

Waardenburg Syndrome: The Contribution of Next-Generation Sequencing to the Identification of Novel Causative Variants

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
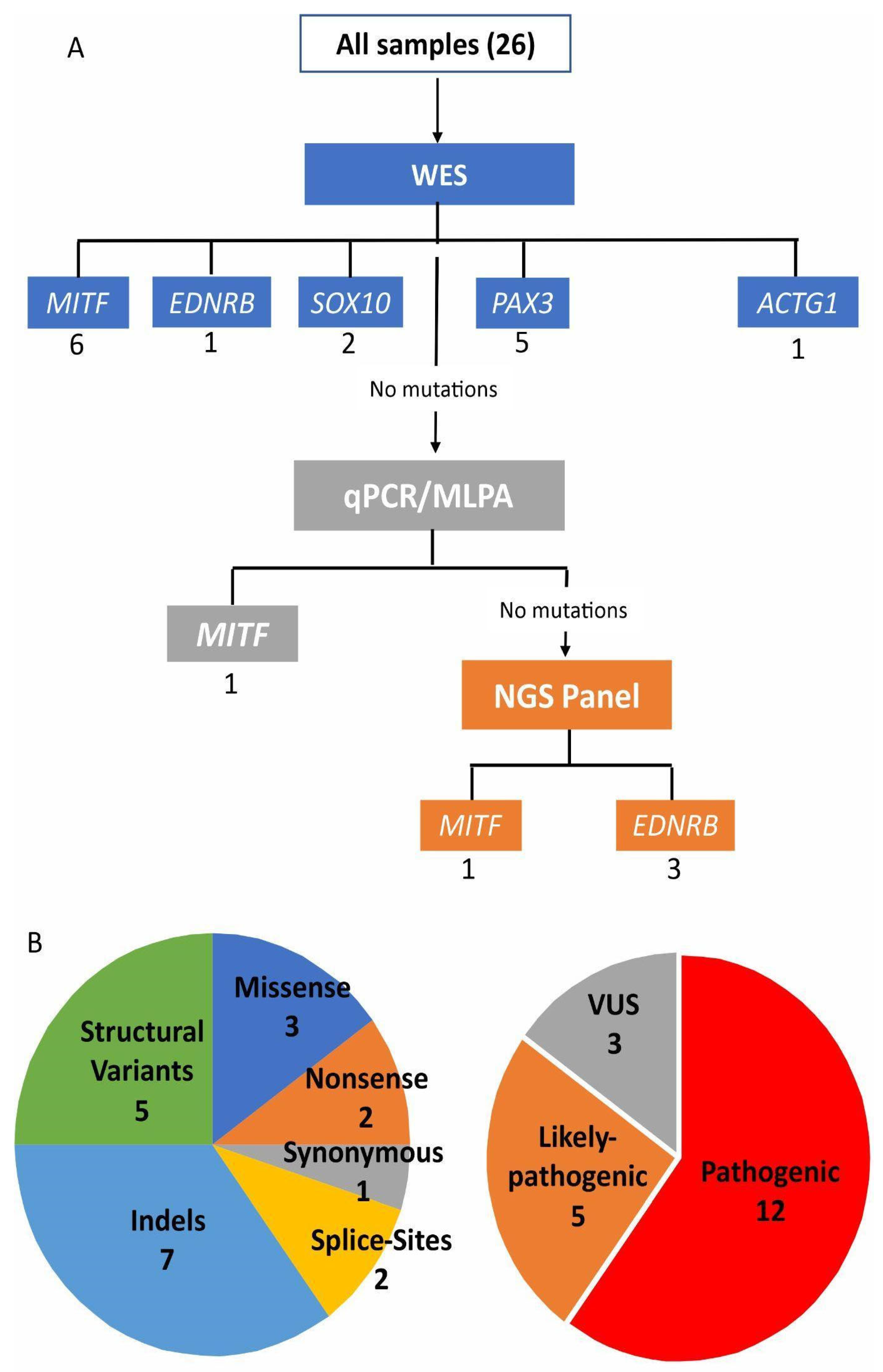
3. Results

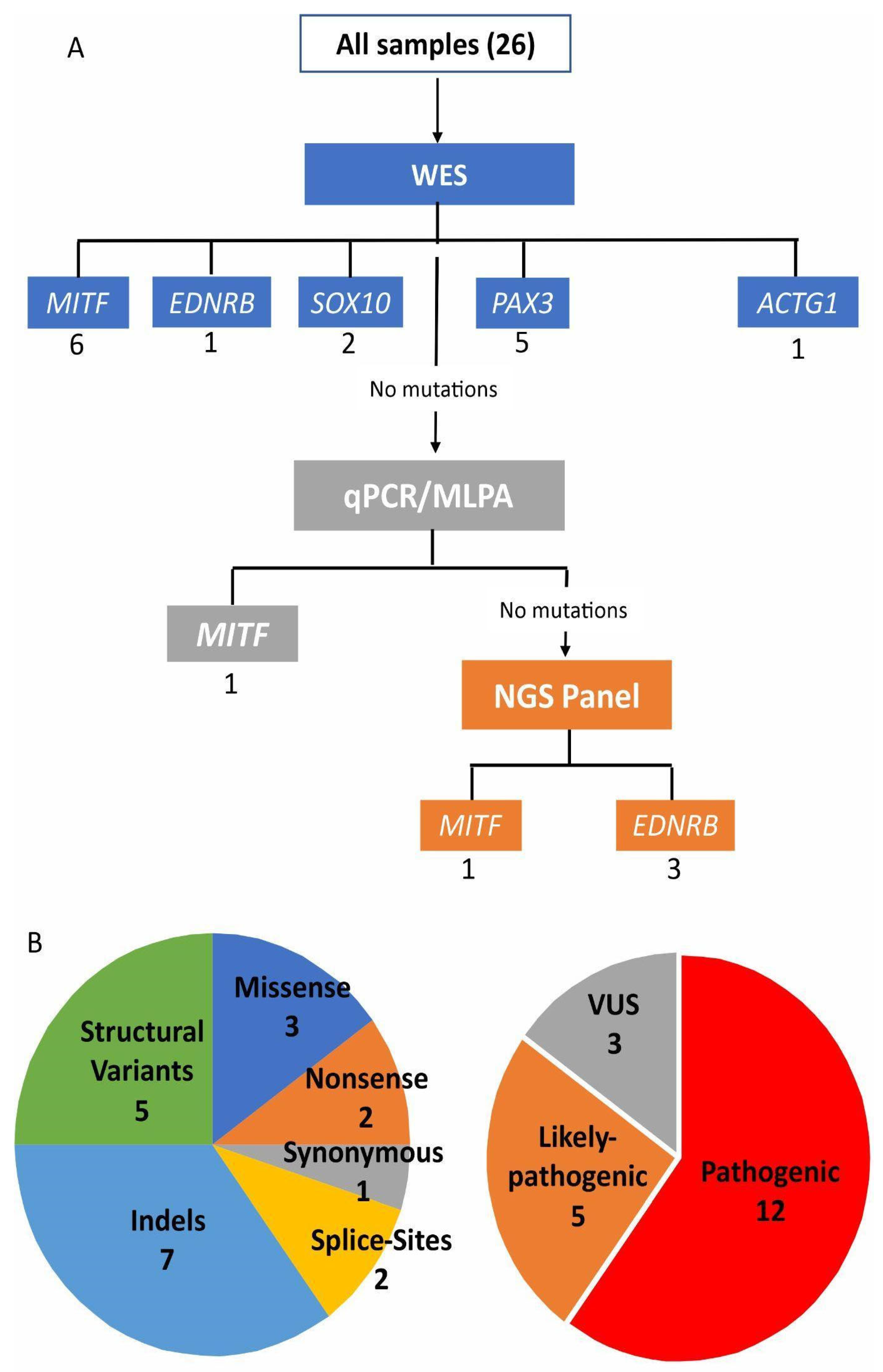{kind=link}
{kind=link}
{kind=link}
| Index Cases | Gene | Clinical Phenotype | Variant | Prediction Protein | dbSNP | Technique $ | ClinVar | Deafness Variation Database | Mutation Taster | ACMG | Inheritance | Segregation Analysis | Previous Description of Variants |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LGH16 | MITF—NM_000248.4 | WS2 | c.33+5G>A | - | - | WES | ND | ND | NA | Uncertain significance (PM2, BP4, PP1) | Familial | Segregates in the family | ND |
| LGH3 | WS2 | c.258del | p.Glu87Argfs*19 | rs1576005420 | WES | Likely pathogenic | ND | Disease causing | Pathogenic (PVS1, PP5, PM2) | Sporadic | Inherited from unaffected mother | ND | |
| LGH18 | WS2 | c.607_608delAG | p.Arg203Alafs*10 | - | WES | ND | ND | Disease causing | Likely pathogenic (PVS1, PM2, PP1) | Familial | Segregates in the family | ND | |
| LGH26 | WS2 | c.610C>T | p.Gln204* | rs1559745185 | WES | Likely pathogenic | Likely pathogenic | Disease causing | Pathogenic (PVS1, PP5, PP3 PM2, PP1) | Familial | Segregates in the family | ND | |
| LGH14 | WS2 | Exon 5 and 6 deletion | - | - | qPCR/MLPA | ND | ND | NA | Pathogenic (PVS1) | Isolated | de novo | Same patient described in [22] as W14 | |
| LGH15 | WS2 | c.763C>T | p.Arg255* | rs1057517966 | WES | Pathogenic/Likely pathogenic | Pathogenic | Disease causing | Pathogenic (PVS1, PM2, PP3, PP5, PP1) | Familial | Segregates in the family | [38]—Patient P44 | |
| LGH12 | WS1 > WS2 | c.909G>A | p.Thr303= | rs1057521096 | WES | Pathogenic/Likely pathogenic | Pathogenic | Disease causing | Pathogenic (PP5, PM2, BP4, PS3) | Sporadic | NA | [37] | |
| LGH25 | WS2 | Exon 8 deletion | - | - | NGS panel | ND | ND | NA | Pathogenic (PVS1) | Familial | Segregates in the family | ND | |
| LGH5 | SOX10—NM_006941 | WS2 | c.12_13delinsAT | p.Gln5* | - | WES | ND | ND | Disease causing | Pathogenic (PVS1, PM2, PP3) | Sporadic | NA | [22]—Patient W6 |
| LGH10 | WS2 | c.271_275dup | p.Arg93Profs*18 | - | WES | ND | ND | Disease causing | Likely pathogenic (PVS1, PM2) | Sporadic | NA | ND | |
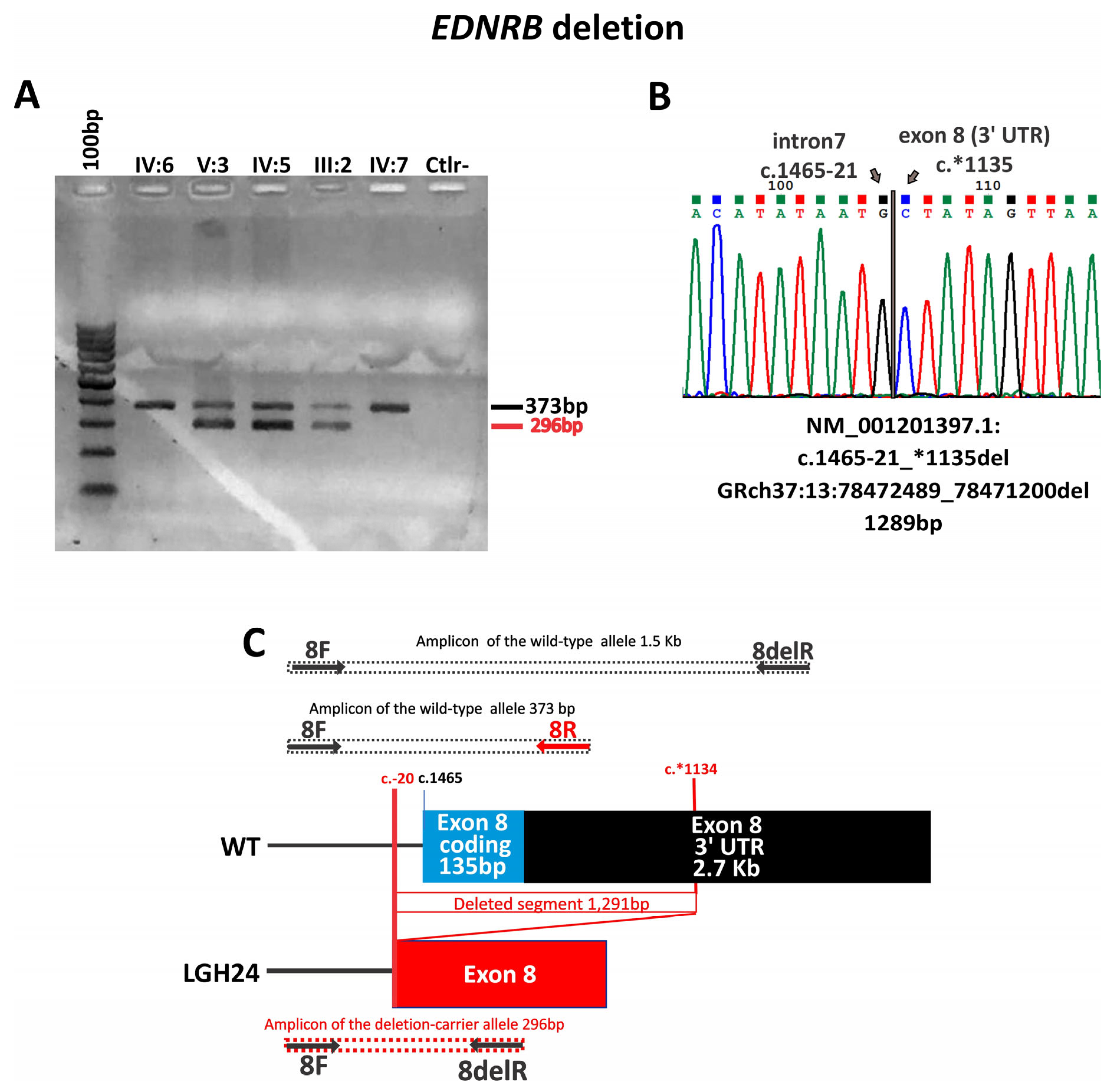
| LGH9 | EDNRB—NM_000115 | WS2 | Whole gene deletion | - | - | WES | NA | NA | NA | Pathogenic (PVS1_Stand-alone) | Sporadic | NA | ND |
| LGH11 | WS1 > WS2 | c.484-1G>A | - | - | NGS panel | ND | ND | Disease causing | Likely pathogenic (PVS1, PM2) | Sporadic | Inherited from unaffected mother | ND | |
| LGH17 | WS2 | c.898A>G | p.Met300Val | - | NGS panel | ND | ND | Disease causing | VUS (PM1, PM2) | Familial | Inherited from unaffected father | ND | |
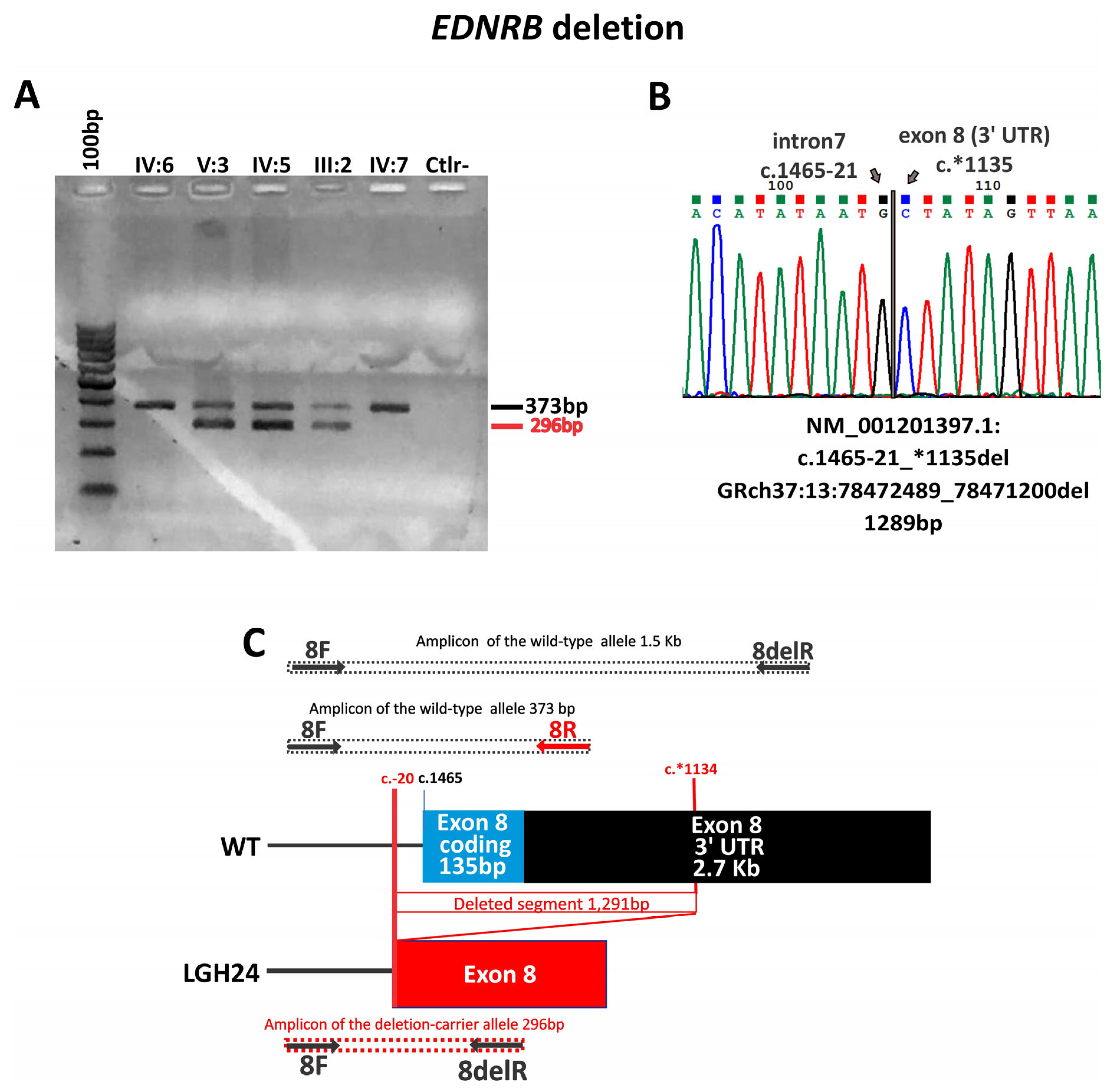
| LGH24 | WS2 | c.1465-21_*1135del Exon 8 deletion | - | - | NGS panel | ND | ND | NA | Pathogenic (PVS1, PP1) | Familial | Segregates in the family | ND | |
| LGH22 | PAX3—NM_181459 | WS1 | c.85_85+12delGGTAAGGGAGGGC | p.Val29Cysfs*81 | - | WES | ND | ND | Disease causing | Likely pathogenic (PVS1, PM2) | Familial | NA | ND |
| LGH13 | WS1 | c.115A>G | p.Asn39Asp | - | WES trio | ND | ND | Disease causing | Pathogenic (PP3, PM1, PM5, PM2, PS2) | Isolated | de novo | ND | |
| LGH21 | WS1 | c.896dup | p.Met299Ilefs*111 | - | WES | ND | ND | Disease causing | Pathogenic (PVS1, PM2, PP1) | Familial | Segregates in the family | ND | |
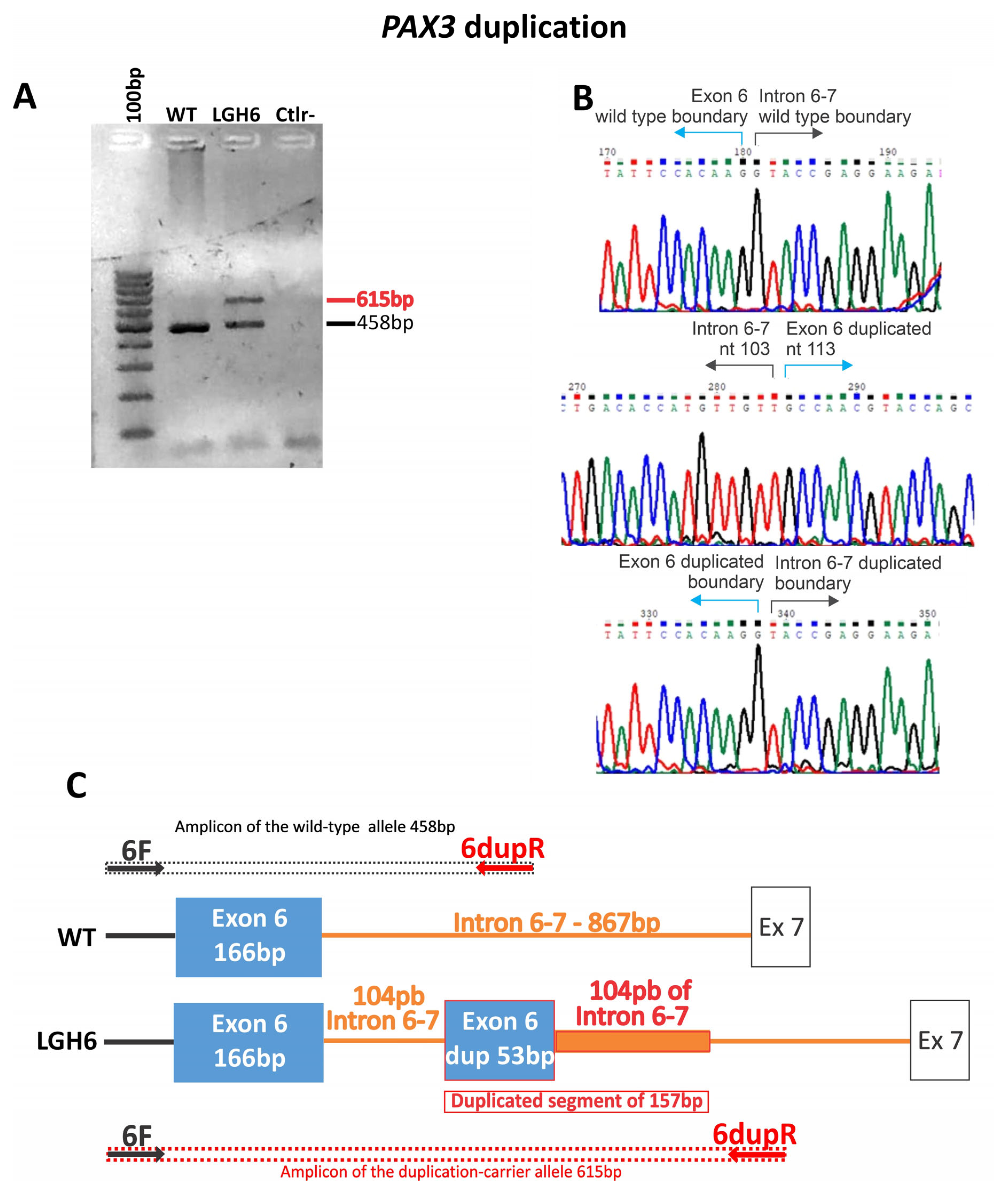
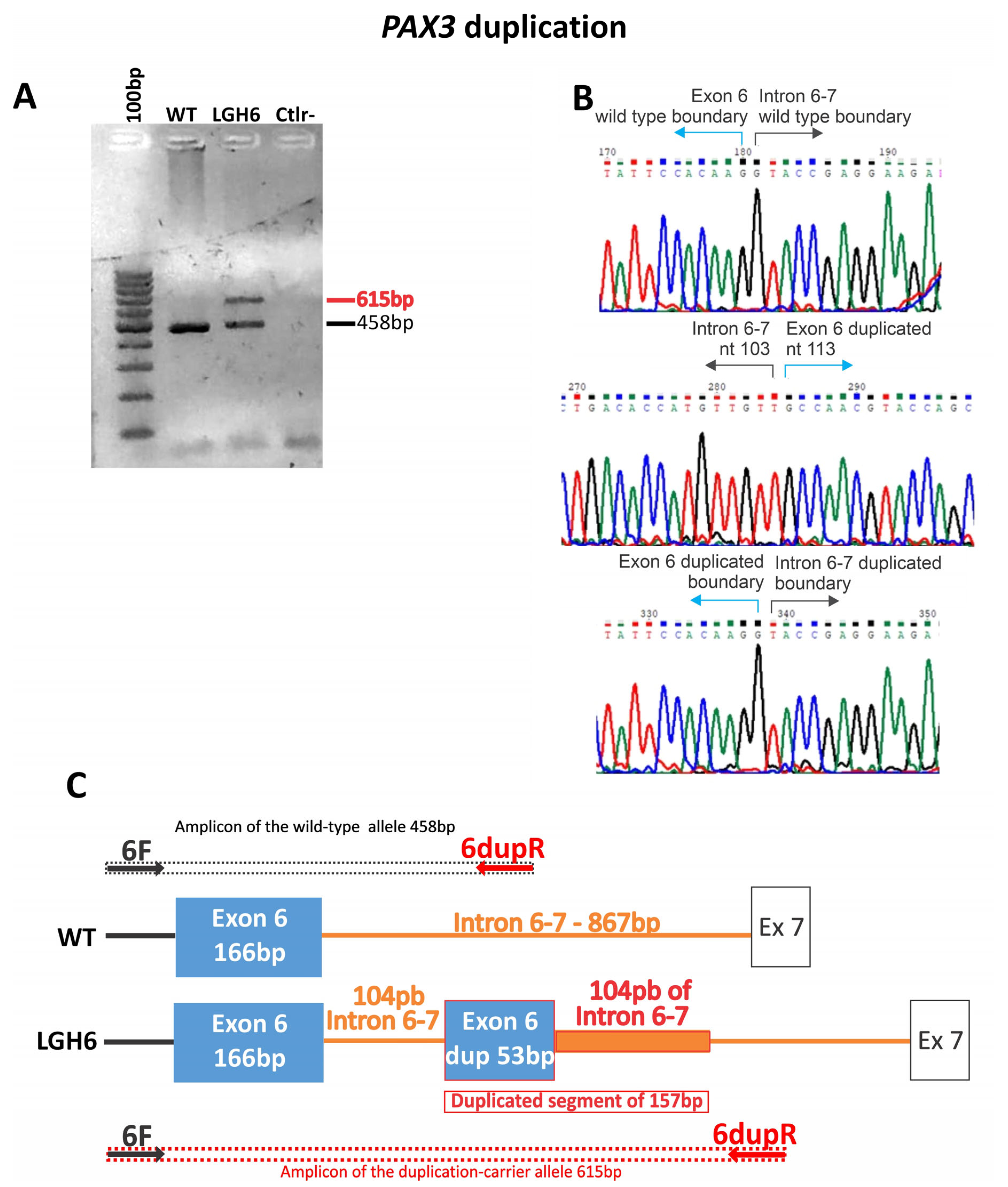
| LGH6 | WS1 | NC_000002.12 (NM_181459) c.958+104 (g.222221118_222221274dup) | ? | - | WES | ND | ND | NA | VUS (PM2, PM4, PP4) | Sporadic | NA | ND | |
| LGH23 | WS1 | c.1253del | p.Gly418Valfs*16 | rs778236891 | WES | ND | Unknown effect | Disease causing | Likely pathogenic (PVS1, PM2) | Familial | NA | ND | |
| LGH1 | ACTG1—NM_001614 | WS1 > BWS2 | c.277G>A | p.Glu93Lys | rs1568062529 | WES trio | Likely pathogenic | Likely pathogenic | Disease causing | Pathogenic (PS2, PM1, PM2, PP2, PP3, PP5) | Isolated | de novo | ND |
| Clinical Features | Additional Molecular Analysis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID | Dystopia Canthorum | Eye Pigmentation Abnormality | Hair Pigmentation Abnormality | Skin Pigmentation Abnormality | Hearing Impairment | WS Type | Proband | Mutation Segregation | NGS Panel | Exome Trio | MLPA (PAX3, MITF, SOX10) | Array-CGH | Comments |
| LGH1 | + | Blue eyes | − | − | + | 1 > BWS2 | Sporadic | de novo | − | + | + | − | |
| LGH2 | − | Bright blue iridis | − | − | + | 2 | Sporadic | Unsolved case | + | + | + | + | |
| LGH3 | − | Bright blue iridis | + | − | + | 2 | Sporadic | Inherited unaffected mother | − | − | + | − | |
| LGH4 | − | Heterochromia iridis | + | + | + | 2 | Sporadic | Unsolved case | + | + | + | − | |
| LGH5 | − | Heterochromia iridis | + | − | + | 2 | Sporadic | NA | − | − | + | − | |
| LGH6 | + | Bilateral heterochromia iridis | + | − | + | 1 | Sporadic | NA | − | − | + | + | Ala nasi hipoplasia |
| LGH7 | − | Bright blue iridis with brown spotting | + | − | + | 2 | Sporadic | Unsolved case | + | + | + | + | |
| LGH8 | − | − | + | − | + | 2 | Sporadic | Unsolved case | − | − | + | − | Moderate mixed (R) and conductive (L) HL and not included for the NGS panel |
| LGH9 | − | Heterochromia iridis and bright blue iridis | + | + | + | 2 | Sporadic | NA | − | − | + | + | Deletion suspicion by WES and confirmed with array-CGH |
| LGH10 | − | Bright blue iridis | − | − | + | 2 | Sporadic | NA | − | − | + | − | |
| LGH11 | Apparent | Heterochromia iridis and bright blue iridis | − | − | + | 1 > 2 | Sporadic | Inherited from unaffected mother | + | + | − | + | |
| LGH12 | Apparent | Heterochromia iridis and bright blue iridis | − | − | + | 1 > 2 | Sporadic | NA | − | − | − | − | |
| LGH13 | + | Heterochromia iridis | + | − | + | 1 | Sporadic | de novo | − | + | − | − | Ala nasi hipoplasia |
| LGH14 | − | Heterochromia iridis | − | + | + | 2 | Sporadic | de novo | − | + | + | − | Nasal root hyperplasia. Normal MRI, CT-scan. Patient W14 [22] |
| LGH15 | − | Bright blue iridis | − | − | + | 2 | Familial | + | − | − | + | − | |
| LGH16 | − | Heterochromia iridis | − | − | + | 2 | Familial | + | − | − | + | + | |
| LGH17 | − | Heterochromia iridis | − | − | + | 2 | Familial | Inherited from unaffected father | + | − | + | + | |
| LGH18 | − | Bright blue iridis | + | − | + | 2 | Familial | + | − | − | + | − | |
| LGH19 | − | − | + | + | + | 2 | Familial | Unsolved case | + | − | + | + | |
| LGH20 | − | Bright blue iridis | + | − | + | 2 | Familial | Unsolved case | + | − | + | + | |
| LGH21 | + | Bright blue iridis | + | − | + | 1 | Familial | + | − | − | − | − | Ala nasi hipoplasia |
| LGH22 | + | Heterochromia iridis and bright blue iridis | + | − | + | 1 | Familial | NA | − | − | + | − | Ala nasi hipoplasia |
| LGH23 | + | Heterochromia iridis and bright blue iridis | + | + | 1 | Familial | NA | − | − | + | − | Ala nasi hipoplasia | |
| LGH24 | − | Heterochromia iridis and bright blue iridis | − | − | + | 2 | Familial | + | + | − | − | + | Normal MRI and CT. Patient W12 [22] |
| LGH25 | − | Bright blue iridis | − | − | + | 2 | Familial | + | + | − | − | + | Normal MRI and CT. Patient W13 [22] |
| LGH26 | − | Heterochromia iridis and bright blue iridis | − | − | + | 2 | Familial | + | − | − | + | − | |
4. Discussion
4.1. PAX3 Variants
4.2. MITF Variants
4.3. SOX10 Variants
4.4. EDNRB Variants
4.5. Final Considerations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waardenburg, P.J. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am. J. Hum. Genet. 1951, 3, 195–253. [Google Scholar] [PubMed]
- Read, A.P.; Newton, V.E.E. Syndrome of the month. J. Med. Genet. 1997, 34, 744. [Google Scholar]
- Jones, M.C. The Neurocristopathies: Reinterpretation Based Upon the Mechanism of Abnormal Morphogenesis. Cleft Palate J. 1990, 27, 136–140. [Google Scholar] [PubMed]
- Farrer, L.A.; Grundfast, K.M.; Amos, J.; Arnos, K.S.; Asher, J.H.; Beighton, P.; Diehl, S.R.; Fex, J.; Foy, C.; Friedman, T.B. Waardenberg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: First report of the WS consortium. Am. J. Hum. Genet. 1992, 50, 902–913. [Google Scholar] [PubMed]
- Liu, X.Z.; Newton, V.E.; Read, A.P. Waardenburg syndrome type II: Phenotypic findings and diagnostic criteria. Am. J. Med. Genet. 1995, 55, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Pardono, E.; van Bever, Y.; van den Ende, J.; Havrenne, P.C.; Iughetti, P.; Maestrelli, S.R.P.; Costa, F.O.; Richieri-Costa, A.; Frota-Pessoa, O.; Otto, P.A. Waardenburg syndrome: Clinical differentiation between types I and II. Am. J. Med. Genet. Part A 2003, 117A, 223–235. [Google Scholar] [CrossRef]
- Shah, K.N.; Dalal, S.J.; Desai, M.P.; Sheth, P.N.; Joshi, N.C.; Ambani, L.M. White forelock, pigmentary disorder of irides, and long segment Hirschsprung disease: Possible variant of Waardenburg syndrome. J. Pediatr. 1981, 99, 432–435. [Google Scholar] [CrossRef]
- Baldwin, C.T.; Hoth, C.F.; Amos, J.A.; Elias, O.; Milunsky, A. An exonic mutation in the HuP2 paired domain gene causes Waardenburg’s syndrome. Nature 1992, 355, 637–638. [Google Scholar] [CrossRef]
- Hoth, C.F.; Milunsky, A.; Lipsky, N.; Sheffer, R.; Clarren, S.K.; Baldwin, C.T. Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I). Am. J. Hum. Genet. 1993, 52, 455–462. [Google Scholar]
- Pingault, V.; Ente, D.; Dastot-Le Moal, F.; Goossens, M.; Marlin, S.; Bondurand, N. Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 2010, 31, 391–406. [Google Scholar] [CrossRef]
- Wollnik, B.; Tukel, T.; Uyguner, O.; Ghanbari, A.; Kayserili, H.; Emiroglu, M.; Yuksel-Apak, M. Homozygous and heterozygous inheritance of PAX3 mutations causes different types of Waardenburg syndrome. Am. J. Med. Genet. Part A 2003, 122A, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Zlotogora, J.; Lerer, I.; Bar-David, S.; Ergaz, Z.; Abeliovich, D. Homozygosity for Waardenburg syndrome. Am. J. Hum. Genet. 1995, 56, 1173–1178. [Google Scholar] [PubMed]
- Issa, S.; Bondurand, N.; Faubert, E.; Poisson, S.; Lecerf, L.; Nitschke, P.; Deggouj, N.; Loundon, N.; Jonard, L.; David, A.; et al. EDNRB mutations cause Waardenburg syndrome type II in the heterozygous state. Hum. Mutat. 2017, 38, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Seco, C.Z.; de Castro, L.S.; van Nierop, J.W.; Morín, M.; Jhangiani, S.; Verver, E.J.J.; Schraders, M.; Maiwald, N.; Wesdorp, M.; Venselaar, H.; et al. Allelic mutations of KITLG, encoding KIT ligand, cause asymmetric and unilateral hearing loss and Waardenburg syndrome type 2. Am. J. Hum. Genet. 2015, 97, 647–660. [Google Scholar]
- Vona, B.; Schwartzbaum, D.; Rodriguez, A.; Lewis, S.; Toosi, M.; Radhakrishnan, P.; Bozan, N.; Akın, R.; Doosti, M.; Manju, R.; et al. Biallelic KITLG variants lead to a distinct spectrum of hypomelanosis and sensorineural hearing loss. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Bondurand, N.; Moal, F.D.-L.; Stanchina, L.; Collot, N.; Baral, V.; Marlin, S.; Attie-Bitach, T.; Giurgea, I.; Skopinski, L.; Reardon, W.; et al. Deletions at the SOX10 Gene Locus Cause Waardenburg Syndrome Types 2 and 4. Am. J. Hum. Genet. 2007, 81, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- Fernández, R.M.; Núñez-Ramos, R.; Enguix-Riego, M.A.V.; Román-Rodríguez, F.J.; Galán-Gómez, E.; Blesa-Sánchez, E.; Antiñolo, G.; Núñez-Núñez, R.; Borrego, S. Waardenburg Syndrome Type 4: Report of Two New Cases Caused by SOX10 Mutations in Spain. Am. J. Med. Genet. Part A 2014, 164, 542–547. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, Y.; Shen, N.; Peng, J.; Wang, C.; Liu, H.; Lu, Y. A de novo deletion mutation in SOX10 in a Chinese family with Waardenburg syndrome type 4. Sci. Rep. 2017, 7, 41513. [Google Scholar] [CrossRef]
- Puffenberger, E.G.; Hosoda, K.; Washington, S.S.; Nakao, K.; Dewit, D.; Yanagisawa, M.; Chakravarti, A. A missense mutation of the endothelin-B receptor gene in multigenic hirschsprung’s disease. Cell 1994, 79, 1257–1266. [Google Scholar] [CrossRef]
- Inoue, K.; Khajavi, M.; Ohyama, T.; Hirabayashi, S.-I.; Wilson, J.; Reggin, J.D.; Mancias, P.; Butler, I.J.; Wilkinson, M.F.; Wegner, M.; et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 2004, 36, 361–369. [Google Scholar] [CrossRef]
- Pingault, V.; Zerad, L.; Bertani-Torres, W.; Bondurand, N. SOX10: 20 years of phenotypic plurality and current understanding of its developmental function. J. Med. Genet. 2021, 59, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Batissoco, A.C.; Pedroso-Campos, V.; Pardono, E.; Sampaio-Silva, J.; Sonoda, C.Y.; Vieira-Silva, G.A.; Longati, E.U.d.S.d.O.; Mariano, D.; Hoshino, A.C.H.; Tsuji, R.K.; et al. Molecular and genetic characterization of a large Brazilian cohort presenting hearing loss. Hum. Genet. 2022, 141, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Bocángel, M.A.P.; Melo, U.S.; Alves, L.U.; Pardono, E.; Lourenço, N.C.V.; Marcolino, H.V.C.; Otto, P.A.; Mingroni-Netto, R.C. Waardenburg syndrome: Novel mutations in a large Brazilian sample. Eur. J. Med. Genet. 2018, 61, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Falah, N.; Posey, J.E.; Thorson, W.; Benke, P.; Tekin, M.; Tarshish, B.; Lupski, J.R.; Harel, T. 22q11.2q13 Duplication Including SOX10 causes Sex-reversal and Peripheral Demyelinating Neuropathy, Central Dysmyelinating Leukodystrophy, Waardenburg Syndrome and Hirschsprung Disease. Am. J. Med. Genet. Part A 2017, 173, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, A.; Okamura, K.; Tazawa, R.; Abe, Y.; Hayashi, M.; Izumi, S.; Tohyama, J.; Shimomura, Y.; Hozumi, Y.; Suzuki, T. Waardenburg syndrome type IIE in a Japanese patient caused by a novel non-frame-shift duplication mutation in the SOX10 gene. J. Dermatol. 2018, 45, e110–e111. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Mei, L.; Chen, H.; Cai, X.; Liu, Y.; Men, M.; Liu, X.Z.; Yan, D.; Ling, J.; Feng, Y. New Genotypes and Phenotypes in Patients with 3 Subtypes of Waardenburg Syndrome Identified by Diagnostic Next-Generation Sequencing. Neural Plast. 2019, 2019, 7143458. [Google Scholar] [CrossRef]
- Milunsky, J.; Maher, T.; Ito, M.; Milunsky, A. The Value of MLPA in Waardenburg Syndrome. Genet. Test. 2007, 11, 179–182. [Google Scholar] [CrossRef]
- Schwarzbraun, T.; Ofner, L.; Gillessen-Kaesbach, G.; Schaperdoth, B.; Preisegger, K.-H.; Windpassinger, C.; Wagner, K.; Petek, E.; Kroisel, P.M. A New 3p Interstitial Deletion Including the Entire MITF Gene Causes a Variation of Tietz/Waardenburg Type IIA Syndromes. Am. J. Med. Genet. Part A 2007, 624, 619–624. [Google Scholar] [CrossRef]
- Stevenson, R.E.; Vincent, V.; Spellicy, C.J.; Friez, M.J.; Chaubey, A. Biallelic deletions of the Waardenburg II syndrome gene, SOX10, cause a recognizable arthrogryposis syndrome. Am. J. Med. Genet. Part A 2018, 176, 1968–1971. [Google Scholar] [CrossRef]
- Wildhardt, G.; Zirn, B.; Graul-Neumann, L.M.; Wechtenbruch, J.; Suckfüll, M.; Buske, A.; Bohring, A.; Kubisch, C.; Vogt, S.; Strobl-Wildemann, G.; et al. Spectrum of novel mutations found in Waardenburg syndrome types 1 and 2: Implications for molecular genetic diagnostics. BMJ Open 2013, 3, e001917. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2009, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Teo, S.M.; Pawitan, Y.; Ku, C.S.; Chia, K.S.; Salim, A. Sequence analysis Statistical challenges associated with detecting copy number variations with next-generation sequencing. Bioinformatics 2012, 28, 2711–2718. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Naslavsky, M.S.; Yamamoto, G.L.; de Almeida, T.F.; Ezquina, S.A.M.; Sunaga, D.Y.; Pho, N.; Bozoklian, D.; Sandberg, T.O.M.; Brito, L.A.; Lazar, M.; et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Agathe, J.-M.d.S.; Filser, M.; Isidor, B.; Besnard, T.; Gueguen, P.; Perrin, A.; Van Goethem, C.; Verebi, C.; Masingue, M.; Rendu, J.; et al. SpliceAI-visual: A free online tool to improve SpliceAI splicing variant interpretation. Hum. Genom. 2023, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Brenner, L.; Burke, K.; LeDuc, C.A.; Guha, S.; Guo, J.; Chung, W.K. Novel Splice Mutation in Microthalmia-Associated Transcription Factor in Waardenburg Syndrome. Genet. Test. Mol. Biomark. 2011, 15, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiang, J.; Chen, L.; Luo, H.; Xu, X.; Li, N.; Cui, C.; Xu, J.; Song, N.; Peng, J.; et al. Molecular Diagnosis of Non-syndromic Hearing Loss patients using a stepwise approach. Sci. Rep. 2021, 11, 4036. [Google Scholar] [CrossRef]
- Sánchez-Martín, M.; Rodríguez-García, A.; Pérez-Losada, J.; Sagrera, A.; Read, A.P.; Sánchez-García, I. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum. Mol. Genet. 2002, 11, 3231–3236. [Google Scholar] [CrossRef]
- Mirhadi, S.; Spritz, R.A.; Moss, C. Does SNAI2 mutation cause human piebaldism and Waardenburg syndrome? Am. J. Med. Genet. Part A 2020, 182, 3074–3075. [Google Scholar] [CrossRef]
- Morimoto, N.; Mutai, H.; Namba, K.; Kaneko, H.; Kosaki, R.; Matsunaga, T. Homozygous EDNRB mutation in a patient with Waardenburg syndrome type 1. Auris Nasus Larynx 2018, 45, 222–226. [Google Scholar] [CrossRef]
- Suzuki, N.; Mutai, H.; Miya, F.; Tsunoda, T.; Terashima, H.; Morimoto, N. A case report of reversible generalized seizures in a patient with Waardenburg syndrome associated with a novel nonsense mutation in the penultimate exon of SOX10. BMC Pediatr. 2018, 18, 171. [Google Scholar] [CrossRef] [PubMed]
- Arias, S.; Mota, M. Apparent non-penetrance for dystopia in Waardenburg syndrome type I, with some hints on the diagnosis of dystopia canthorum. J. Genet. Hum. 1978, 26, 103–131. [Google Scholar] [PubMed]
- Minami, S.B.; Nara, K.; Mutai, H.; Morimoto, N.; Sakamoto, H.; Takiguchi, T.; Kaga, K.; Matsunaga, T. A clinical and genetic study of 16 Japanese families with Waardenburg syndrome. Gene 2019, 704, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Gao, X.; Su, Y.; Han, M.; Gao, B.; Guo, C.; Kang, D.; Huang, S.; Yuan, Y.; et al. Analysis of genotype–phenotype relationships in 90 Chinese probands with Waardenburg syndrome. Hum. Genet. 2021, 141, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Baraitser, M.; Winter, R.M. Iris coloboma, ptosis, hypertelorism, and mental retardation: A new syndrome. J. Med. Genet. 1988, 25, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Verloes, A.; Di Donato, N.; Masliah-Planchon, J.; Jongmans, M.; Abdul-Raman, O.A.; Albrecht, B.; Allanson, J.; Brunner, H.; Bertola, D.; Chassaing, N.; et al. Baraitser–Winter cerebrofrontofacial syndrome: Delineation of the spectrum in 42 cases. Eur. J. Hum. Genet. 2015, 23, 292–301. [Google Scholar] [CrossRef]
- Lautenschlager, N.T.; Milunsky, A.; DeStefano, A.; Farrer, L.; Baldwin, C.T. A novel mutation in the MITF gene causes Waardenburg Syndrome. Genet. Anal. Biomol. Eng. 1996, 13, 43–44. [Google Scholar] [CrossRef]
- Carlson, R.J.; Walsh, T.; Mandell, J.B.; Aburayyan, A.; Lee, M.K.; Gulsuner, S.; Horn, D.L.; Ou, H.C.; Sie, K.C.Y.; Mancl, L.; et al. Association of Genetic Diagnoses for Childhood-Onset Hearing Loss with Cochlear Implant Outcomes. JAMA Otolaryngol. Head Neck Surg. 2023, 149, 212–222. [Google Scholar] [CrossRef]
- Haddad, N.; Ente, D.; Chouery, E.; Jalkh, N.; Mehawej, C.; Khoueir, Z.; Pingault, V.; Mégarbané, A. Molecular Study of Three Lebanese and Syrian Patients with Waardenburg Syndrome and Report of Novel Mutations in the EDNRB and MITF Genes. Mol. Syndr. 2010, 1, 169–175. [Google Scholar] [CrossRef]
- Rauschendorf, M.-A.; Zimmer, A.D.; Laut, A.; Demmer, P.; Rösler, B.; Happle, R.; Sartori, S.; Fischer, J. Homozygous intronic MITF mutation causes severe Waardenburg syndrome type 2A. Pigment. Cell Melanoma Res. 2019, 32, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Pingault, V.; Bodereau, V.; Baral, V.; Marcos, S.; Watanabe, Y.; Chaoui, A.; Fouveaut, C.; Leroy, C.; Vérier-Mine, O.; Francannet, C.; et al. Loss-of-Function Mutations in SOX10 Cause Kallmann Syndrome with Deafness. Am. J. Hum. Genet. 2013, 92, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Pingault, V.; Bondurand, N.; Lemort, N.; Sancandi, M.; Ceccherini, I.; Hugot, J.P.; Jouk, P.S.; Goossens, M. A heterozygous endothelin 3 mutation in Waardenburg-Hirschsprung disease: Is there a dosage e V ect of EDN3/EDNRB gene mutations on neurocristopathy phenotypes? J. Med. Genet. 2001, 38, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Pingault, V.; Girard, M.; Bondurand, N.; Dorkins, H.; Van Maldergem, L.; Mowat, D.; Shimotake, T.; Verma, I.; Baumann, C.; Goossens, M. SOX10 mutations in chronic intestinal pseudo-obstruction suggest a complex physiopathological mechanism. Hum. Genet. 2002, 111, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Syrris, P.; Carter, N.D.; Patton, M.A. Novel nonsense mutation of the endothelin-B receptor gene in a family with Waardenburg-Hirschsprung disease. Am. J. Med. Genet. 1999, 87, 69–71. [Google Scholar] [CrossRef]
- Abe, Y.; Sakurai, T.; Yamada, T.; Nakamura, T.; Yanagisawa, M.; Goto, K. Functional Analysis of Five Endothelin-B Receptor Mutations Found in Human Hirschsprung Disease Patients. Biochem. Biophys. Res. Commun. 2000, 531, 524–531. [Google Scholar] [CrossRef]
- Fuchs, S.; Amiel, J.; Claudel, S.; Lyonnet, S.; Corvol, P.; Pinet, F. Functional Characterization of Three Mutations of the Endothelin B Receptor Gene in Patients With Hirschsprung’s Disease: Evidence for Selective Loss of G i Coupling. Mol. Med. 2001, 7, 115–124. [Google Scholar] [CrossRef]
- Tanaka, H.; Moroi, K.; Iwai, J.; Takahashi, H.; Ohnuma, N.; Hori, S.; Takimoto, M.; Nishiyama, M.; Masaki, T.; Yanagisawa, M.; et al. Novel Mutations of the Endothelin B Receptor Gene in Patients with Hirschsprung’s Disease and Their Characterization. J. Biol. Chem. 1998, 273, 11378–11383. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertani-Torres, W.; Lezirovitz, K.; Alencar-Coutinho, D.; Pardono, E.; da Costa, S.S.; Antunes, L.d.N.; de Oliveira, J.; Otto, P.A.; Pingault, V.; Mingroni-Netto, R.C. Waardenburg Syndrome: The Contribution of Next-Generation Sequencing to the Identification of Novel Causative Variants. Audiol. Res. 2024, 14, 9-25. https://doi.org/10.3390/audiolres14010002
Bertani-Torres W, Lezirovitz K, Alencar-Coutinho D, Pardono E, da Costa SS, Antunes LdN, de Oliveira J, Otto PA, Pingault V, Mingroni-Netto RC. Waardenburg Syndrome: The Contribution of Next-Generation Sequencing to the Identification of Novel Causative Variants. Audiology Research. 2024; 14(1):9-25. https://doi.org/10.3390/audiolres14010002
Chicago/Turabian StyleBertani-Torres, William, Karina Lezirovitz, Danillo Alencar-Coutinho, Eliete Pardono, Silvia Souza da Costa, Larissa do Nascimento Antunes, Judite de Oliveira, Paulo Alberto Otto, Véronique Pingault, and Regina Célia Mingroni-Netto. 2024. "Waardenburg Syndrome: The Contribution of Next-Generation Sequencing to the Identification of Novel Causative Variants" Audiology Research 14, no. 1: 9-25. https://doi.org/10.3390/audiolres14010002
APA StyleBertani-Torres, W., Lezirovitz, K., Alencar-Coutinho, D., Pardono, E., da Costa, S. S., Antunes, L. d. N., de Oliveira, J., Otto, P. A., Pingault, V., & Mingroni-Netto, R. C. (2024). Waardenburg Syndrome: The Contribution of Next-Generation Sequencing to the Identification of Novel Causative Variants. Audiology Research, 14(1), 9-25. https://doi.org/10.3390/audiolres14010002