
Which Came First? When Usher Syndrome Type 1 Couples with Neuropsychiatric Disorders

 , , , ,
, , , ,  and
and 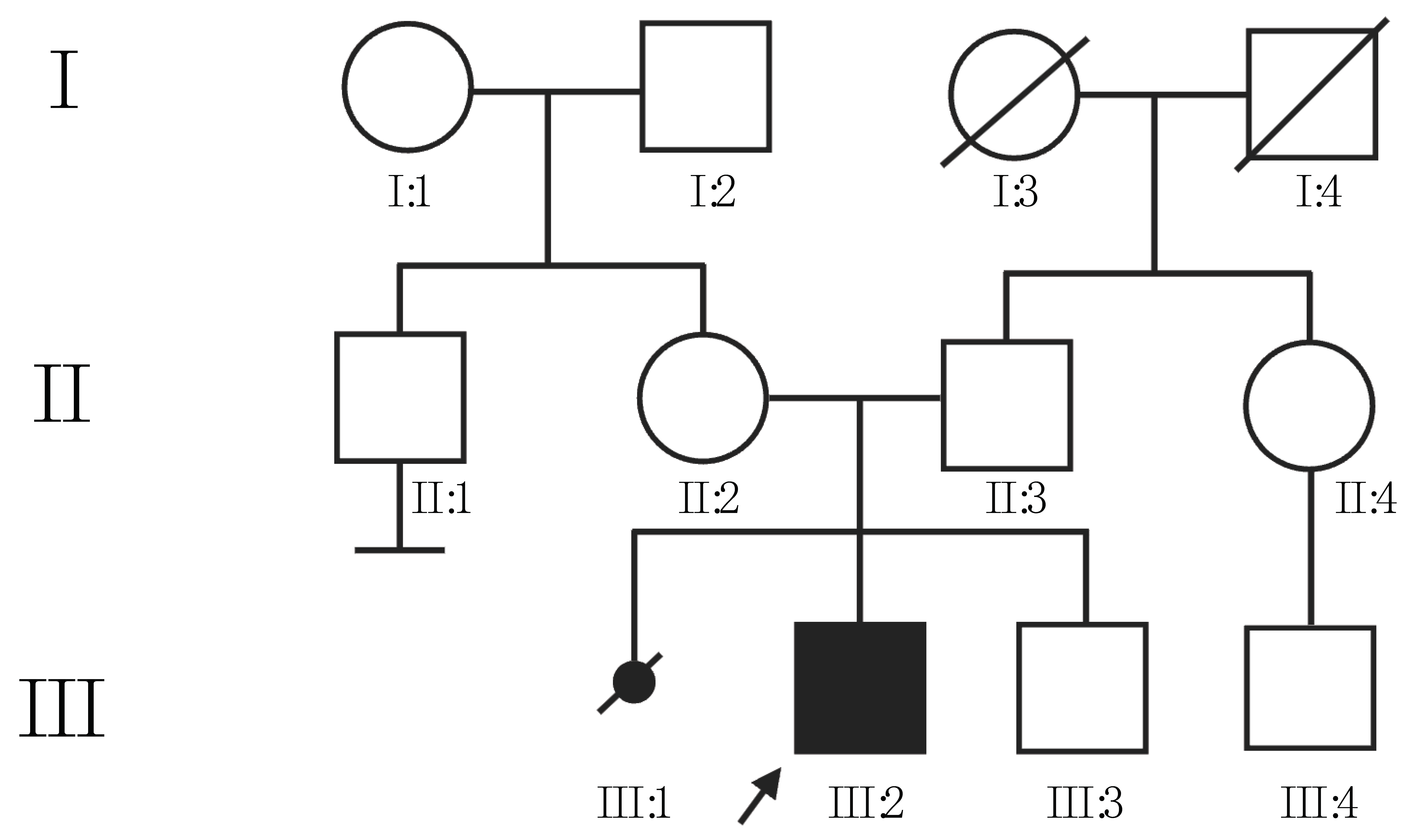{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
- Variant frequency—Minor allele frequency cut off: 0.1%;
- Variant effect: coding and splicing variants;
- Pathogenicity: ACMG/AMP criteria;
- Family inheritance: autosomal recessive, X-linked recessive;
- Quality of the variant call: ≥20.
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Domanico, D.; Fragiotta, S.; Cutini, A.; Grenga, P.L.; Vingolo, E.M. Psychosis, Mood and Behavioral Disorders in Usher Syndrome: Review of the Literature. Med. Hypothesis Discov. Innov. Ophthalmol. 2015, 4, 50–55. [Google Scholar]
- Delmaghani, S.; El-Amraoui, A. The Genetic and Phenotypic Landscapes of Usher Syndrome: From Disease Mechanisms to a New Classification. Hum. Genet. 2022, 141, 709–735. [Google Scholar] [CrossRef]
- Koenekoop, R.K.; Arriaga, M.A.; Trzupek, K.M.; Lentz, J.J. Usher Syndrome Type I. GeneReviews 2020. [Google Scholar]
- Domanico, D.; Fragiotta, S.; Trabucco, P.; Nebbioso, M.; Vingolo, E.M. Genetic Analysis for Two Italian Siblings with Usher Syndrome and Schizophrenia. Case Rep. Ophthalmol. Med. 2012, 2012, 380863. [Google Scholar] [CrossRef]
- Arcous, M.; Putois, O.; Dalle-Nazébi, S.; Kerbourch, S.; Cariou, A.; Ben Aissa, I.; Marlin, S.; Potier, R. Psychosocial Determinants Associated with Quality of Life in People with Usher Syndrome. A Scoping Review. Disabil. Rehabil. 2020, 42, 2809–2820. [Google Scholar] [CrossRef] [PubMed]
- Dean, G.; Orford, A.; Staines, R.; McGee, A.; Smith, K.J. Psychosocial Well-Being and Health-Related Quality of Life in a UK Population with Usher Syndrome. BMJ Open 2017, 7, e013261. [Google Scholar] [CrossRef]
- Roborel de Climens, A.; Tugaut, B.; Piscopo, A.; Arnould, B.; Buggage, R.; Brun-Strang, C. Living with Type I Usher Syndrome: Insights from Patients and Their Parents. Ophthalmic Genet. 2020, 41, 240–251. [Google Scholar] [CrossRef]
- Waldeck, T.; Wyszynski, B.; Medalia, A. The Relationship between Usher’s Syndrome and Psychosis with Capgras Syndrome. Psychiatry 2001, 64, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Hess-Röver, J.; Crichton, J.; Byrne, K.; Holland, A.J. Case Report: Diagnosis and Treatment of a Severe Psychotic Illness in a Man with Dual Severe Sensory Impairments Caused by the Presence of Usher Syndrome. J. Intellect. Disabil. Res. 1999, 43, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Rijavec, N.; Grubic, V.N. Usher Syndrome and Psychiatric Symptoms: A Challenge in Psychiatric Management. Psychiatr. Danub. 2009, 21, 68–71. [Google Scholar] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing Its Use in a Clinical Diagnostic or Research Setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Shearer, A.E.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T.; et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, I.; Marini, S.; Bellazzi, R. PaPI: Pseudo Amino Acid Composition to Score Human Protein-Coding Variants. BMC Bioinform. 2015, 16, 123. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. DANN: A Deep Learning Approach for Annotating the Pathogenicity of Genetic Variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ma, J.; Ma, X.; Lin, K.; Huang, R.; Bi, X.; Ming, C.; Li, L.; Li, X.; Li, G.; Zhao, L.; et al. Genetic Screening of a Chinese Cohort of Children with Hearing Loss Using a Next-Generation Sequencing Panel. Hum. Genom. 2023, 17, 1. [Google Scholar] [CrossRef]
- Vaché, C.; Besnard, T.; Blanchet, C.; Baux, D.; Larrieu, L.; Faugère, V.; Mondain, M.; Hamel, C.; Malcolm, S.; Claustres, M.; et al. Nasal Epithelial Cells Are a Reliable Source to Study Splicing Variants in Usher Syndrome. Hum. Mutat. 2010, 31, 734–741. [Google Scholar] [CrossRef]
- Viala, A.; Nicot, T.; Levy, F.; Vacheron, M.N. A Case of Usher’s Syndrome Associated with Psychotic Symptoms: Diagnosis and Follow-up in a Psychiatric Unit. Encephale 2009, 35, 286–291. [Google Scholar] [CrossRef]
- Dammeyer, J. Children with Usher Syndrome: Mental and Behavioral Disorders. Behav. Brain Funct. 2012, 8, 16. [Google Scholar] [CrossRef]
- Müller, U. Cadherins and Mechanotransduction by Hair Cells. Curr. Opin. Cell Biol. 2008, 20, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Balan, S.; Ohnishi, T.; Watanabe, A.; Ohba, H.; Iwayama, Y.; Toyoshima, M.; Hara, T.; Hisano, Y.; Miyasaka, Y.; Toyota, T.; et al. Role of an Atypical Cadherin Gene, Cdh23 in Prepulse Inhibition, and Implication of CDH23 in Schizophrenia. Schizophr. Bull. 2021, 47, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, A.; Sanna, S.; Uda, M.; Deiana, B.; Usala, G.; Busonero, F.; Maschio, A.; Scally, M.; Patriciu, N.; Chen, W.M.; et al. Genome-Wide Association Scan for Five Major Dimensions of Personality. Mol. Psychiatry 2008, 15, 647–656. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Braff, D.L.; Geyer, M.A. Sensorimotor Gating of the Startle Reflex: What We Said 25 Years Ago, What Has Happened since Then, and What Comes Next. J. Psychopharmacol. 2016, 30, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Kumari, V.; Das, M.; Hodgins, S.; Zachariah, E.; Barkataki, I.; Howlett, M.; Sharma, T. Association between Violent Behaviour and Impaired Prepulse Inhibition of the Startle Response in Antisocial Personality Disorder and Schizophrenia. Behav. Brain Res. 2005, 158, 159–166. [Google Scholar] [CrossRef]
- Koizumi, J.; Ofuku, K.; Sakuma, K.; Shiraishi, H.; Saakio, M.; Nawano, S. CNS Changes in Usher’s Syndrome with Mental Disorder: CT, MRI and PET Findings. J. Neurol. Neurosurg. Psychiatry 1988, 51, 987–990. [Google Scholar] [CrossRef]
- Tamayo, M.L.; Maldonado, C.; Plaza, S.L.; Alvira, G.M.; Tamayo, G.E.; Zambrano, M.; Frias, J.L.; Bernai, J.E. Neuroradiology and Clinical Aspects of Usher Syndrome. Clin. Genet. 1996, 50, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, T.A.; Lazzeroni, L.C.; Maihofer, A.X.; Swerdlow, N.R.; Calkins, M.E.; Freedman, R.; Green, M.F.; Light, G.A.; Nievergelt, C.M.; Nuechterlein, K.H.; et al. Genome-Wide Association of Endophenotypes for Schizophrenia From the Consortium on the Genetics of Schizophrenia (COGS) Study. JAMA Psychiatry 2019, 76, 1274–1284. [Google Scholar] [CrossRef]
- Han, M.R.; Han, K.M.; Kim, A.; Kang, W.; Kang, Y.; Kang, J.; Won, E.; Tae, W.S.; Cho, Y.; Ham, B.J. Whole-Exome Sequencing Identifies Variants Associated with Structural MRI Markers in Patients with Bipolar Disorders. J. Affect. Disord. 2019, 249, 159–168. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tesolin, P.; Santin, A.; Morgan, A.; Lenarduzzi, S.; Rubinato, E.; Girotto, G.; Spedicati, B. Which Came First? When Usher Syndrome Type 1 Couples with Neuropsychiatric Disorders. Audiol. Res. 2023, 13, 989-995. https://doi.org/10.3390/audiolres13060086
Tesolin P, Santin A, Morgan A, Lenarduzzi S, Rubinato E, Girotto G, Spedicati B. Which Came First? When Usher Syndrome Type 1 Couples with Neuropsychiatric Disorders. Audiology Research. 2023; 13(6):989-995. https://doi.org/10.3390/audiolres13060086
Chicago/Turabian StyleTesolin, Paola, Aurora Santin, Anna Morgan, Stefania Lenarduzzi, Elisa Rubinato, Giorgia Girotto, and Beatrice Spedicati. 2023. "Which Came First? When Usher Syndrome Type 1 Couples with Neuropsychiatric Disorders" Audiology Research 13, no. 6: 989-995. https://doi.org/10.3390/audiolres13060086
APA StyleTesolin, P., Santin, A., Morgan, A., Lenarduzzi, S., Rubinato, E., Girotto, G., & Spedicati, B. (2023). Which Came First? When Usher Syndrome Type 1 Couples with Neuropsychiatric Disorders. Audiology Research, 13(6), 989-995. https://doi.org/10.3390/audiolres13060086