Abstract
This is a report of a novel variant of the α1-globin gene—(α1) α51 Gly > Cys (CE9), c.154 GGC > TGC, named Hb Mazandaran, which was observed in an Iranian family. This variant gives rise to a previously undescribed haemoglobin variant that was undetectable by capillary haemoglobin electrophoresis (CE). This variant was detected in two cases in combination with β-globin mutation, and it does not seem to be associated with severe haematological abnormalities in the carriers.
1. Introduction
Haemoglobin (Hb) disorders are a major worldwide global health concern, and it is estimated that around 7% of the world population are carriers of different kinds of haemoglobinopathies. Alpha-thalassemia (α-thal) is the most common Hb disorder around the world, with the highest incidence rate in South-East Asia, the Mediterranean and Middle Eastern populations, India, and sub-Saharan Africa [1]. In the north of Iran, about 15% of neonates are carriers of α-globin mutations [2]. Depending on the number of deleted copies or non-deletional mutations, the clinical presentations of the disease are varied, ranging from almost asymptomatic to lethal haemolytic anaemia [3].
More than 400 different structural variations of α-globin protein have been introduced, most of which are not associated with noticeable clinical manifestation [3]; however, some can affect the function of the Hb molecule and its stability, leading to erythrocytosis and haemolytic anaemia.
There is no genotype–phenotype correlation, even in the presence of a similar genotype in patients with α-thalassemia [3,4]. Hence, introduction of novel variants of the α-globin gene helps to collect comprehensive knowledge about thalassemia disease. The present study aimed to introduce a new H. variant caused as a result of mutation of the α1-globin gene.
2. Case Presentation
A 25-year-old male was referred to Fajr Medical Genetics Laboratory (Sari, Iran) for routine haematological analysis as part of a national screening program for beta thalassemia [5].
Complete blood count (CBC) and Hb capillary electrophoresis (CE) were carried out. After obtaining written informed consent from the patient, molecular analysis was conducted on genomic DNA extracted from peripheral blood using a QIAamp DNA Mini Kit (Qiagen, Germany). To identify common Mediterranean α-Globin gene deletions (-α3.7, -α4.2, --MED, and --20.5), multiplex Gap-PCR was performed, and to detect other mutations of the α and β-Globin genes, the PCR sequencing method was used.
Haematological indices and CE results were compatible with being a β-thalassemia carrier (Figure 1, Table 1). Molecular analysis of α and β-globin genes indicated that the subject carried c.315 + 1G>A (IVSII-IG > A) and c.154G > T (p.Gly52Cys) mutations of the β and α1-globin genes, respectively (Figure 2). DNA analysis of the subject’s parents showed that the father did not carry the identified mutations; however, his mother carried both of the detected mutations. To the best of our knowledge, the c.154G > T (p.Gly52Cys) variant has not been reported in databases (Clin Var and USSC), and we named it Hb Mazandaran. In silico analysis using PolyPhen-2 software predicted the altered protein to be probably damaging (with a score of 1).
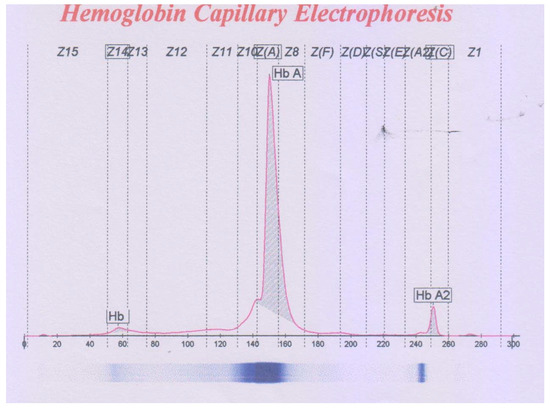
Figure 1.
The capillary electrophoresis pattern of the case with co-inheritance of HBB: c.315 + 1G > A and HBA1: c.154G > T mutations.

Table 1.
Haematological indices of a case with co-inheritance of c.315 + 1G > A mutation of the β-globin gene and c.154G > T mutation of the α1-globin gene.
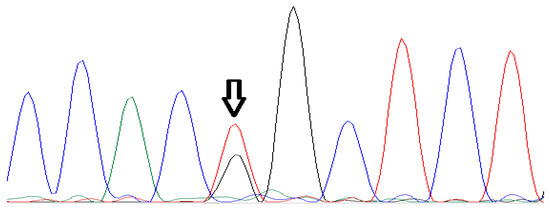
Figure 2.
Sanger sequencing analysis of HbA1 gene in the case with Hb Mazandaran variant (c.154G > T).
3. Discussion
Mazandaran is a province of Iran located on the southern coastline of the Caspian Sea, and thalassemia is a common hereditary genetic disorder in that region [6]. Several variants of Hb, such as Hb D [7], Hb J-Toronto [8], Hb Fontainebleau [9], and Hb S [10], have also been reported in Mazandaran. In the present study, we report a novel variant of the α1-globin gene, Hb Mazandaran (α1) α51 Gly > Cys(CE9), c.154 GGC > TGC, detected in a family from Mazandaran, Iran. This mutation gives rise to a previously undescribed Hb variant that was undetectable via the CE technique.
To date, three different variants of Hb have been identified at codon 52 of the HbA1 gene, including Hb Riccarton (p.Glys51Ser) [11], Hb Russ (p.Gly51Arg) [12], and Hb J Abidjan (p.Gly51Asp) [13]. The presented case is the fourth identified Hb variant as a result of an amino acid change at codon 52 of the HbA1 gene. This variant was detected in two cases in combination with β-globin mutation, and it does not seem to be associated with severe haematological abnormalities in the carriers. Identification of all variants of Hb in different regions helps to collect comprehensive knowledge about thalassemia disease, and it can be used in preventive programmes and in prenatal diagnosis.
Author Contributions
M.M and H.J.; methodology, M.M.; software, A.A.; formal analysis, M.M, and A.A.; data curation, H.J and A.A.; writing—original draft preparation, M.R.M., writing—review and editing, M.R.M.; project administration. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no extra funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data can be made available upon request to the corresponding author.
Acknowledgments
The authors are grateful to Fatemeh Alizadeh, Bita Talebi and Maryam Rahimifor their assistance in sampling and writing of the manuscript. This case report was coordinated by Fajr Genetics and Pathobiology Laboratory.
Conflicts of Interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Weatherall, D.J. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012, 26, S3–S6. [Google Scholar] [CrossRef]
- Jalali, H.; Mahdavi, M.R.; Roshan, P.; Kosaryan, M.; Karami, H.; Mahdavi, M. Alpha thalassemia gene mutations in neonates from Mazandaran, Iran, 2012. Hematology 2014, 19, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R.; Cao, A. Gene test review. Alpha-thalassemia. Genet. Med. Off. J. Am. Coll. Med. Genet. 2011, 13, 83–88. [Google Scholar]
- Akhtar, M.S.; Qaw, F.; Borgio, J.F.; Albuali, W.; Suliman, A.; Nasserullah, Z.; Al-Jarrash, S.; Al-Ali, A. Spectrum of alpha-thalassemia mutations in transfusion-dependent beta-thalassemia patients from the Eastern Province of Saudi Arabia. Hemoglobin 2013, 37, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Khorasani, G.; Kosaryan, M.; Vahidshahi, K.; Shakeri, S.; Nasehi, M.M. Results of the national program for prevention of ²خ-thalassemia major in the Iranian Province of Mazandaran. Hemoglobin 2008, 32, 263–271. [Google Scholar] [CrossRef]
- Kosaryan, M.; Karami, H.; Darvishi-Khezri, H.; Akbarzadeh, R.; Aliasgharian, A.; Bromand, K. Treatment Status of Patients with Β-Thalassemia Major in Northern Iran: Thalassemia Registry System. Iran. J. Public Health 2019, 48, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, M.R.; Jalali, H.; Kosaryan, M.; Roshan, P.; Mahdavi, M. β-Globin gene cluster haplotypes of Hb D-Los Angeles in Mazandaran province, Iran. Genes Genet. Syst. 2015, 90, 55–57. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mahdavi, M.R.; Bayat, N.; Hadavi, V.; Karami, H.; Roshan, P.; Najmabadi, H.; Rohanizadeh, H. Report of Haemoglobin J-Toronto and alpha thalassemia in a family from North of Iran. JPMA-J. Pak. Med. Assoc. 2012, 62, 396. [Google Scholar] [PubMed]
- Jalali, H.; Rasouli, S.T.; Najafi, M.; Karami, H.; Mahdavi, M.R.; Mahdavi, M. A report of Hb Fontainebleau [±21خ (B2) Ala> Pro] as a result of founder effect phenomenon. Gene Rep. 2020, 19, 100587. [Google Scholar] [CrossRef]
- Aghajani, F.; Mahdavi, M.R.; Kosaryan, M.; Mahdavi, M.; Hamidi, M.; Jalali, H. Identification of β-globin haplotypes linked to sickle hemoglobin (Hb S) alleles in Mazandaran province, Iran. Genes Genet. Syst. 2017, 91, 311–313. [Google Scholar] [CrossRef] [PubMed][Green Version]
- van den Ouweland, J.M.; van Daal, H.; Klaassen, C.H.; van Aarssen, Y.; Harteveld, C.L.; Giordano, P.C. The silent hemoglobin α chain variant Hb Riccarton [α51 (CE9) Gly→Ser] may affect HbA1c determination on the HLC-723 G7 analyzer. Clin. Chem. Lab. Med. 2008, 46, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.A.; Huisman, T.H. Hemoglobin Russ or alpha-2-51-arg-beta-2. Biochim. Et Biophys. Acta 1966, 130, 541–543. [Google Scholar] [CrossRef]
- Cabannes, R.; Renaud, R.; Mauran, A.; Pennors, H.; Charlesworth, D.; Price, B.G.; Lehmann, H. Two fast hemoglobins in Ivory-Coast: Hb K Woolwich and a new hemoglobin Hb J Abidjan (alpha-51 Gly-Asp). Nouv. Rev. Fr. D’hematologie 1972, 12, 289–300. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).