
Cardiovascular Complications in β-Thalassemia: Getting to the Heart of It

 , ,
, , 
Abstract
1. Introduction
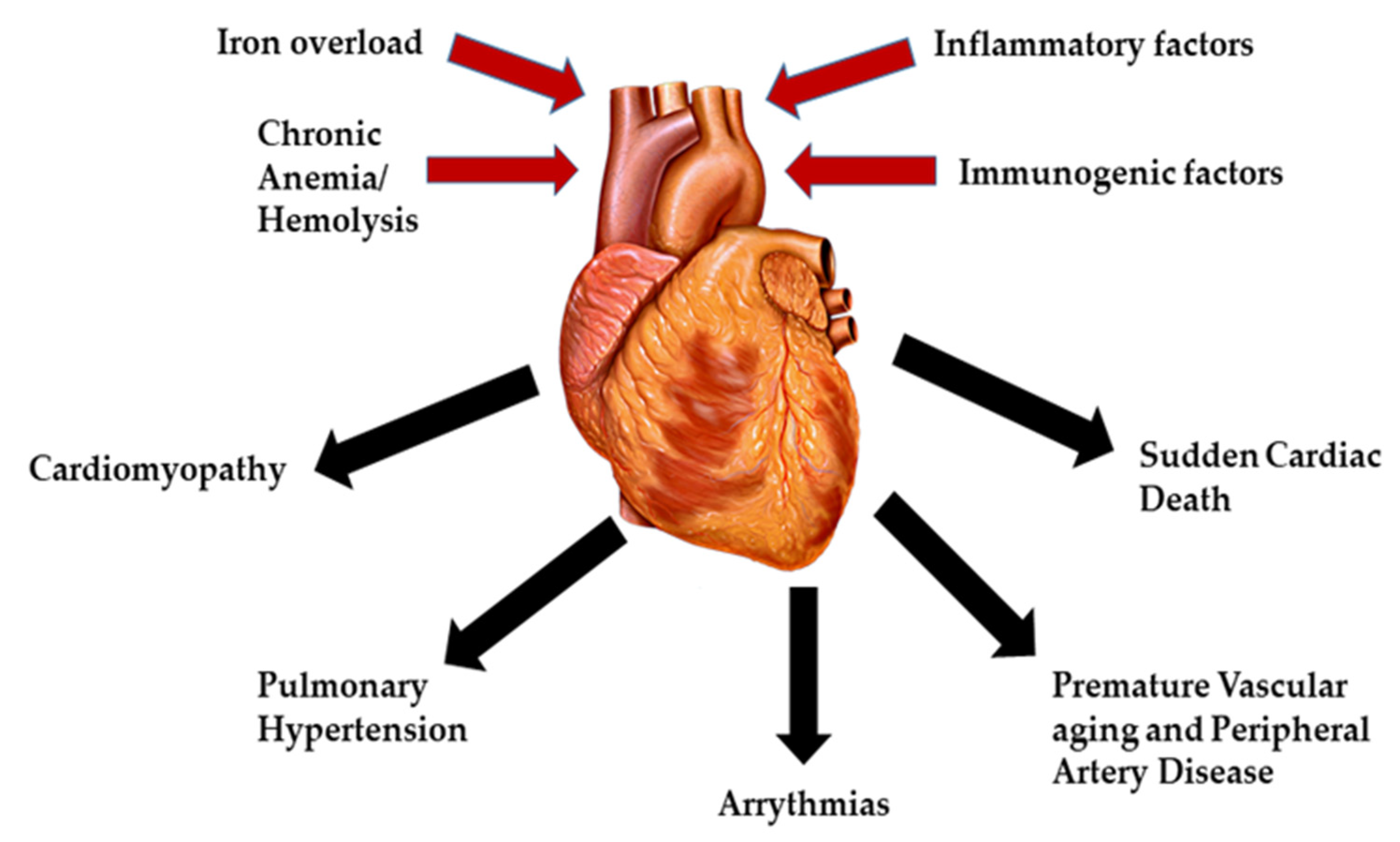
2. Pathophysiology of Cardiac Complications in β-Thalassemia
2.1. Cardiomyopathy
2.2. Pulmonary Hypertension
2.3. Arrhythmias
2.4. Premature Vascular Aging and Peripheral Artery Disease
2.5. Sudden Cardiac Death
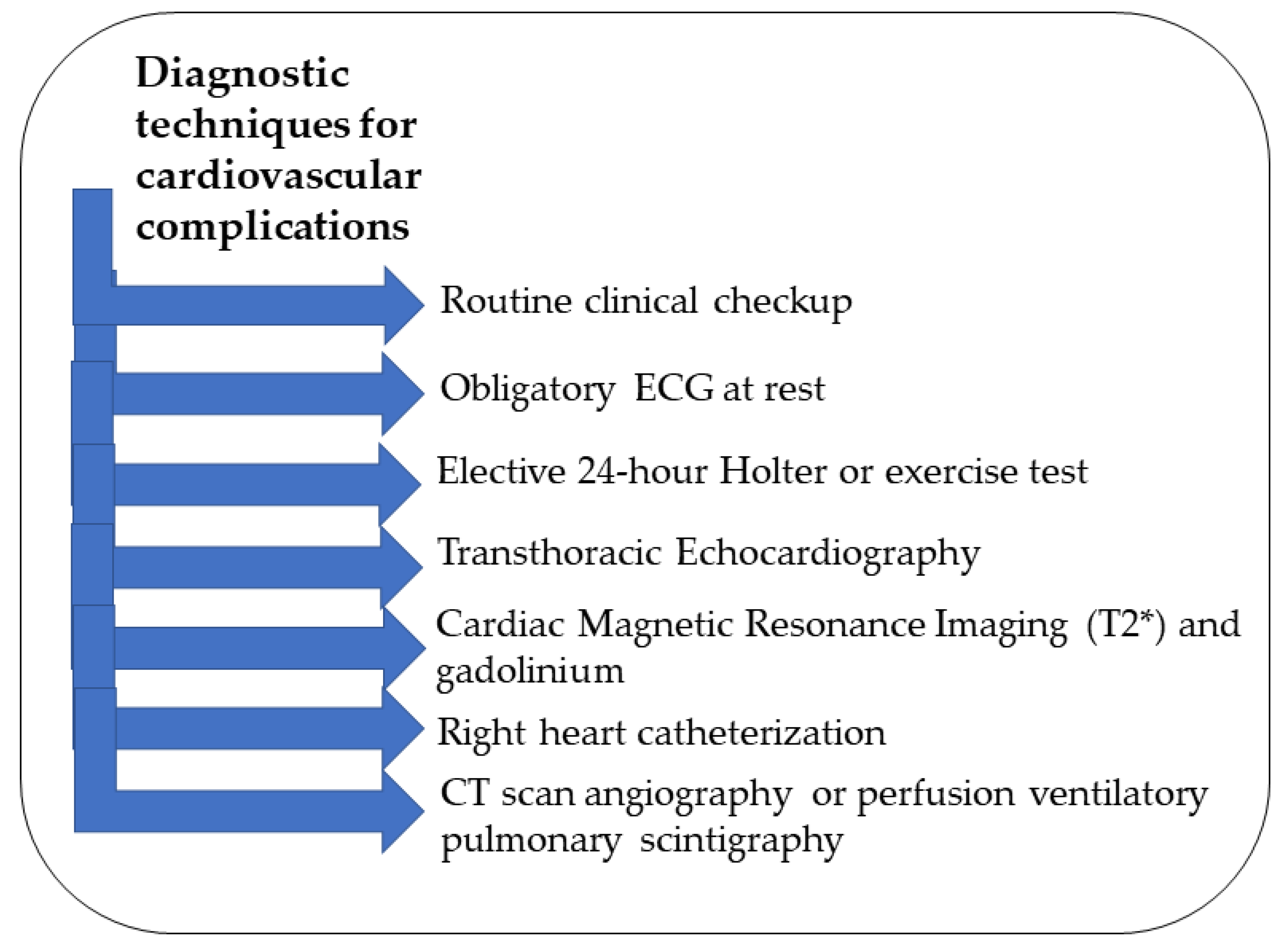
3. Diagnosis of Cardiovascular Complications in β-Thalassemia
4. Prevention and Management
5. Conclusions
Funding
Conflicts of Interest
References
- Russo, V.; Melillo, E.; Papa, A.A.; Rago, A.; Chamberland, C.; Nigro, G. Arrhythmias and Sudden Cardiac Death in Beta-Thalassemia Major Patients: Noninvasive Diagnostic Tools and Early Markers. Cardiol. Res. Pract. 2019, 2019, 9319832. [Google Scholar] [CrossRef] [PubMed]
- Motta, I.; Mancarella, M.; Marcon, A.; Vicenzi, M.; Cappellini, M.D. Management of age-associated medical complications in patients with beta-thalassemia. Expert Rev. Hematol. 2020, 13, 85–94. [Google Scholar] [CrossRef]
- Angastiniotis, M.; Lobitz, S. Thalassemias: An Overview. Int. J. Neonatal Screen. 2019, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. β-Thalassemias. N. Engl. J. Med. 2021, 384, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Cappellini, M.D.; Viprakasit, V.; Kattamis, A.; Rivella, S.; Taher, A.T. Revisiting the non-transfusion-dependent (NTDT) vs. transfusion-dependent (TDT) thalassemia classification 10 years later. Am. J. Hematol. 2021, 96, E54–E56. [Google Scholar] [CrossRef]
- Ali, S.; Mumtaz, S.; Shakir, H.A.; Khan, M.; Tahir, H.M.; Mumtaz, S.; Mughal, T.A.; Hassan, A.; Kazmi, S.A.R.; Sadia; et al. Current status of beta-thalassemia and its treatment strategies. Mol. Genet. Genom. Med. 2021, 9, e1788. [Google Scholar] [CrossRef]
- Barbero, U.; Fornari, F.; Guarguagli, S.; Gaglioti, C.M.; Longo, F.; Doronzo, B.; Anselmino, M.; Piga, A. Atrial fibrillation in β-thalassemia Major Patients: Diagnosis, Management and Therapeutic Options. Hemoglobin 2018, 42, 189–193. [Google Scholar] [CrossRef]
- Pinto, V.M.; Forni, G.L. Management of Iron Overload in Beta-Thalassemia Patients: Clinical Practice Update Based on Case Series. Int. J. Mol. Sci. 2020, 21, 8771. [Google Scholar] [CrossRef]
- Adramerina, A.; Printza, N.; Hatzipantelis, E.; Symeonidis, S.; Tarazi, L.; Teli, A.; Economou, M. Use of Deferasirox Film-Coated Tablets in Pediatric Patients with Transfusion Dependent Thalassemia: A Single Center Experience. Biology 2022, 11, 247. [Google Scholar] [CrossRef]
- Aydinok, Y.; Porter, J.B.; Piga, A.; Elalfy, M.; El-Beshlawy, A.; Kilinc, Y.; Viprakasit, V.; Yesilipek, A.; Habr, D.; Quebe-Fehling, E.; et al. Prevalence and distribution of iron overload in patients with transfusion-dependent anemias differs across geographic regions: Results from the CORDELIA study. Eur. J. Haematol. 2015, 95, 244–253. [Google Scholar] [CrossRef]
- Taher, A.T.; Cappellini, M.D. How I manage medical complications of β-thalassemia in adults. Blood 2018, 132, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Koohi, F.; Kazemi, T.; Miri-Moghaddam, E. Cardiac complications and iron overload in beta thalassemia major patients—A systematic review and meta-analysis. Ann. Hematol. 2019, 98, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Adjimani, J.P.; Asare, P. Antioxidant and free radical scavenging activity of iron chelators. Toxicol. Rep. 2015, 2, 721–728. [Google Scholar] [CrossRef]
- Kremastinos, D.T.; Farmakis, D.; Aessopos, A.; Hahalis, G.; Hamodraka, E.; Tsiapras, D.; Keren, A. Beta-thalassemia cardiomyopathy: History, present considerations, and future perspectives. Circ. Heart Fail. 2010, 3, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Nomani, H.; Bayat, G.; Sahebkar, A.; Fazelifar, A.F.; Vakilian, F.; Jomezade, V.; Johnston, T.P.; Mohammadpour, A.H. Atrial fibrillation in β-thalassemia patients with a focus on the role of iron-overload and oxidative stress: A review. J. Cell. Physiol. 2019, 234, 12249–12266. [Google Scholar] [CrossRef]
- Advani, N.; Advani, N.; Andriastuti, M. The corrected QT interval prolongation in adolescents with cardiac iron overload β-thalassemia major. Turk. J. Pediatr. 2020, 62, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Hershko, C.; Link, G.; Cabantchik, I. Pathophysiology of iron overload. Ann. N. Y. Acad. Sci. 1998, 850, 191–201. [Google Scholar] [CrossRef]
- Wood, J.C. Cardiac iron across different transfusion-dependent diseases. Blood Rev. 2008, 22, S14–S21. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.A.; Sellan, M.; Simpson, J.A.; Izaddoustdar, F.; Cifelli, C.; Panama, B.K.; Davis, M.; Zhao, D.; Markhani, M.; Murphy, G.G.; et al. Iron overload decreases CaV1.3-dependent L-type Ca2+ currents leading to bradycardia, altered electrical conduction, and atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2011, 4, 733–742. [Google Scholar] [CrossRef]
- Wood, J.C.; Enriquez, C.; Ghugre, N.; Otto-Duessel, M.; Aguilar, M.; Nelson, M.D.; Moats, R.; Coates, T.D. Physiology and pathophysiology of iron cardiomyopathy in thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 386–395. [Google Scholar] [CrossRef]
- Russo, V.; Rago, A.; Pannone, B.; Papa, A.A.; Di Meo, F.; Mayer, M.C.; Spasiano, A.; Russo, M.G.; Golino, P.; Calabro, R.; et al. Dispersion of repolarization and beta-thalassemia major: The prognostic role of QT and JT dispersion for identifying the high-risk patients for sudden death. Eur. J. Haematol. 2011, 86, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Hahalis, G.; Manolis, A.S.; Gerasimidou, I.; Alexopoulos, D.; Sitafidis, G.; Kourakli, A.; Körfer, R.; Koerner, M.M.; Vagenakis, A.G.; Zoumbos, N.C. Right ventricular diastolic function in β-thalassemia major: Echocardiographic and clinical correlates. Am. Heart J. 2001, 141, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Ciarambino, T.; Menna, G.; Sansone, G.; Giordano, M. Cardiomyopathies: An Overview. Int. J. Mol. Sci. 2021, 22, 7722. [Google Scholar] [CrossRef] [PubMed]
- Wexler, R.K.; Elton, T.; Pleister, A.; Feldman, D. Cardiomyopathy: An overview. Am. Fam. Physician 2009, 79, 778–784. [Google Scholar]
- Debonnaire, P.; Katsanos, S.; Joyce, E.; Van Den Brink, O.V.W.; Atsma, D.E.; Schalij, M.J.; Bax, J.J.; Delgado, V.; Marsan, N.A. QRS Fragmentation and QTc Duration Relate to Malignant Ventricular Tachyarrhythmias and Sudden Cardiac Death in Patients with Hypertrophic Cardiomyopathy. J. Cardiovasc. Electrophysiol. 2015, 26, 547–555. [Google Scholar] [CrossRef]
- Farmakis, D.; Triposkiadis, F.; Lekakis, J.; Parissis, J. Heart failure in haemoglobinopathies: Pathophysiology, clinical phenotypes, and management. Eur. J. Heart Fail. 2017, 19, 479–489. [Google Scholar] [CrossRef]
- Kremastinos, D.T. Heart failure in β-thalassemia. Congest. Heart Fail. 2001, 7, 312–314. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Meloni, A.; Guerrini, G.; Gulino, L.; Filosa, A.; Ruffo, G.B.; Casini, T.; Chiodi, E.; Lombardi, M.; Pepe, A. Myocardial iron overload in thalassaemia major. How early to check? Br. J. Haematol. 2014, 164, 579–585. [Google Scholar] [CrossRef]
- Meloni, A.; Gulino, L.; Rossi, G.; Pitrolo, L.; De Marchi, D.; Vallone, A.; Resta, M.; Positano, V.; Lombardi, M.; Pepe, A. Prognostic CMR parameters for heart failure and arrhythmias in large cohort of well treated thalssemia major patients. Eur. Heart J. 2013, 34, 1509. [Google Scholar] [CrossRef]
- Du, Z.D.; Roguin, N.; Milgram, E.; Saab, K.; Koren, A. Pulmonary hypertension in patients with thalassemia major. Am. Heart J. 1997, 134, 532–537. [Google Scholar] [CrossRef]
- Jabbar, D.A.; Davison, G.; Muslin, A.J. Getting the iron out: Preventing and treating heart failure in transfusion-dependent thalassemia. Cleve. Clin. J. Med. 2007, 74, 807–810, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Rund, D. Myocardial infarction in a patient with beta-thalassemia major: First report. Am. J. Hematol. 2004, 75, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.F.; Chan, G.C.; Ha, S.Y. Arterial stiffness and endothelial function in patients with beta-thalassemia major. Circulation 2002, 106, 2561–2566. [Google Scholar] [CrossRef]
- Maioli, M.; Vigna, G.B.; Tonolo, G.; Brizzi, P.; Ciccarese, M.; Donegà, P.; Maioli, M.; Fellin, R. Plasma lipoprotein composition, apolipoprotein(a) concentration and isoforms in β-thalassemia. Atherosclerosis 1997, 131, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Rhee, T.-M.; Jeon, K.; Cho, Y.; Lee, S.-W.; Han, K.-D.; Seong, M.-W.; Park, S.-S.; Lee, Y.K. Epidemiologic Trends of Thalassemia, 2006–2018: A Nationwide Population-Based Study. J. Clin. Med. 2022, 11, 2289. [Google Scholar]
- Helmi, N.; Choudhry, H.; Qari, M.; Kumosani, T.A.; Al-Malki, A.L.; Moselhy, S.S.; Kumosani, A.T. Association of serum asymmetric dimethyl-arginine and troponin I levels as a risk of myocardial infarction in thalassemia. Afr. Health Sci. 2018, 18, 720–726. [Google Scholar] [CrossRef]
- Derchi, G.; Galanello, R.; Bina, P.; Cappellini, M.D.; Piga, A.; Lai, M.E.; Quarta, A.; Casu, G.; Perrotta, S.; Pinto, V.; et al. Prevalence and risk factors for pulmonary arterial hypertension in a large group of β-thalassemia patients using right heart catheterization: A Webthal study. Circulation 2014, 129, 338–345. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef]
- Taher, A.; Vichinsky, E.; Musallam, K.; Cappellini, M.D.; Viprakasit, V. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT); Weatherall, D., Ed.; Thalassaemia International Federation: Nicosia, Cyprus, 2013. [Google Scholar]
- Musallam, K.M.; Taher, A.T.; Rachmilewitz, E.A. β-thalassemia intermedia: A clinical perspective. Cold Spring Harb. Perspect. Med. 2012, 2, a013482. [Google Scholar] [CrossRef]
- Mokhtar, G.M.; Adly, A.A.; El Alfy, M.S.; Tawfik, L.M.; Khairy, A.T. N-terminal natriuretic peptide and ventilation-perfusion lung scan in sickle cell disease and thalassemia patients with pulmonary hypertension. Hemoglobin 2010, 34, 78–94. [Google Scholar] [CrossRef]
- Taher, A.T.; Cappellini, M.D.; Bou-Fakhredin, R.; Coriu, D.; Musallam, K.M. Hypercoagulability and Vascular Disease. Hematol. Oncol. Clin. North Am. 2018, 32, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Fraidenburg, D.R.; Machado, R.F. Pulmonary hypertension associated with thalassemia syndromes. Ann. N. Y. Acad. Sci. 2016, 1368, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, J.; Tarhini, A.; Bou-Fakhredin, R.; Saliba, A.N.; Cappellini, M.D.; Taher, A.T. Non-Transfusion-Dependent Thalassemia: An Update on Complications and Management. Int. J. Mol. Sci. 2018, 19, 182. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Rago, A.; Pannone, B.; Mayer, M.C.; Spasiano, A.; Calabro, R.; Russo, M.G.; Gerardo, N.; Papa, A.A. Atrial Fibrillation and Beta Thalassemia Major: The Predictive Role of the 12-lead Electrocardiogram Analysis. Indian Pacing Electrophysiol. J. 2014, 14, 121–132. [Google Scholar] [CrossRef]
- Pennell, D.J.; Udelson, J.E.; Arai, A.E.; Bozkurt, B.; Cohen, A.R.; Galanello, R.; Hoffman, T.M.; Kiernan, M.S.; Lerakis, S.; Piga, A.; et al. Cardiovascular function and treatment in β-thalassemia major: A consensus statement from the American Heart Association. Circulation 2013, 128, 281–308. [Google Scholar] [CrossRef]
- Malagù, M.; Marchini, F.; Fiorio, A.; Sirugo, P.; Clò, S.; Mari, E.; Gamberini, M.R.; Rapezzi, C.; Bertini, M. Atrial Fibrillation in β-Thalassemia: Overview of Mechanism, Significance and Clinical Management. Biology 2022, 11, 148. [Google Scholar] [CrossRef]
- Guerra, F.; Brambatti, M.; Nieuwlaat, R.; Marcucci, M.; Dudink, E.; Crijns, H.; Matassini, M.V.; Capucci, A. Symptomatic atrial fibrillation and risk of cardiovascular events: Data from the Euro Heart Survey. Europace 2017, 19, 1922–1929. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.; Mohamed, S.; Ako, E.; Chatterjee, R.; Bajoria, R.; Porter, J.; Walker, J. The prevalence and risk factors for atrial fibrillation in beta-thalassemia major: A cross-sectional study in a UK specialist cardio-haematology clinic. Eur. Heart J. 2015, 36, 916. [Google Scholar]
- Stoyanova, E.; Trudel, M.; Felfly, H.; Lemsaddek, W.; Garcia, D.; Cloutier, G. Vascular endothelial dysfunction in beta-thalassemia occurs despite increased eNOS expression and preserved vascular smooth muscle cell reactivity to NO. PLoS ONE 2012, 7, e38089. [Google Scholar] [CrossRef]
- Hahalis, G.; Kremastinos, D.T.; Terzis, G.; Kalogeropoulos, A.P.; Chrysanthopoulou, A.; Karakantza, M.; Kourakli, A.; Adamopoulos, S.; Tselepis, A.D.; Grapsas, N.; et al. Global vasomotor dysfunction and accelerated vascular aging in β-thalassemia major. Atherosclerosis 2008, 198, 448–457. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Cohen, A.; Porter, J.; Taher, A.; Viprakasit, V. (Eds.) Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT); Thalassaemia International Federation: Nicosia, Cyprus, 2021. [Google Scholar]
- Nassef, S.; El Shenoufy, M.; Rawi, R.; El Demerdash, D.; Hassan, M.; Mustafa, H.; Mattar, M.; El Husseiny, N. Assessment of Atherosclerosis in Peripheral and Central Circulation in Adult β Thalassemia Intermedia Patients by Color Doppler Ultrasound: Egyptian Experience. J. Vasc. Res. 2020, 57, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Adly, A.A.; El-Sherif, N.H.; Ismail, E.A.; El-Zaher, Y.A.; Farouk, A.; El-Refaey, A.M.; Wahba, M.S. Vascular dysfunction in patients with young β-thalassemia: Relation to cardiovascular complications and subclinical atherosclerosis. Clin. Appl. Thromb. Hemost. 2015, 21, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Zhang, L.; Kim, C.; Uy-Evanado, A.; Teodorescu, C.; Reinier, K.; Zheng, Z.J.; Gunson, K.; Jui, J.; Chugh, S.S. QRS fragmentation and sudden cardiac death in the obese and overweight. J. Am. Heart Assoc. 2015, 4, e001654. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, S. Abnormal QT Dispersion Predicts Unexpected Sudden Death in Young Patients with Thalassemia Major. Ann. Noninvasive Electrocardiol. 1999, 4, 295–300. [Google Scholar] [CrossRef]
- Cogliandro, T.; Derchi, G.; Mancuso, L.; Mayer, M.C.; Pannone, B.; Pepe, A.; Pili, M.; Bina, P.; Cianciulli, P.; De Sanctis, V.; et al. Guideline recommendations for heart complications in thalassemia major. J. Cardiovasc. Med. 2008, 9, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Najimi, M.; Ghandi, Y.; Mehrabi, S.; Eghbali, A.; Habibi, D. Correlation between Myocardial Iron Overload Detected by CMRT2* and Left Ventricular Function Assessed by Tissue Doppler Imaging in Patients with Thalassemia Major. J. Cardiovasc. Echogr. 2022, 32, 17–22. [Google Scholar]
- Ramazzotti, A.; Pepe, A.; Positano, V.; Scattini, B.; Santarelli, M.F.; Landini, L.; De Marchi, D.; Keilberg, P.; Derchi, G.; Formisano, F.; et al. Standardized T2* map of a normal human heart to correct T2* segmental artefacts; myocardial iron overload and fibrosis in thalassemia intermedia versus thalassemia major patients and electrocardiogram changes in thalassemia major patients. Hemoglobin 2008, 32, 97–107. [Google Scholar] [CrossRef]
- Anderson, L.J.; Holden, S.; Davis, B.; Prescott, E.; Charrier, C.C.; Bunce, N.H.; Firmin, D.N.; Wonke, B.; Porter, J.; Walker, J.M.; et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur. Heart J. 2001, 22, 2171–2179. [Google Scholar] [CrossRef]
- Chaosuwannakit, N.; Makarawate, P.; Wanitpongpun, C. The Importance of Cardiac T2* Magnetic Resonance Imaging for Monitoring Cardiac Siderosis in Thalassemia Major Patients. Tomography 2021, 7, 130–138. [Google Scholar] [CrossRef]
- Pennell, D.J.; Porter, J.B.; Cappellini, M.D.; Chan, L.L.; El-Beshlawy, A.; Aydinok, Y.; Ibrahim, H.; Li, C.K.; Viprakasit, V.; Elalfy, M.S.; et al. Deferasirox for up to 3 years leads to continued improvement of myocardial T2* in patients with β-thalassemia major. Haematologica 2012, 97, 842–848. [Google Scholar] [CrossRef]
- Gupta, V.; Kumar, I.; Raj, V.; Aggarwal, P.; Agrawal, V. Comparison of the effects of calcium channel blockers plus iron chelation therapy versus chelation therapy only on iron overload in children and young adults with transfusion-dependent thalassemia: A randomized double-blind placebo-controlled trial. Pediatr. Blood Cancer 2022, 69, e29564. [Google Scholar] [CrossRef] [PubMed]
- Zargari, A.; Wu, S.; Greenway, A.; Cheng, K.; Kaplan, Z. Effects of dual chelation therapy with deferasirox and deferoxamine in patients with beta thalassaemia major. Vox Sang. 2022, 117, 733–737. [Google Scholar] [CrossRef]
- Elfaituri, M.K.; Ghozy, S.; Ebied, A.; Morra, M.E.; Hassan, O.G.; Alhusseiny, A.; Abbas, A.S.; Sherif, N.A.; Fernandes, J.L.; Huy, N.T. Amlodipine as adjuvant therapy to current chelating agents for reducing iron overload in thalassaemia major: A systematic review, meta-analysis and simulation of future studies. Vox Sang. 2021, 116, 887–897. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Trivieri, M.G.; Khaper, N.; Liu, P.P.; Backx, P.H. Role of L-type Ca2+ channels in iron transport and iron-overload cardiomyopathy. J. Mol. Med. 2006, 84, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Bahrani, S.; Teimouri-Jervekani, Z.; Sadeghi, M. Thrombotic Events and Anticoagulants in Beta-thalassemia Patients with Focus on Anticoagulants for Atrial Fibrillation: A Brief Review. Curr. Probl. Cardiol. 2022, 47, 100912. [Google Scholar] [CrossRef]
- Gažová, A.; Leddy, J.J.; Rexová, M.; Hlivák, P.; Hatala, R.; Kyselovič, J. Predictive value of CHA2DS2-VASc scores regarding the risk of stroke and all-cause mortality in patients with atrial fibrillation (CONSORT compliant). Medicine 2019, 98, e16560. [Google Scholar] [CrossRef]
- Hahalis, G.; Alexopoulos, D.; Kremastinos, D.T.; Zoumbos, N.C. Heart failure in beta-thalassemia syndromes: A decade of progress. Am. J. Med. 2005, 118, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Koerner, M.M.; Tenderich, G.; Minami, K.; zu Knyphausen, E.; Mannebach, H.; Kleesiek, K.; Meyer, H.; Koerfer, R. Heart transplantation for end-stage heart failure caused by iron overload. Br. J. Haematol. 1997, 97, 293–296. [Google Scholar] [CrossRef]
- Pepe, A.; Pistoia, L.; Gamberini, M.R.; Cuccia, L.; Peluso, A.; Messina, G.; Spasiano, A.; Allò, M.; Bisconte, M.G.; Putti, M.C.; et al. The Close Link of Pancreatic Iron With Glucose Metabolism and With Cardiac Complications in Thalassemia Major: A Large, Multicenter Observational Study. Diabetes Care 2020, 43, 2830–2839. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Right Ventricular Enlargement and Dilation with basal RV/LV ratio > 1.0 |
| interventricular septum Flattening |
| IVC distention and decreased collapsibility |
| RVOT acceleration time of pulmonary ejection < 105 ms mid-systolic notch |
| RV fractional area change < 35% |
| TAPSE < 18 mm |
| Peak systolic velocity of tricuspid annulus < 9.5 cm/s |
| Right atrial area >18 cm2 |
| Systolic peak tricuspid regurgitation velocity > 2.8 m/s |
| Pericardial effusion |
| Type of Complication | Causes and Risk Factors | Prevention and Management |
|---|---|---|
| Cardiomyopathy |
|
|
| Pulmonary Hypertension |
|
|
| Arrhythmias |
|
(Warfarin or DOACS) |
| Vasculopathies |
|
|
| Sudden Cardiac Death |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akiki, N.; Hodroj, M.H.; Bou-Fakhredin, R.; Matli, K.; Taher, A.T. Cardiovascular Complications in β-Thalassemia: Getting to the Heart of It. Thalass. Rep. 2023, 13, 38-50. https://doi.org/10.3390/thalassrep13010005
Akiki N, Hodroj MH, Bou-Fakhredin R, Matli K, Taher AT. Cardiovascular Complications in β-Thalassemia: Getting to the Heart of It. Thalassemia Reports. 2023; 13(1):38-50. https://doi.org/10.3390/thalassrep13010005
Chicago/Turabian StyleAkiki, Nathalie, Mohammad H. Hodroj, Rayan Bou-Fakhredin, Kamal Matli, and Ali T. Taher. 2023. "Cardiovascular Complications in β-Thalassemia: Getting to the Heart of It" Thalassemia Reports 13, no. 1: 38-50. https://doi.org/10.3390/thalassrep13010005
APA StyleAkiki, N., Hodroj, M. H., Bou-Fakhredin, R., Matli, K., & Taher, A. T. (2023). Cardiovascular Complications in β-Thalassemia: Getting to the Heart of It. Thalassemia Reports, 13(1), 38-50. https://doi.org/10.3390/thalassrep13010005