VEGF-mediated prostate cancer metastasis is not yet completely understood. But it has been hypothesized that the bone stroma-secreted VEGF interacts with cancer cells’ VEGFR and subsequently enhances cancer cells nesting to the bone [116]. This hypothesis is supported by the fact that elevated levels of VEGFR-2 were consistently found on prostate cancer cells [117]. More importantly, both VEGF and VEGFR-2 are notably higher at the sites of bone metastasis than at the primary prostate tumors of the same individual, further demonstrating the importance of the VEGF/VEGFR-2 axis in prostate cancer bone metastasis [117]. Interestingly, RSV and its analog HS-1793 are capable of inhibiting both hypoxia-induced VEGF expression and prostate cancer cell migration [115] (Figure 3). In addition, RSV, either alone or in combination with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), can inhibit the VEGF/VEGFR2 axis in vivo [104]. RSV’s inhibitory effect in bone stroma–prostate cancer cell communication is an ongoing research topic [116,117,118].
4.4. RSV Inhibits Immune Cell-Mediated Prostate Cancer Metastasis
Although the origin of tumor-associated macrophages is not completely known, different immune cells, especially the macrophages, are found ubiquitously in solid tumors. One school of thought is that they are tissue-resident macrophages, and another postulates that the tumor macrophages are derived from monocytes [80,119]. Nevertheless, it is widely accepted that immune cells not only exist in the TME but also affect the development of prostate cancer, and multiple lines of evidence indicate that RSV is capable of inhibiting the immune cell–mediated prostate cancer metastasis. Based on the number of macrophages found in prostate cancer tissues derived from different patients, Lewis and colleagues concluded that macrophages could lead to either favorable or unfavorable prognosis in prostate cancer [120]. Later, it was found that higher tumor-associated macrophage (TAM) count is correlated with a reduced survival time but an increased disease-free survival after treatment [121,122]. The authors attribute this inconsistency to not only the methodological differences between studies but also the complex functions of macrophages.
While direct evidence of RSV’s effects on macrophage activity in prostate cancer is limited, evidence of RSV’s effects on macrophages in other cancers is well documented. Based on their roles in tumor development, the macrophages in the tumor tissues can be divided into either M1- or M2-like subtypes. The M1-like macrophages possess an antitumor property by promoting Th1 response, and the M2-like macrophages usually promote Th2 response, tissue remodeling, immune tolerance, and tumor progression [123]. TAMs are phenotypically similar to M2 macrophages [124]. Recently, Kimura et al. experimentally demonstrated that RSV can prevent tumor cell metastasis by inhibiting M2-like macrophage differentiation [125]. Consistent with the findings that prostate cancer can secret certain factors, including IL-10, to induce M2 polarization [126], the authors further demonstrated that RSV is capable of inhibiting M2 differentiation by downregulating IL-10 secretion. Finally, oral administration of RSV (25 mg/kg, twice daily) for 30 days to mice injected with the highly metastatic osteosarcoma LM8 cells significantly reduced the number of metastatic tumors in both the lungs and the liver. These in vitro and in vivo data altogether unambiguously demonstrate that RSV is indeed capable of inhibiting cancer cell metastasis. However, whether RVS has a similar effect on prostate cancer cell metastasis still needs to be determined experimentally.
5. Conclusions and Perspectives
Since its anti-inflammation and anti-oxidation activities were demonstrated experimentally in 1997 [22], RSV and its derivatives have attracted great attention in both basic science and clinical settings. Because of its beneficial effects on improved metabolism, cardio protection, and cancer prevention, RSV has even been widely accepted as an anti-aging reagent. The general beneficial effects of RSV on many diseases have been thoroughly reviewed by Baur and Sinclair [23]. Discussion of clinical literature, along with the limitations of preclinical and in vitro RSV studies, can be found in the review article by Tome-Carneiro et al. [127]. The in vivo effects of resveratrol on different cancers, including breast, colorectal, liver, pancreatic, and prostate cancer have been systematically reviewed by Lindsay G. Carter [128]. More importantly, data from both in vitro and in vivo experiments indicate that RSV is one of a few natural products with minimal adverse effects. These findings make RSV an ideal preventive and therapeutic reagent.
Because of the ubiquitous role of the androgen/AR axis in each process of prostate cancer development, including initiation, progression, and metastasis, special attention has been paid to the effects of RSV on AR. Relatively little attention has been paid to the RSV effect on epithelium–stroma interaction, although it is well known that RSV can exert its effects on prostate cancer in an AR-independent manner [129]. RSV has differential effects on growth, cell cycle arrest, and induction of apoptosis by RSV in human prostate cancer cell lines [130,131]. Overwhelming evidence suggests that RSV’s anti-prostate cancer effect is through targeting the AR and subsequent AR-regulated genes [50,132,133]. Harada et al. reported that RSV represses AR target gene expression, at least partially, by enhancing AR degradation in a time- and dose-dependent manner [134]. Along with other groups, we have demonstrated that RSV can repress AR function by inhibiting AR transcriptional activity [135]. In addition, a group of ~20 AR variants (ARVs) has been identified in prostate cancer, especially the metastatic castration-resistant prostate cancer (mCRPC). Among these variants, ARV7 has attracted special attention because of its androgen-independent transcriptional activity [136]. More recent findings suggest that approximately 10–30% of men with mCRPC are positive for AR-V7 [137]. We have also demonstrated that RSV is capable of repressing mCRPC cell growth by enhancing ARV7 poly-ubiquitination and subsequent degradation [138]. These findings suggest that resveratrol could be used as a prostate cancer preventive or therapeutic reagent.
Given the emerging roles of the stromal cells in cancer initiation and progression, great attention has been paid to the communication between stromal and cancer cells in recent years. Mounting evidence indicates that the stromal cells play indispensable roles in prostate cancer initiation, proliferation, and metastasis. As we have discussed in this review, by interrupting the epithelial and stromal cell communication, RSV can inhibit prostate cancer development through different mechanisms. However, there are still many unanswered questions about how RSV affects stromal–cancer cell communication. For example, it is well established that AR is expressed and functional in both epithelial and stromal cells [11]. But compared to our understanding of RSV’s effect on epithelial cells, very little is known about the role of RSV in stromal cells. Besides, ARV7 plays an indispensable role in mCRPC, and resveratrol can inhibit the proliferation of AVR7-positive cells. Our unpublished data indicate that RSV can affect the E-cadherin and N-cadherin ratio in ARV7-positive prostate cancer cells, suggesting that RSV could potentially be able to affect the metastasis of these cells. Whether ARV7 is simultaneously or subsequently expressed in cancer and stromal cells is unknown. If ARV7 is indeed expressed in stromal cells, will RSV also affect the stromal cells through targeting ARV7 as it did in the cancer cells? Answers to these questions and a better understanding of the underlying molecular mechanisms in how RSV inhibits prostate cancer progression by interrupting stromal–cancer cell communication will pave the road toward the application of RSV as both a cancer preventive and therapeutic reagent.
Funding
This research was funded by the Philadelphia College of Osteopathic Medicine.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ferlay, J.; Soejomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Humphrey, P.A. Histopathology of prostate cancer. Cold Spring Harbor Perspect. Med. 2017, 7, a030411. [Google Scholar] [CrossRef] [PubMed]
- Lu-Yao, G.L.; Albertsen, P.C.; Moore, D.F.; Shih, W.; Lin, Y.; DiPaola, R.S.; Barry, M.J.; Zietman, A.; O’Leary, M.; Walker-Corkery, E.; et al. Outcomes of localized prostate cancer following conservative management. JAMA J. Am. Med Assoc. 2009, 302, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Docherty, F.E.; Brown, H.K.; Reeves, K.J.; Fowles, A.C.M.; Ottewell, P.D.; Dear, T.N.; Holen, I.; Croucher, P.I.; Eaton, C.L. Prostate cancer cells preferentially home to osteoblast rich areas in the early stages of bone metastasis: Evidence from in vivo models. J. Bone Miner. Res. 2014, 29, 2688–2696. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef]
- Pandini, G.; Mineo, R.; Frasca, F.; Roberts, C.T., Jr.; Marcelli, M.; Vigneri, R.; Belfiore, A. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res. 2005, 65, 1849–1857. [Google Scholar] [CrossRef]
- Cao, R.; Ke, M.; Wu, Q.; Tian, Q.; Liu, L.; Dai, Z.; Lu, S.; Liu, P. AZGP1 is androgen responsive and involved in AR induced prostate cancer cell proliferation and metastasis. J. Cell Physiol. 2019, 234, 17444–17458. [Google Scholar] [CrossRef]
- Wen, S.; Niu, Y.; Lee, S.O.; Chang, C. Androgen receptor (AR) positive vs negative roles in prostate cancer cell deaths including apoptosis, anoikis, entosis, necrosis and autophagic cell death. Cancer Treat Rev. 2014, 40, 31–40. [Google Scholar] [CrossRef]
- Thalmann, G.N.; Rhee, H.; Sikes, R.A.; Pathak, S.; Multani, A.; Zhau, H.E.; Marshall, F.F.; Chunge, L.W.K. Human prostate fibroblasts induce growth and confer castration resistance and metastatic potential in LNCaP cells. Eur. Urol. 2009, 58, 162–172. [Google Scholar] [CrossRef]
- Bonollo, F.; Thalmann, G.N.; Kruithof-de Julio, M.; Karkampouna, S. The role of cancer-associated fibroblasts in prostate cancer tumorigenesis. Cancers 2020, 12, 1887. [Google Scholar] [CrossRef]
- Cioni, B.; Zwart, W.; Bergman, A.M. Androgen receptor moonlighting in the prostate cancer microenvironment. Endocr. Relat. Cancer 2018, 25, R331–R349. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar]
- Lai, K.; Yamashita, S.; Huang, C.; Yeh, S.; Chang, C. Loss of stromal androgen receptor leads to suppressed prostate tumourigenesis via modulation of pro-inflammatory cytokines/chemokines. EMBO Mol. Med. 2012, 4, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Wikström, P.; Marusic, J.; Stattin, P.; Bergh, A. Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. Prostate 2009, 69, 799–809. [Google Scholar] [CrossRef]
- Herden, J.; Weissbach, L. Utilization of active surveillance and watchful waiting for localized prostate cancer in the daily practice. World J. Urol. 2018, 36, 383–391. [Google Scholar] [CrossRef]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen-targeted therapy in men with prostate cancer: Evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2018, 22, 24–38. [Google Scholar] [CrossRef]
- De Amicis, F.; Chimento, A.; Montalto, F.I.; Casaburi, I.; Sirianni, R.; Pezzi, V. Steroid receptor signallings as targets for resveratrol actions in breast and prostate cancer. Int. J. Mol. Sci. 2019, 20, 1087. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Auchus, R.J. The backdoor pathway to dihydrotestosterone. Trends Endocrinol. Metab. 2004, 15, 432–438. [Google Scholar] [CrossRef]
- Guerrero, J.; Alfaro, I.E.; Gómez, F.; Protter, A.A.; Bernales, S. Enzalutamide, an androgen receptor signaling inhibitor, induces tumor regression in a mouse model of castration-resistant prostate cancer. Prostate 2013, 73, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.W.; Fong, H.H.S.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Kumar, A.; Butt, N.A.; Zhang, L.; Williams, R.; Rimando, A.M.; Biswas, P.K.; Levenson, A.S. Molecular insight into the differential anti-androgenic activity of resveratrol and its natural analogs: In silico approach to understand biological actions. Mol. Biosyst. 2016, 12, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Kundu, J.K.; Keum, Y.S.; Cho, Y.Y.; Surh, Y.J.; Choi, B.Y. Resveratrol inhibits IL-6-induced transcriptional activity of AR and STAT3 in human prostate cancer LNCaP-FGC cells. Biomol. Ther. 2014, 22, 426–430. [Google Scholar] [CrossRef]
- Vancauwenberghe, E.; Noyer, L.; Derouiche, S.; Lemonnier, L.; Gosset, P.; Sadofsky, L.R.; Mariot, P.; Warnier, M.; Bokhobza, A.; Slomianny, C.; et al. Activation of mutated TRPA1 ion channel by resveratrol in human prostate cancer associated fibroblasts (CAF). Mol. Carcinog. 2017, 56, 1851–1867. [Google Scholar] [CrossRef]
- Martínez-Martínez, D.; Soto, A.; Gil-Araujo, B.; Gallego, B.; Chiloeches, A.; Lasa, M. Resveratrol promotes apoptosis through the induction of dual specificity phosphatase 1 and sensitizes prostate cancer cells to cisplatin. Food Chem. Toxicol. 2019, 124, 273–279. [Google Scholar] [CrossRef]
- Kai, L.; Levenson, A.S. Combination of resveratrol and antiandrogen flutamide has synergistic effect on androgen receptor inhibition in prostate cancer cells. Anticancer Res. 2011, 31, 3323. [Google Scholar]
- Franks, L.M.; Riddle, P.N.; Carbonell, A.W.; Gey, G.O. A comparative study of the ultrastructure and lack of growth capacity of adult human prostate epithelium mechanically separated from its stroma. J. Pathol. 1970, 100, 113–119. [Google Scholar] [CrossRef]
- Chung, L.W.; Chang, S.M.; Bell, C.; Zhau, H.E.; Ro, J.Y.; von Eschenbach, A.C. Co-inoculation of tumorigenic rat prostate mesenchymal cells with non-tumorigenic epithelial cells results in the development of carcinosarcoma in syngeneic and athymic animals. Int. J. Cancer 1989, 43, 1179–1187. [Google Scholar] [CrossRef]
- Marker, P.C.; Donjacour, A.A.; Dahiya, R.; Cunha, G.R. Hormonal, cellular, and molecular control of prostatic development. Dev. Biol. 2003, 253, 165–174. [Google Scholar] [CrossRef]
- Miah, S.; Catto, J. BPH and prostate cancer risk. Indian J. Urol. 2014, 30, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Ao, M.; Franco, O.E.; Park, D.; Raman, D.; Williams, K.; Hayward, S.W. Cross-talk between paracrine-acting cytokine and chemokine pathways promotes malignancy in benign human prostatic epithelium. Cancer Res. 2007, 67, 4244–4253. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.; Buchanan, G. Stromal androgen receptor in prostate cancer development and progression. Cancers 2017, 9, 10. [Google Scholar] [CrossRef]
- Barron, D.A.; Rowley, D.R. The reactive stroma microenvironment and prostate cancer progression. Endocr. Relat. Cancer 2012, 19, R187–R204. [Google Scholar] [CrossRef]
- Cunha, G.R.; Hayward, S.W.; Wang, Y.Z. Role of stroma in carcinogenesis of the prostate. Differentiation 2002, 70, 473–485. [Google Scholar] [CrossRef]
- Singh, S.; Singh, U.P.; Grizzle, W.E.; Lillard, J.W., Jr. CXCL12-CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab. Investig. 2004, 84, 1666–1676. [Google Scholar] [CrossRef]
- Tao, L.; Huang, G.; Song, H.; Chen, Y.; Chen, L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol. Lett. 2017, 14, 2611–2620. [Google Scholar] [CrossRef]
- Shaw, A.K.; Pickup, M.W.; Chytil, A.; Aakre, M.; Owens, P.; Moses, H.L.; Novitskiy, S.V. TGFβ signaling in myeloid cells regulates mammary carcinoma cell invasion through fibroblast interactions. PLoS ONE 2015, 10, e0117908. [Google Scholar] [CrossRef]
- Schoonen, W.M.; Salinas, C.A.; Kiemeney, L.A.; Stanford, J.L. Alcohol consumption and risk of prostate cancer in middle-aged men. Int. J. Cancer 2005, 113, 133–140. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, H.; Yu, Y.; Zhang, J.; Liu, Q.; Yang, B. Resveratrol improved detrusor fibrosis induced by mast cells during progression of chronic prostatitis in rats. Eur. J. Pharmacol. 2017, 815, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhou, J.; Zhao, Y.; Liu, P.Y.; Yao, H.J.; Da, J.; Zhang, M.; Zhou, Z.; Chen, Q.; Peng, Y.B.; et al. Normal peripheral prostate stromal cells stimulate prostate cancer development: Roles of c-kit signal. Am. J. Transl. Res. 2015, 7, 502–512. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.; Chung, L.W.K. Prostate tumor-stroma interaction: Molecular mechanisms and opportunities for therapeutic targeting. Differentiation 2002, 70, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Liyanarachchi, S.; Davuluri, R.V.; Auer, H.; Martin Jr, E.W.; de la Chapelle, A.; Frankel, W.L. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene 2004, 23, 7366–7377. [Google Scholar] [CrossRef]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Wang, T.T.; Hudson, T.S.; Wang, T.C.; Remsberg, C.M.; Davies, N.M.; Takahashi, Y.; Kim, Y.S.; Seifried, H.; Vinyard, B.T.; Perkins, S.N.; et al. Differential effects of resveratrol on androgen-responsive LNCaP human prostate cancer cells in vitro and in vivo. Carcinogenesis 2008, 29, 2001–2010. [Google Scholar] [CrossRef]
- Harper, C.E.; Patel, B.B.; Wang, J.; Arabshahi, A.; Eltoum, I.A.; Lamartiniere, C.A. Resveratrol suppresses prostate cancer progression in transgenic mice. Carcinogenesis 2007, 28, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.W. STAT3 target genes relevant to human cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Zha, H.; Aime-Sempe, C.; Takayama, S.; Wang, H.G. Structure-function analysis of bcl-2 family proteins. regulators of programmed cell death. Adv. Exp. Med. Biol. 1996, 406, 99–112. [Google Scholar] [PubMed]
- Twillie, D.A.; Eisenberger, M.A.; Carducci, M.A.; Hseih, W.S.; Kim, W.Y.; Simons, J.W. Interleukin-6: A candidate mediator of human prostate cancer morbidity. Urology 1995, 45, 542–549. [Google Scholar] [CrossRef]
- Chung, L.W.; Baseman, A.; Assikis, V.; Zhau, H.E. Molecular insights into prostate cancer progression: The missing link of tumor microenvironment. J. Urol. 2005, 173, 10–20. [Google Scholar] [CrossRef]
- Cheteh, E.H.; Sarne, V.; Ceder, S.; Bianchi, J.; Augsten, M.; Rundqvist, H.; Egevad, L.; Östman, A.; Wiman, K.G. Interleukin-6 derived from cancer-associated fibroblasts attenuates the p53 response to doxorubicin in prostate cancer cells. Cell Death Discov. 2020, 6, 42–45. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef]
- Hobisch, A.; Eder, I.E.; Putz, T.; Horninger, W.; Bartsch, G.; Klocker, H.; Culig, Z. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998, 58, 4640–4645. [Google Scholar]
- van der Poel, H.G.; Zevenhoven, J.; Bergman, A.M. Pim1 regulates androgen-dependent survival signaling in prostate cancer cells. Urol. Int. 2010, 84, 212–220. [Google Scholar] [CrossRef]
- Ha, S.; Iqbal, N.J.; Mita, P.; Ruoff, R.; Gerald, W.L.; Lepor, H.; Taneja, S.S.; Lee, P.; Melamed, J.; Garabedian, M.J.; et al. Phosphorylation of the androgen receptor by PIM1 in hormone refractory prostate cancer. Oncogene 2013, 32, 3992–4000. [Google Scholar] [CrossRef] [PubMed]
- Zemskova, M.; Sahakian, E.; Bashkirova, S.; Lilly, M. The PIM1 kinase is a critical component of a survival pathway activated by docetaxel and promotes survival of docetaxel-treated prostate cancer cells. J. Biol. Chem. 2008, 283, 20635–20644. [Google Scholar] [CrossRef] [PubMed]
- Kampan, N.C.; Xiang, S.D.; McNally, O.M.; Stephens, A.N.; Quinn, M.A.; Plebanski, M. Immunotherapeutic interleukin-6 or interleukin-6 receptor blockade in cancer: Challenges and opportunities. Curr. Med. Chem. 2018, 25, 4785–4806. [Google Scholar] [CrossRef] [PubMed]
- Gharaee-Kermani, M.; Moore, B.B.; Macoska, J.A. Resveratrol-mediated repression and reversion of prostatic myofibroblast phenoconversion. PLoS ONE 2016, 11, e0158357. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Yen, M.L.; Lin, C.Y.; Kuo, M.L. Inhibition of vascular endothelial growth factor-induced angiogenesis by resveratrol through interruption of src-dependent vascular endothelial cadherin tyrosine phosphorylation. Mol. Pharmacol. 2003, 64, 1029–1036. [Google Scholar] [CrossRef]
- Al Aameri, R.F.H.; Sheth, S.; Alanisi, E.M.A.; Borse, V.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Tonic suppression of PCAT29 by the IL-6 signaling pathway in prostate cancer: Reversal by resveratrol. PLoS ONE 2017, 12, e0177198. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.-M.; Yang-Yen, H.-F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased mcl-1 expression. J. Clin. Investig. 2001, 107, 351–362. [Google Scholar] [CrossRef]
- Sinibaldi, D.; Wharton, W.; Turkson, J.; Bowman, T.; Pledger, W.J.; Jove, R. Induction of p21WAF1/CIP1 and cyclin D1 expression by the src oncoprotein in mouse fibroblasts: Role of activated STAT3 signaling. Oncogene 2000, 19, 5419–5427. [Google Scholar] [CrossRef]
- Parkins, C.S.; Stratford, M.R.; Dennis, M.F.; Stubbs, M.; Chaplin, D.J. The relationship between extracellular lactate and tumour pH in a murine tumour model of ischaemia-reperfusion. Br. J. Cancer 1997, 75, 319–323. [Google Scholar] [CrossRef]
- Deep, G.; Panigrahi, G.K. Hypoxia-induced signaling promotes prostate cancer progression: Exosomes role as messenger of hypoxic response in tumor microenvironment. Crit. Rev. Oncog. 2015, 20, 419–434. [Google Scholar] [CrossRef]
- Fraga, A.; Ribeiro, R.; Principe, P.; Lopes, C.; Medeiros, R. Hypoxia and prostate cancer aggressiveness: A tale with many endings. Clin. Genitourin Cancer 2015, 13, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G. The mitochondrial production of reactive oxygen species: Mechanisms and implications in human pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.L.; Salter, J.D.; Brookes, P.S. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: Implications for hypoxic cell signaling. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, 101. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Kietzmann, T. Superoxide and derived reactive oxygen species in the regulation of hypoxia-inducible factors. Methods Enzymol. 2007, 435, 421–446. [Google Scholar] [CrossRef]
- Rodriguez-Enriquez, S.; Pacheco-Velazquez, S.C.; Marin-Hernandez, A.; Gallardo-Pérez, J.C.; Robledo-Cadena, D.X.; Hernández-Reséndiz, I.; García-García, J.D.; Belmont-Díaz, J.; López-Marure, R.; Hernández-Esquivel, L.; et al. Resveratrol inhibits cancer cell proliferation by impairing oxidative phosphorylation and inducing oxidative stress. Toxicol. Appl. Pharmacol. 2019, 370, 65–77. [Google Scholar] [CrossRef]
- Kovacic, P.; Somanathan, R. Multifaceted approach to resveratrol bioactivity: Focus on antioxidant action, cell signaling and safety. Oxid. Med. Cell Longev. 2010, 3, 86–100. [Google Scholar] [CrossRef]
- Mancuso, R.; del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Núñez, O.; Pallás, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar] [CrossRef]
- Shi, Y.; Zhou, J.; Jiang, B.; Miao, M. Resveratrol and inflammatory bowel disease. Ann. N. Y. Acad. Sci. 2017, 1403, 38–47. [Google Scholar] [CrossRef]
- Zhang, J.; Patel, L.; Pienta, K.J. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev. 2010, 21, 41–48. [Google Scholar] [CrossRef]
- Shamim, U.; Hanif, S.; Albanyan, A.; Beck, F.W.J.; Bao, B.; Wang, Z.; Banerjee, S.; Sarkar, F.H.; Mohammad, R.M.; Hadi, S.M.; et al. Resveratrol-induced apoptosis is enhanced in low pH environments associated with cancer. J. Cell Physiol. 2012, 227, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Muqbil, I.; Beck, F.W.; Bao, B.; Sarkar, F.H.; Mohammad, R.M.; Hadi, S.M.; Azmi, A.S. Old wine in a new bottle: The warburg effect and anticancer mechanisms of resveratrol. Curr. Pharm. Des. 2012, 18, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.F.; Wei, Q.Y.; Cai, Y.J.; Fang, J.G.; Zhou, B.; Yang, L.; Liu, Z.-L. DNA damage induced by resveratrol and its synthetic analogues in the presence of cu (II) ions: Mechanism and structure-activity relationship. Free Radic. Biol. Med. 2006, 41, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gao, Z.; Zhang, X. Resveratrol induces apoptosis in murine prostate cancer cells via hypoxia-inducible factor 1-alpha (HIF-1alpha)/reactive oxygen species (ROS)/P53 signaling. Med. Sci. Monit. 2018, 24, 8970–8976. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Low, I.C.; Chen, Z.X.; Pervaiz, S. Bcl-2 modulates resveratrol-induced ROS production by regulating mitochondrial respiration in tumor cells. Antioxid. Redox. Signal. 2010, 13, 807–819. [Google Scholar] [CrossRef]
- Niu, Y.N.; Xia, S.J. Stroma-epithelium crosstalk in prostate cancer. Asian J. Androl. 2009, 11, 28–35. [Google Scholar] [CrossRef]
- Xia, N.; Daiber, A.; Forstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef]
- Li, J.; Chong, T.; Wang, Z.; Chen, H.; Li, H.; Cao, J.; Zhang, P.; Li, H. A novel anti-cancer effect of resveratrol: Reversal of epithelial-mesenchymal transition in prostate cancer cells. Mol. Med. Rep. 2014, 10, 1717–1724. [Google Scholar] [CrossRef]
- Varkaris, A.; Corn, P.G.; Gaur, S.; Dayyani, F.; Logothetis, C.J.; Gallick, G.E. The role of HGF/c-met signaling in prostate cancer progression and c-met inhibitors in clinical trials. Expert Opin. Investig. Drugs 2011, 20, 1677–1684. [Google Scholar] [CrossRef]
- Gmyrek, G.A.; Walburg, M.; Webb, C.P.; Yu, H.M.; You, X.; Vaughan, E.D.; Vande Woude, G.F.; Knudsen, B.S. Normal and malignant prostate epithelial cells differ in their response to hepatocyte growth factor/scatter factor. Am. J. Pathol. 2001, 159, 579–590. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Wu, J.M. Resveratrol suppresses prostate cancer epithelial cell scatter/invasion by targeting inhibition of hepatocyte growth factor (HGF) secretion by prostate stromal cells and upregulation of E-cadherin by prostate cancer epithelial cells. Int. J. Mol. Sci. 2020, 21, 1760. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, S.; Kogure, Y.; Yamamoto, S.; Noguchi, K.; Dai, Y. Modulation of TRP channels by resveratrol and other stilbenoids. Mol. Pain 2013, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Nalli, M.; Ortar, G.; Moriello, A.S.; Morera, E.; Di Marzo, V.; De Petrocellis, L. TRPA1 channels as targets for resveratrol and related stilbenoids. Bioorganic Med. Chem. Lett. 2016, 26, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive stroma in human prostate cancer: Induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 1–22. [Google Scholar] [CrossRef]
- Putzke, A.P.; Ventura, A.P.; Bailey, A.M.; Akture, C.; Opoku-Ansah, J.; Çeliktaş, M.; Hwang, M.S.; Darling, D.S.; Coleman, I.M.; Nelson, P.S.; et al. Metastatic progression of prostate cancer and e-cadherin regulation by zeb1 and SRC family kinases. Am. J. Pathol. 2011, 179, 400–410. [Google Scholar] [CrossRef]
- Jennbacken, K.; Tesan, T.; Wang, W.; Gustavsson, H.; Damber, J.E.; Welén, K. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr. Relat. Cancer 2010, 17, 469–479. [Google Scholar] [CrossRef]
- Wegner, K.A.; Mueller, B.R.; Unterberger, C.J.; Avila, E.J.; Ruetten, H.; Turco, A.E.; Oakes, S.R.; Girardi, N.M.; Halberg, R.B.; Swanson, S.M.; et al. Prostate epithelial-specific expression of activated PI3K drives stromal collagen production and accumulation. J. Pathol. 2020, 250, 231–242. [Google Scholar] [CrossRef]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and cancer associated fibroblasts in the reactive stroma and its relation to cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 115–116. [Google Scholar] [CrossRef]
- Phan, S.H. Biology of fibroblasts and myofibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Lichtinghagen, R.; Musholt, P.B.; Lein, M.; Römer, A.; Rudolph, B.; Kristiansen, G.; Hauptmann, S.; Schnorr, D.; ALoening, S.; Jung, K. Different mRNA and protein expression of matrix metalloproteinases 2 and 9 and tissue inhibitor of metalloproteinases 1 in benign and malignant prostate tissue. Eur. Urol. 2002, 42, 398–406. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Stetler-Stevenson, W.G. Matrix metalloproteinases and metastasis. Cancer Chemother Pharmacol. 1999, 43, S42–S51. [Google Scholar] [CrossRef] [PubMed]
- PLoS ONE Editors. Expression of concern: Resveratrol enhances antitumor activity of TRAIL in prostate cancer xenografts through activation of FOXO transcription factor. PLoS ONE 2019, 14, e0223138. [Google Scholar] [CrossRef]
- Jang, Y.G.; Go, R.E.; Hwang, K.A.; Choi, K.C. Resveratrol inhibits DHT-induced progression of prostate cancer cell line through interfering with the AR and CXCR4 pathway. J. Steroid. Biochem. Mol. Biol. 2019, 192, 105406. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-cadherin and N-cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Sung, S.Y.; Hsieh, C.L.; Law, A.; Zhau, H.E.; Pathak, S.; Multani, A.S.; Lim, S.; Coleman, I.M.; Wu, L.-C.; Figg, W.D. Coevolution of prostate cancer and bone stroma in three-dimensional coculture: Implications for cancer growth and metastasis. Cancer Res. 2008, 68, 9996–10003. [Google Scholar] [CrossRef]
- Dubrovska, A.; Elliott, J.; Salamone, R.J.; Telegeev, G.; Stakhovsky, A.E.; Schepotin, I.B.; Yan, F.; Wang, Y.; Bouchez, L.; Kularatne, S.A.; et al. CXCR4 expression in prostate cancer progenitor cells. PLoS ONE 2012, 7, e31226. [Google Scholar] [CrossRef]
- Conley-LaComb, M.K.; Semaan, L.; Singareddy, R.; Li, Y.; Heath, E.I.; Kim, S.; Cher, M.L.; Chinni, S.R. Pharmacological targeting of CXCL12/CXCR4 signaling in prostate cancer bone metastasis. Mol. Cancer 2016, 15, 68. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Sun, Y.X.; Schneider, A.; Jung, Y.; Wang, J.; Dai, J.; Wang, J.; Cook, K.; Osman, N.I.; Koh-Paige, A.J.; Shim, H.; et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J. Bone Miner. Res. 2005, 20, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Pérez, J.; Monter-Vera, M.D.R.; Barrientos-Alvarado, C.; Toscano-Garibay, J.D.; Cuesta-Mejías, T.; Flores-Estrada, J. Evaluation of VEGF and PEDF in prostate cancer: A preliminary study in serum and biopsies. Oncol. Lett. 2018, 15, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Zhao, H.; Ren, X.B. Relationship of VEGF/VEGFR with immune and cancer cells: Staggering or forward? Cancer Biol. Med. 2016, 13, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Hossain, M.A.; Kim, M.Y.; Kim, J.-A.; Yoon, J.-H.; Suh, H.S.; Kim, G.-Y.; Choi, Y.H.; Chung, H.Y.; Kim, N.D. A novel resveratrol analogue, HS-1793, inhibits hypoxia-induced HIF-1α and VEGF expression, and migration in human prostate cancer cells. Int. J. Oncol. 2013, 43, 1915–1924. [Google Scholar] [CrossRef]
- Walsh, K.; Sriprasad, S.; Hopster, D.; Codd, J.; Mulvin, D. Distribution of vascular endothelial growth factor (VEGF) in prostate disease. Prostate Cancer Prostatic Dis. 2002, 5, 119–122. [Google Scholar] [CrossRef]
- Chen, J.; De, S.; Brainard, J.; Byzova, T.V. Metastatic properties of prostate cancer cells are controlled by VEGF. Cell Commun. Adhes. 2004, 11, 1–11. [Google Scholar] [CrossRef]
- Reynolds, A.R.; Kyprianou, N. Growth factor signalling in prostatic growth: Significance in tumour development and therapeutic targeting. Br. J. Pharmacol. 2006, 147 (Suppl. 2), 144. [Google Scholar] [CrossRef]
- Stephens, T.C.; Currie, G.A.; Peacock, J.H. Repopulation of gamma-irradiated lewis lung carcinoma by malignant cells and host macrophage progenitors. Br. J. Cancer 1978, 38, 573–582. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Lissbrant, I.F.; Stattin, P.; Wikstrom, P.; Damber, J.E.; Egevad, L.; Bergh, A. Tumor associated macrophages in human prostate cancer: Relation to clinicopathological variables and survival. Int. J. Oncol. 2000, 17, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Shimura, S.; Yang, G.; Ebara, S.; Wheeler, T.M.; Frolov, A.; Thompson, T.C. Reduced infiltration of tumor-associated macrophages in human prostate cancer: Association with cancer progression. Cancer Res. 2000, 60, 5857–5861. [Google Scholar] [PubMed]
- Mantovani, A.; Germano, G.; Marchesi, F.; Locatelli, M.; Biswas, S.K. Cancer-promoting tumor-associated macrophages: New vistas and open questions. Eur. J. Immunol. 2011, 41, 2522–2525. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef]
- Kimura, Y.; Sumiyoshi, M. Resveratrol prevents tumor growth and metastasis by inhibiting lymphangiogenesis and M2 macrophage activation and differentiation in tumor-associated macrophages. Nutr. Cancer 2016, 68, 667–678. [Google Scholar] [CrossRef]
- Solís-Martínez, R.; Cancino-Marentes, M.; Hernández-Flores, G.; Ortiz-Lazareno, P.; Mandujano-Álvarez, G.; Cruz-Gálvez, C.; Sierra-Díaz, E.; Rodríguez-Padilla, C.; Jave-Suárez, L.F.; Aguilar-Lemarroy, A.; et al. Regulation of immunophenotype modulation of monocytes-macrophages from M1 into M2 by prostate cancer cell-culture supernatant via transcription factor STAT3. Immunol. Lett. 2018, 196, 140–148. [Google Scholar] [CrossRef]
- Tomé-Carneiro, J.; Larrosa, M.; González-Sarrías, A.; Tomás-Barberán, F.A.; García-Conesa, M.T.; Espín, J.C. Resveratrol and clinical trials: The crossroad from in vitro studies to human evidence. Curr. Pharm. Des. 2013, 19, 6064–6093. [Google Scholar] [CrossRef]
- Kotha, A.; Sekharam, M.; Cilenti, L.; Siddiquee, K.; Khaled, A.; Zervos, A.S.; Carter, B.; Turkson, J.; Jove, R. Resveratrol inhibits src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol. Cancer Ther. 2006, 5, 621–629. [Google Scholar] [CrossRef]
- Sobhani, N.; Generali, D.; D’Angelo, A.; Aieta, M.; Roviello, G. Current status of androgen receptor-splice variant 7 inhibitor niclosamide in castrate-resistant prostate-cancer. Investig. New Drugs 2018, 36, 1133–1137. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Wu, J.M. Grape-derived chemopreventive agent resveratrol decreases prostate-specific antigen (PSA) expression in LNCaP cells by an androgen receptor (AR)-independent mechanism. Anticancer Res. 2000, 20, 225–228. [Google Scholar]
- Benitez, D.A.; Pozo-Guisado, E.; Clementi, M.; Castellón, E.; Fernandez-Salguero, P.M. Non-genomic action of resveratrol on androgen and oestrogen receptors in prostate cancer: Modulation of the phosphoinositide 3-kinase pathway. Br. J. Cancer 2007, 96, 1595–1604. [Google Scholar] [CrossRef]
- Jones, S.B.; DePrimo, S.E.; Whitfield, M.L.; Brooks, J.D. Resveratrol-induced gene expression profiles in human prostate cancer cells. Cancer Epidemiol. Biomark. Prev. 2005, 14, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Seeni, A.; Takahashi, S.; Takeshita, K.; Tang, M.; Sugiura, S.; Sato, S.; Shirai, T. Suppression of prostate cancer growth by resveratrol in the transgenic rat for adenocarcinoma of prostate (TRAP) model. Asian Pac. J. Cancer Prev. 2008, 9, 7–14. [Google Scholar]
- Harada, N.; Murata, Y.; Yamaji, R.; Miura, T.; Inui, H.; Nakano, Y. Resveratrol down-regulates the androgen receptor at the post-translational level in prostate cancer cells. J. Nutr. Sci. Vitaminol. 2007, 53, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.F.; Leong, M.; Cho, E.; Farrell, J.; Chen, H.; Tian, J.; Zhang, D. Repressive effects of resveratrol on androgen receptor transcriptional activity. PLoS ONE 2009, 4, e7398. [Google Scholar] [CrossRef]
- Izumi, K.; Mizokami, A. Suppressive role of androgen/androgen receptor signaling via chemokines on prostate cancer cells. J. Clin. Med. 2019, 8, 354. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.C.; Lu, C.; Antonarakis, E.S.; Luo, J.; Armstrong, A.J. Androgen receptor variant-driven prostate cancer II: Advances in clinical investigation. Prostate Cancer Prostatic Dis. 2020, 23, 367–380. [Google Scholar] [CrossRef]
- Wilson, S.; Cavero, L.; Tong, D.; Liu, Q.; Geary, K.; Talamonti, N.; Xu, J.; Fu, J.; Jiang, J.; Zhang, D. Resveratrol enhances polyubiquitination-mediated ARV7 degradation in prostate cancer cells. Oncotarget 2017, 8, 54683–54693. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).