

Effect of HFE Gene Mutations on Iron Metabolism of Beta-Thalassemia Carriers

Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Hematological and Molecular Analysis
- −
- Forward Primer: 5′-GCTGTC ATCACTTAGACCTCA-3′.
- −
- Reverse Primer: 5′-CACAGTGCAGCTCA CTCAG-3′.
2.3. Statistical Analysis
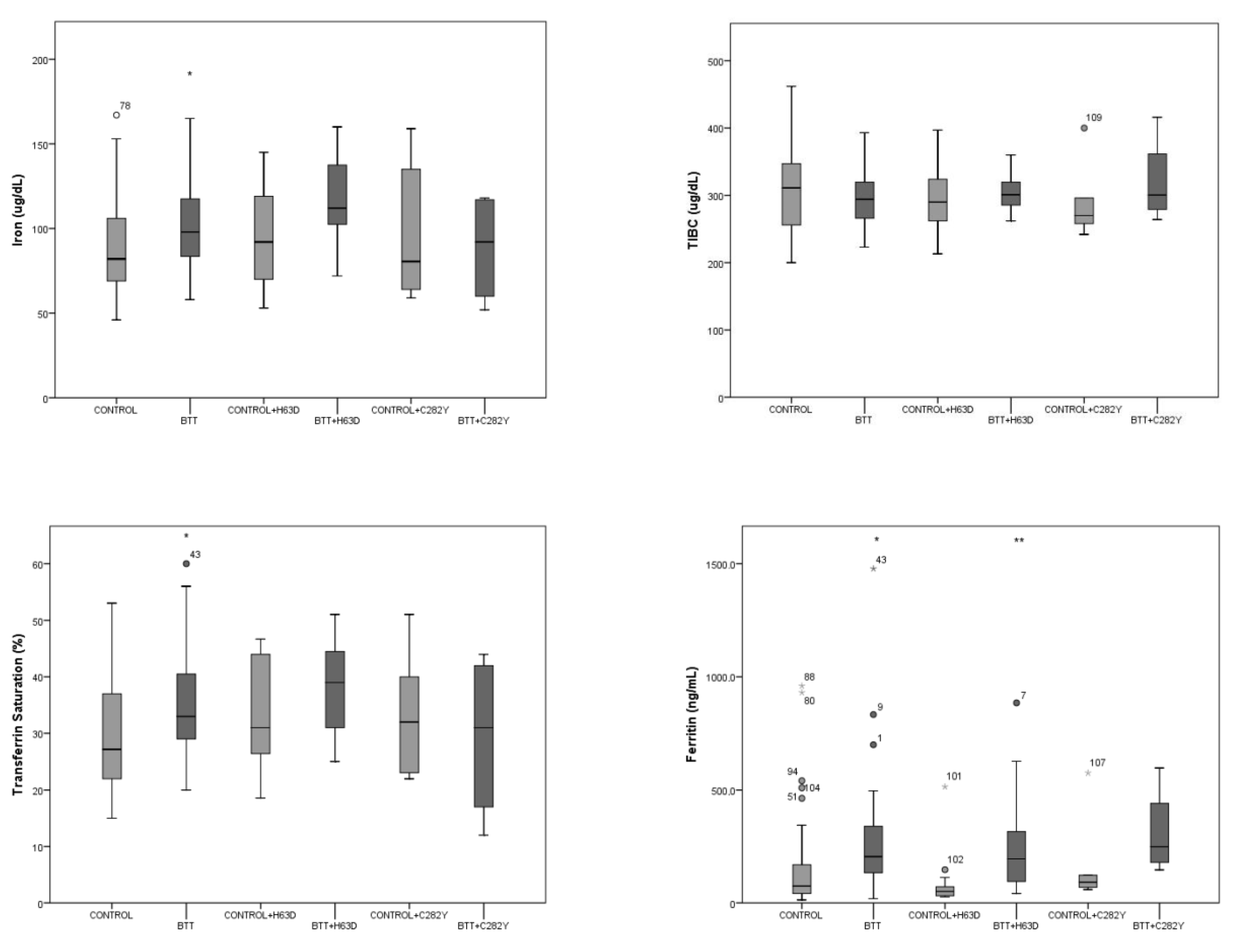
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Camaschella, C.; Pagani, A. Advances in understanding iron metabolism and its crosstalk with erythropoiesis. Br. J. Haematol. 2018, 182, 481–494. [Google Scholar] [CrossRef]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Ribeil, J.A.; Arlet, J.B.; Dussiot, M.; Moura, I.C.; Courtois, G.; Hermine, O. Ineffective erythropoiesis in β- thalassemia. Sci. World J. 2013, 394295. [Google Scholar]
- Jones, E.; Pasricha, S.R.; Allen, A.; Evans, P.; Fisher, C.A.; Wray, K.; Premawardhena, A.; Bandara, D.; Perera, A.; Webster, C.; et al. Hepcidin is suppressed by erythropoiesis in hemoglobin E β-thalassemia and β-thalassemia trait. Blood 2015, 125, 873–880. [Google Scholar] [CrossRef] [PubMed]
- El Beshlawy, A.; Alaraby, I.; Abdel Kader, M.S.; Ahmed, D.H.; Abdelrahman, H.E. Study of serum hepcidin in hereditary hemolytic anemias. Hemoglobin 2012, 36, 555–570. [Google Scholar] [CrossRef]
- Nemeth, E. Hepcidin in beta-thalassemia. Ann. N. Y. Acad. Sci. 2010, 1202, 31–35. [Google Scholar] [CrossRef]
- Powell, L.W.; Seckington, R.C.; Deugnier, Y. Haemochromatosis. Lancet 2016, 388, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R., Jr.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Hollerer, I.; Bachmann, A.; Muckenthaler, M.U. Pathophysiological consequences and benefits of HFE mutations: 20 years of research. Haematologica 2017, 102, 809–817. [Google Scholar] [CrossRef]
- Vrettou, C.; Traeger-Synodinos, J.; Tzetis, M.; Malamis, G.; Kanavakis, E. Rapid screening of multiple beta-globin gene mutations by real-time PCR on the LightCycler: Application to carrier screening and prenatal diagnosis of thalassemia syndromes. Clin. Chem. 2003, 49, 769–776. [Google Scholar] [CrossRef]
- Lazarte, S.S.; Mónaco, M.E.; Haro, A.C.; Jiménez, C.L.; Ledesma Achem, M.E.; Issé, B.A. Molecular characterization and phenotypical study of β-thalassemia in Tucumán, Argentina. Hemoglobin 2014, 38, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Moysés, C.B.; Moreira, E.S.; Asprino, P.F.; Guimarães, G.S.; Alberto, F.L. Simultaneous detection of the C282Y, H63D and S65C mutations in the hemochromatosis gene using quenched-FRET real-time PCR. Braz. J. Med. Biol. Res. 2008, 41, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Melis, M.A.; Cau, M.; Deidda, F.; Barella, S.; Cao, A.; Galanello, R. H63D mutation in the HFE gene increases iron overload in β-thalassemia carriers. Haematologica. 2002, 87, 242–245. [Google Scholar]
- Martins, R.; Picanço, I.; Fonseca, A.; Ferreira, L.; Rodrigues, O.; Coelho, M.; Seixas, T.; Miranda, A.; Nunes, B.; Costa, L.; et al. The role of HFE mutations on iron metabolism in beta-thalassemia carriers. J. Hum. Genet. 2004, 49, 651–655. [Google Scholar] [CrossRef]
- Nadkarni, A.H.; Singh, A.A.; Colaco, S.; Hariharan, P.; Colah, R.B.; Ghosh, K. Effect of the hemochromatosis mutations on iron overload among the indian β thalassemia carriers. J. Clin. Lab. Anal. 2016, 31, e22054. [Google Scholar] [CrossRef]
- AlFadhli, S.; Salem, M.; Shome, D.K.; Mahdi, N.; Nizam, R. The effects of HFE polymorphisms on biochemical parameters of iron status in Arab beta-thalassemia patients. Indian J. Hematol. Blood Transfus. 2017, 33, 545–551. [Google Scholar] [CrossRef]
- Jazayeri, M.; Bakayev, V.; Adibi, P.; Haghighi Rad, H.F.; Zakeri, H.; Kalantar, E.; Zali, M.R. Frequency of HFE gene mutations in Iranian beta thalassaemia minor patients. Eur. J. Haematol. 2003, 71, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Mellouli, F.; El Borgi, W.; Kaabi, H.; Ben Hassen, E.; Sassi, R.; Hmida, H.; Cherif, G.; Maamar, M.; Zouari, B.; Boukef, K.; et al. HFE gene mutations in Tunisian major β-thalassemia and iron overload. Transfus. Clin. Et Biol. 2006, 13, 353–357. [Google Scholar]
- Estevão, I.F.; Peitl Junior, P.; Bonini-Domingos, C.R. Serum ferritin and transferrin saturation levels in β0 and β+ thalassemia patients. Genet Mol. Res. 2011, 10, 632–639. [Google Scholar] [CrossRef]
- López-Escribano, H.; Ferragut, J.F.; Parera, M.M.; Guix, P.; Castro, J.A.; Ramon, M.M.; Picornell, A. Effect of co-inheritance of β-thalassemia and hemochromatosis mutations on iron overload. Hemoglobin 2012, 36, 85–92. [Google Scholar] [CrossRef]
- Enein, A.A.; El Dessouky, N.A.; Mohamed, K.S.; Botros, S.K.A.; Abd El Gawad, M.F.; Hamdy, M.; Dyaa, N. Frequency of hereditary hemochromatosis (HFE) gene mutations in egyptian beta thalassemia patients and its relation to iron overload. Maced. J. Med. Sci. 2016, 4, 226–231. [Google Scholar] [CrossRef]
- Soltanpour, M.S.; Kambiz Davari, K. The correlation of cardiac and hepatic hemosiderosis as measured by T2*MRI technique with ferritin levels and hemochromatosis gene mutations in Iranian patients with beta thalassemia major. Oman Med. J. 2018, 33, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.B.; Fucharoen, S.; Winichagoon, P.; Sirankapracha, P.; Zeder, C.; Gowachirapant, S.; Judprasong, K.; Tanno, T.; Miller, J.L.; Hurrell, R.F. Iron metabolism in heterozygotes for hemoglobin E (HbE), alpha-thalassemia 1, or beta-thalassemia and in compound heterozygotes for HbE/beta-thalassemia. Am. J. Clin Nutr. 2008, 88, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Mehta, B.C.; Pandya, B.G. Iron status of beta thalassemia carriers. Am. J. Hematol. 1987, 24, 137–141. [Google Scholar] [CrossRef]
- Adams, P.C.; Reboussin, D.M.; Barton, J.C.; McLaren, C.E.; Eckfeldt, J.H.; McLaren, G.D.; Sholinsky, P.; Walker, A.P.; Gordeuk, V.R. Hemochromatosis and Iron Overload Screening (HEIRS) Study Research Investigators. Hemochromatosis and iron-overload screening in a racially diverse population. N. Engl. J. Med. 2005, 352, 1769–1778. [Google Scholar] [CrossRef]
- Aranda, N.; Viteri, F.E.; Montserrat, C.; Arija, V. Effects of C282Y, H63D, and S65C HFE gene mutations, diet, and life-style factors on iron status in a general Mediterranean population from Tarragona, Spain. Ann. Hematol. 2010, 89, 767–773. [Google Scholar] [CrossRef]
- Kaczorowska-Hac, B.; Luszczyk, M.; Antosiewicz, J.; Ziolkowski, W.; Adamkiewicz-Drozynska, E.; Mysliwiec, M.; Milosz, E.; Kaczor, J.J. HFE gene mutations and iron status in 100 healthy polish children. J. Pediatr. Hematol. Oncol. 2017, 39, e240–e243. [Google Scholar] [CrossRef]
- Jackson, H.A.; Carter, K.; Darke, C.; Guttridge, M.G.; Ravine, D.; Hutton, R.D.; Napier, J.A.; Worwood, M. HFE mutations, iron deficiency and overload in 10,500 blood donors. Br. J. Haematol. 2001, 114, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Worwood, M. Iron deficiency anaemia and iron overload. In Dacie and Lewis Practical Haematology, 10th ed.; Meloni, D., Oberle, K., Saltzberg, D., Eds.; Churchil Livingstone Elsevier: Philadelphia, PA, USA, 2006; pp. 131–160. [Google Scholar]
- Rosatelli, M.C.; Tuveri, T.; Scalas, M.T.; Leoni, G.B.; Sardu, R.; Faà, V.; Meloni, A.; Pischedda, M.A.; Demurtas, M.; Monni, G.; et al. Molecular screening and fetal diagnosis of beta-thalassemia in the Italian population. Hum. Genet. 1992, 89, 585–589. [Google Scholar]
- Rigoli, L.; Meo, A.; Miceli, M.R.; Alessio, K.; Caruso, R.A.; La Rosa, M.A.; Salpietro, D.C.; Ricca, M.; Barberi, I. Molecular analysis of beta-thalassaemia patients in a high incidence area of southern Italy. Clin. Lab. Haematol. 2001, 23, 373–378. [Google Scholar] [CrossRef]
- Murad, H.; Moassas, F.; Ghoury, I.; Mukhalalaty, Y. Haplotype Analysis of Three Common β-Thalassemia Mutations in Syrian Patients. Hemoglobin 2018, 42, 302–305. [Google Scholar] [CrossRef]
- Makhoul, N.J.; Wells, R.S.; Kaspar, H.; Shbaklo, H.; Taher, A.; Chakar, N.; Zalloua, P.A. Genetic heterogeneity of Beta thalassemia in Lebanon reflects historic and recent population migration. Ann. Hum. Genet. 2005, 69, 55–66. [Google Scholar] [CrossRef]
- Madani, H.A.; Afify, R.A.; Abd El-Aal, A.A.; Salama, N.; Ramy, N. Role of HFE gene mutations on developing iron overload in beta-thalassaemia carriers in Egypt. East Mediterr. Health J. 2011, 17, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.M.; Souza, F.P.; Jardim, A.C.; Cordeiro, J.A.; Pinho, J.R.; Sitnik, R.; Estevão, I.F.; Bonini-Domingos, C.R.; Rahal, P. HFE gene mutations in Brazilian thalassemic patients. Braz. J. Med. Biol. Res. 2006, 39, 1575–1580. [Google Scholar] [CrossRef]
- Beutler, E. The significance of the 187G (H63D) mutation in hemochromatosis. Am. J. Hum. Genet. 1997, 61, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Jaing, T.H.; Chang, T.Y.; Chen, S.H.; Lin, C.W.; Wen, Y.C.; Chiu, C.C. Molecular genetics of β-thalassemia: A narrative review. Medicine 2021, 100, e27522. [Google Scholar] [CrossRef]
- Kawabata, H. The mechanisms of systemic iron homeostasis and etiology, diagnosis, and treatment of hereditary hemochromatosis. Int. J. Hematol. 2018, 107, 31–43. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Genotype | Control (n = 69) | BTT (n = 50) |
|---|---|---|
| H63D/wt | 11 (16%) | 14 (28%) |
| H63D/H63D | 2 (3%) | 0 |
| C282Y/wt | 6 (9%) | 3 (6%) |
| S65C/wt | 0 | 1 (2%) |
| H63D/C282Y | 0 | 1 (2%) |
| wt/wt | 50 (72%) | 30 (60%) |
| Ethnicity | Beta-thalassemia Mutation | Total | HFE Mutation | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CD39 | IVSI-1 | IVSI-110 | IVSII-1 | IVSII-745 | ND | C282Y | H63D | S65C | WT | ||
| Spanish | 1 | 3 | 1 | 0 | 0 | 2 | 7 | 0 | 0 | 0 | 7 |
| Italian | 6 | 1 | 5 | 1 | 0 | 1 | 14 | 1 * | 5 | 1 | 7 |
| Arab | 0 | 4 | 0 | 0 | 0 | 1 | 5 | 0 | 2 | 0 | 3 |
| Spanish–Italian | 2 | 0 | 2 | 0 | 1 | 1 | 6 | 1 | 2 | 0 | 3 |
| Spanish–Arab | 0 | 3 | 1 | 0 | 0 | 1 | 5 | 1 | 1 | 0 | 3 |
| Creole | 1 | 2 | 1 | 0 | 0 | 0 | 4 | 0 | 1 | 0 | 3 |
| Arab–French | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| Spanish–Italian–Arab | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 1 |
| Spanish–Italian–Arab–German | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 |
| Spanish–Italian–Bulgarian | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
| French | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 |
| Spanish–Arab–Bulgarian | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 |
| Unknown | 0 | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 2 | 0 | 0 |
| Total | 10 | 16 | 14 | 1 | 1 | 8 | 50 | 4 | 15 | 1 | 30 |
| β-thalassemia Mutation Type | HFE Gene Mutation | Fe (µg/dL) | TIBC (µg/dL) | Transferrin Saturation (%) | Ferritin (ng/mL) |
|---|---|---|---|---|---|
| β0 (IVSI-1, CD39, IVSII-1) n = 27 | Present, n = 9 | 112 (72–139) | 286 (262–325) | 39 (25–48) | 203.0 (41.6–626.0) |
| Absent, n = 18 | 102 (73–165) | 287 (223–392) | 33 (26–56) | 196.7 (32.6–833.0) | |
| β+ (IVSI-110, IVSII-745) n = 15 | Present, n = 8 | 120 (52–160) | 311 (264–416) * | 42 (12–51) | 203.7 (45.1–884.7) |
| Absent, n = 7 | 98 (63–156) | 299 (276–321) | 33 (21–48) | 254.2 (18.6–359.0) | |
| Not determined n = 8 | Present, n = 2 | 105 (105–105) | 331 (302–359) | 32 (29–35) | 148.8 (45.3–252.4) |
| Absent, n = 6 | 98 (58–154) | 299 (259–393) | 32 (20–60) | 258.4 (55.7–1478.0) |
| Parameter | Control without HFE Gene Mutation | Control with HFE Gene Mutation | ||
|---|---|---|---|---|
| Female (n = 32) | Male (n = 18) | Female (n = 16) | Male (n = 3) | |
| Age (years) | 28 (22–64) | 31 (21–63) | 26 (24–37) | 39 (21–42) |
| RBC (×1012/L) | 4.42 (3.97–5.02) | 4.95 (4.37–5.43) a | 4.38 (3.84–5.03) | 4.96 (4.36–5.65) |
| Ht (L/L) | 0.40 (0.37–0.44) | 0.44 (0.41–0.50) a | 0.41 (0.35–0.44) | 0.44 (0.39–0.47) |
| Hb (g/L) | 126 (114–141) | 146 (133–164) a | 130 (113–137) | 148 (127–161) |
| MCV (fL) | 90.0 (83.0–95.0) | 89.5 (83.2–96.0) | 91.4 (82.0–98.3) a | 88.0 (82.8–89.7) |
| MCH (pg) | 29 (26–31.2) | 30 (28–33) | 29.4 (25.0–31.2) | 29.0 (28.5–29.8) |
| MCHC (g/L) | 321 (308–338) | 329 (317–372) | 318 (298–338) | 333 (326–344) |
| RDW (%) | 12.8 (11.2–17.4) | 12.7 (12.0–13.7) | 12.7 (11.5–13.5) | 12.4 (12.3–12.9) |
| Iron (µg/dL) | 76.5 (46–153) | 98.5 (57–167) a | 94 (53–159) | 72 (64–135) |
| TIBC (µg/dL) | 314 (200–462) | 284 (209–372) | 288 (213–400) | 264 (263–296) |
| Sat (%) | 24 (15–53) b | 32 (21–50) a | 32 (19–47) | 27 (22–51) |
| Ferritin (ng/mL) | 57.6 (13.2–959.9) | 200.6 (47–930.5)a | 52.8 (26.3–147) | 514.9 (83.8–574.7) |
| BTT without HFE gene mutation | BTT with HFE gene mutation | |||
| Female (n = 19) | Male (n = 12) | Female (n = 12) | Male (n = 7) | |
| Age (years) | 38 (21–79) | 36 (16–75) | 35 (16–69) | 52 (29–65) |
| RBC (×1012/L) | 5.52 (4.40–6.10) a,b | 6.12 (5.06–7.14) c,d | 5.62 (4.89–7.39) a,b | 6.32 (5.46–7.10) c,d |
| Ht (L/L) | 0.35 (0.30–0.40) a,b | 0.41 (0.35–0.46) | 0.36 (0.33–0.47) a,b | 0.41 (0.35–0.46) |
| Hb (g/L) | 107 (89–123) a,b | 122 (105–137) c,d | 111 (99–135) a,b | 121 (103–137) c,d |
| MCV (fL) | 65.3 (62.1–72.7) a,b | 66.4 (60.8–69.5) c,d | 64.9 (61.0–74.0) a,b | 64.1 (60.8–69.6) c,d |
| MCH (pg) | 19.5 (17.9–22.3) a,b | 198 (17.9–21.8) c,d | 19.6 (18.1–22.7) a,b | 19.0 (17.6–21.6) c,d |
| MCHC (g/L) | 296 (216–316) a,b | 301 (294–317) | 302 (205–317) a,b | 296 (289–310) |
| RDW (%) | 15.8 (13.2–18.4) a,b | 16.8 (14.4–19.9) c,d | 16.1 (14.2–17.0) a,b | 16.4 (15.7–18.0) c,d |
| Iron (µg/dL) | 98 (63–131) a | 104.5 (58–165) | 114 (52–152) | 102 (68–160) |
| TIBC (µg/dL) | 280 (223–393) | 298 (259–353) | 301 (264–416) | 307 (262–360) |
| Sat (%) | 32 (21–56) a | 34 (20–60) | 40 (12–51) | 39 (22–48) |
| Ferritin (ng/mL) | 165.6 (18.6–833) a | 298.2 (97.7–1478) | 137.8 (41.6–308) b,e | 510 (163–884.7) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mónaco, M.E.; Alvarez Asensio, N.S.; Haro, C.; Terán, M.M.; Ledesma Achem, M.E.; Issé, B.A.; Lazarte, S.S. Effect of HFE Gene Mutations on Iron Metabolism of Beta-Thalassemia Carriers. Thalass. Rep. 2023, 13, 113-121. https://doi.org/10.3390/thalassrep13010010
Mónaco ME, Alvarez Asensio NS, Haro C, Terán MM, Ledesma Achem ME, Issé BA, Lazarte SS. Effect of HFE Gene Mutations on Iron Metabolism of Beta-Thalassemia Carriers. Thalassemia Reports. 2023; 13(1):113-121. https://doi.org/10.3390/thalassrep13010010
Chicago/Turabian StyleMónaco, María E., Natalia S. Alvarez Asensio, Cecilia Haro, Magdalena M. Terán, Miryam E. Ledesma Achem, Blanca A. Issé, and Sandra S. Lazarte. 2023. "Effect of HFE Gene Mutations on Iron Metabolism of Beta-Thalassemia Carriers" Thalassemia Reports 13, no. 1: 113-121. https://doi.org/10.3390/thalassrep13010010
APA StyleMónaco, M. E., Alvarez Asensio, N. S., Haro, C., Terán, M. M., Ledesma Achem, M. E., Issé, B. A., & Lazarte, S. S. (2023). Effect of HFE Gene Mutations on Iron Metabolism of Beta-Thalassemia Carriers. Thalassemia Reports, 13(1), 113-121. https://doi.org/10.3390/thalassrep13010010