
A Rare Case of Perrault Syndrome with Auditory Neuropathy Spectrum Disorder: Cochlear Implantation Treatment and Literature Review


 ,
,  ,
,
Abstract
:1. Introduction
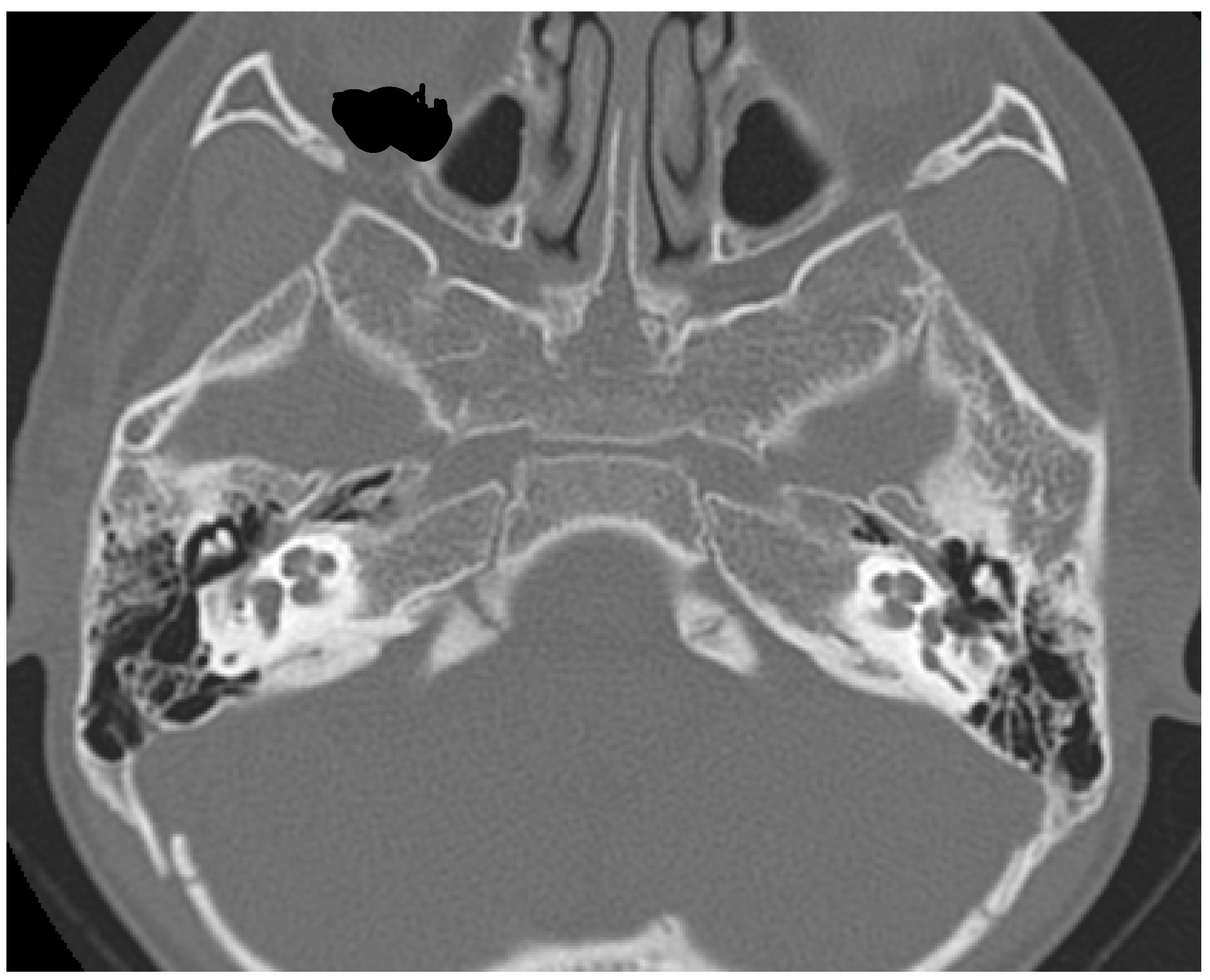
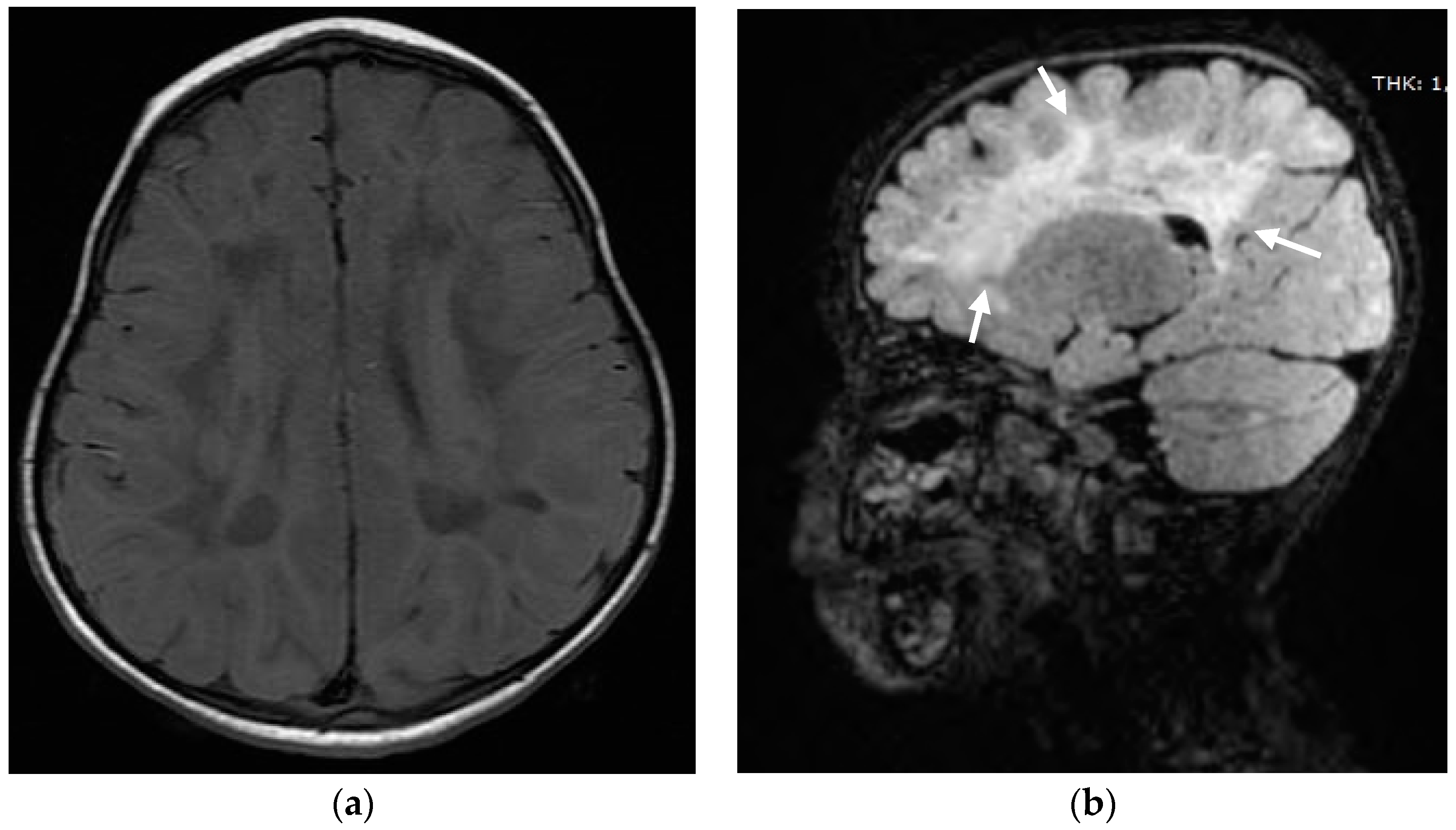
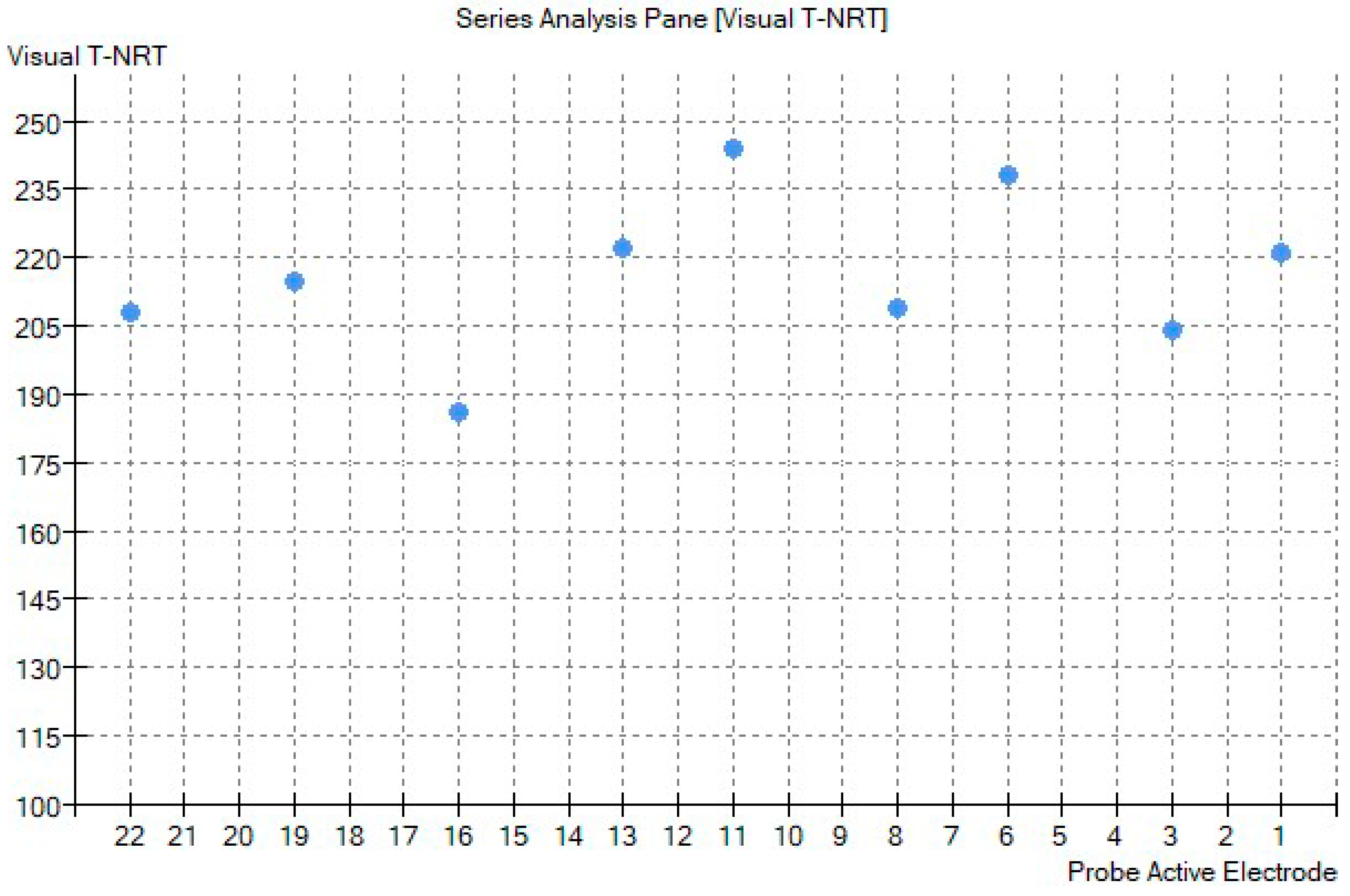
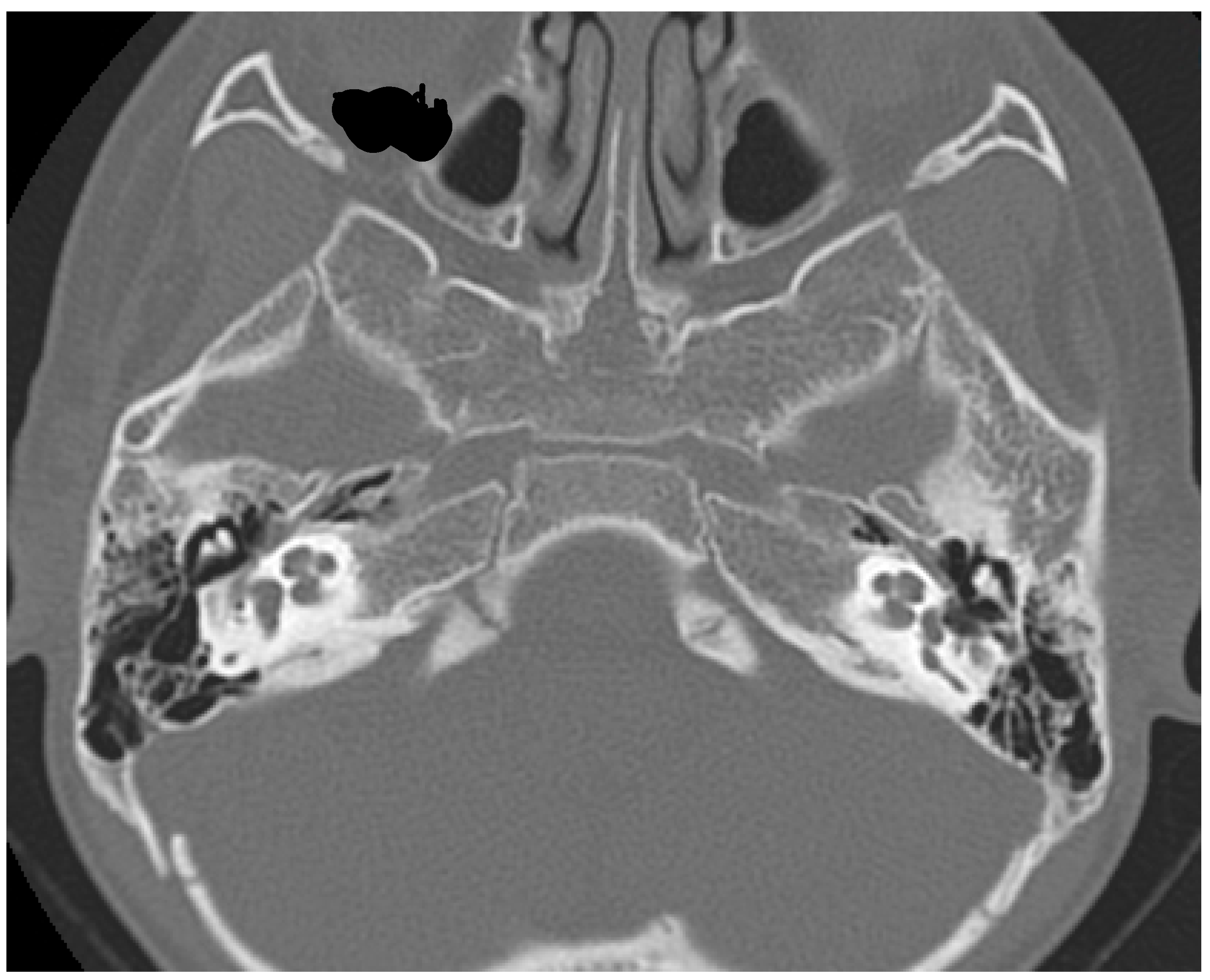
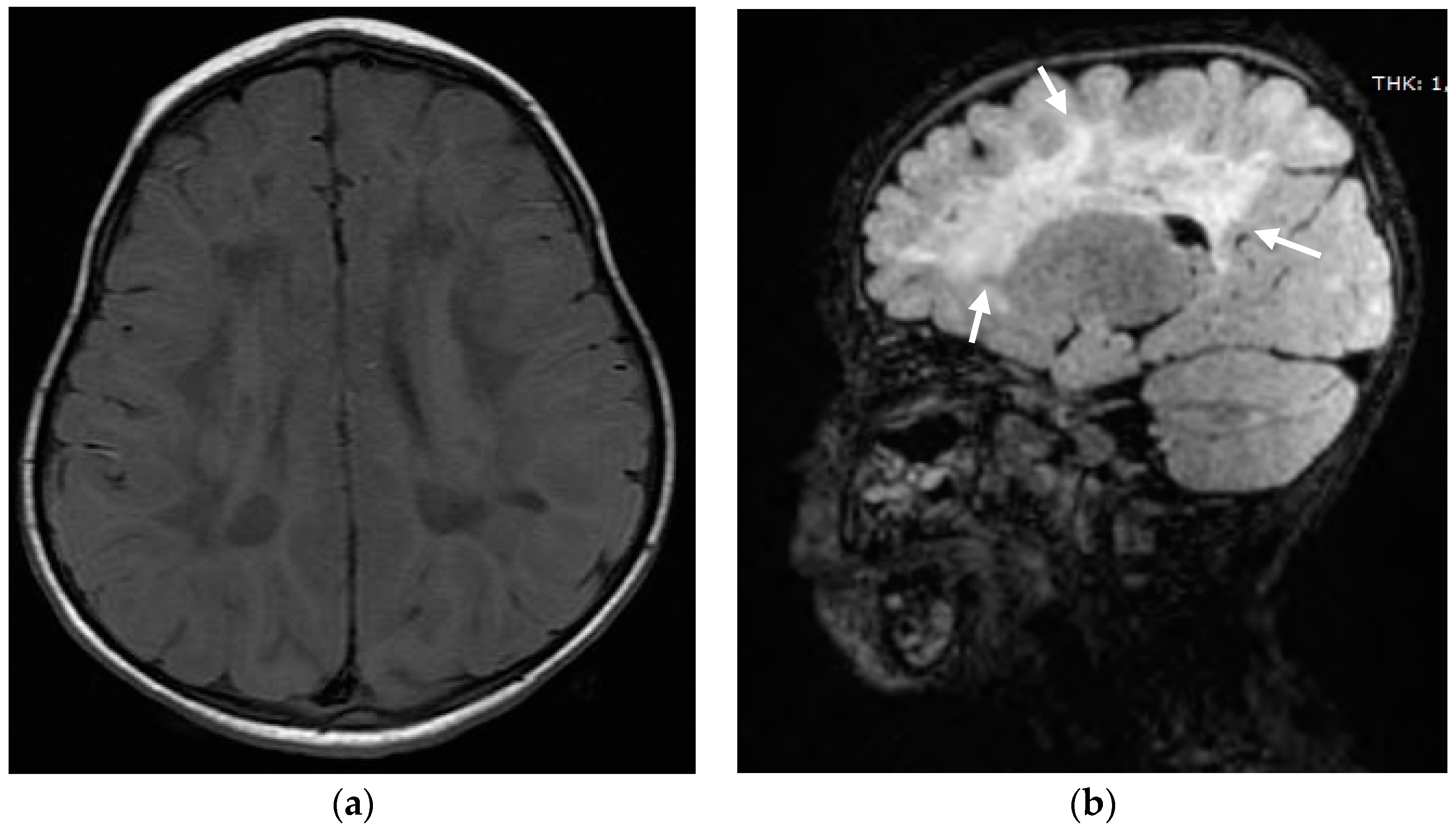
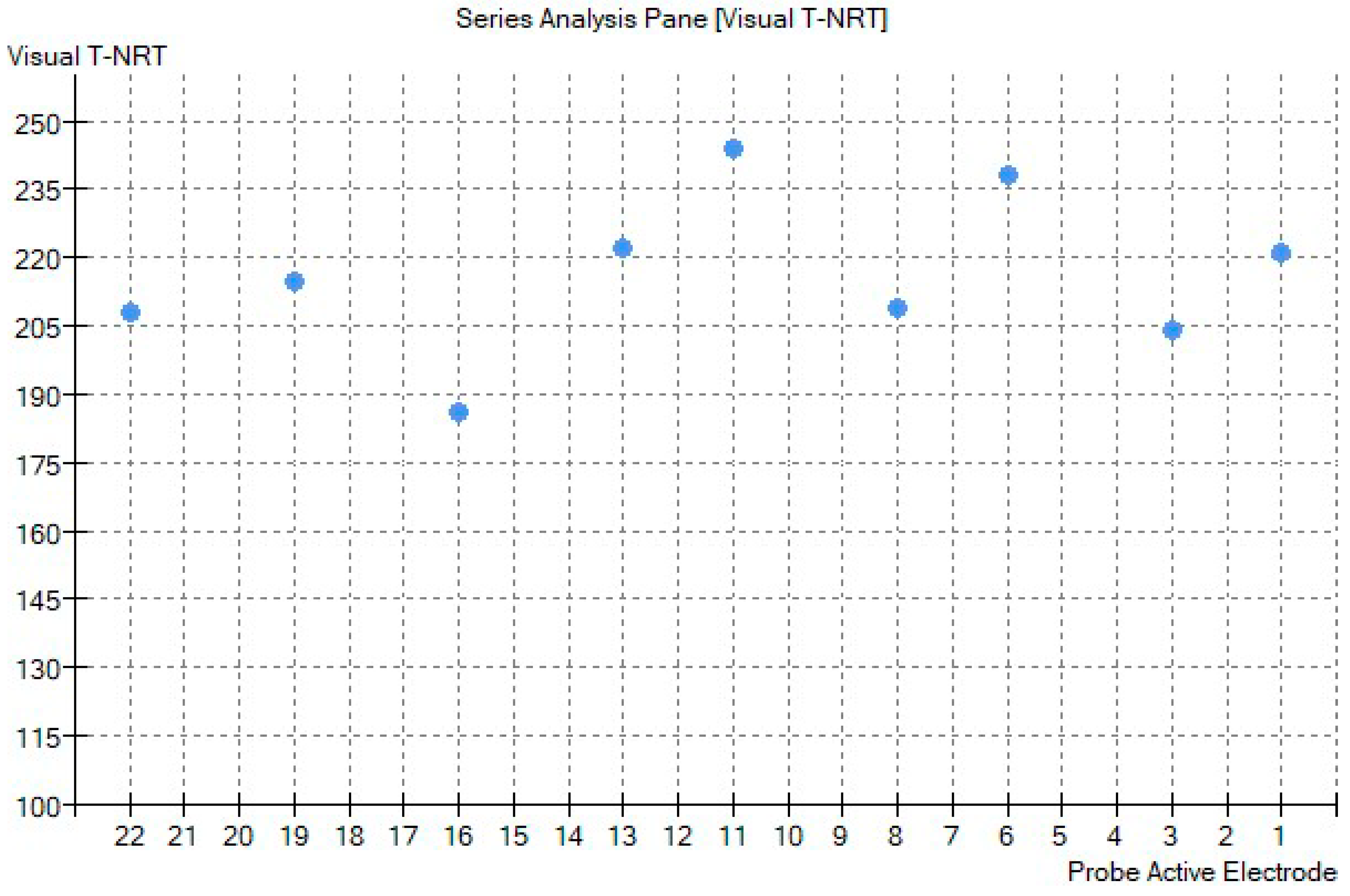
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Disclosure Statement
References
- Marlin, S.; Lacombe, D.; Jonard, L.; Leboulanger, N.; Bonneau, D.; Goizet, C.; de Villemeur, T.B.; Cabrol, S.; Houang, M.; Moatti, L.; et al. Perrault syndrome: Report of four new cases, review and exclusion of candidate genes. Am. J. Med. Genet. Part A 2008, 146A, 661–664. [Google Scholar] [CrossRef]
- Jenkinson, E.M.; Rehman, A.U.; Walsh, T.; Clayton-Smith, J.; Lee, K.; Morell, R.J.; Drummond, M.C.; Khan, S.N.; Naeem, M.A.; Rauf, B.; et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013, 92, 605–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiumara, A.; Sorge, G.; Toscano, A.; Parano, E.; Pavone, L.; Opitz, J.M. Perrault syndrome: Evidence for progressive nervous system involvement. Am. J. Med. Genet. Part A 2004, 128A, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Kobe, C.; Kracht, L.W.; Timmermann, L.; Bachmann, J.; Schmidt, M.C. Perrault Syndrome with progressive nervous system involvement. Clin. Nucl. Med. 2008, 33, 922–924. [Google Scholar] [CrossRef]
- Pan, Z.; Xu, H.; Tian, Y.; Liu, D.; Liu, H.; Li, R.; Dou, Q.; Zuo, B.; Zhai, R.; Tang, W.; et al. Perrault syndrome: Clinical report and retrospective analysis. Mol. Genet. Genom. Med. 2020, 8, e1445. [Google Scholar] [CrossRef]
- Roberts, L.M.; Carnivale, B. Perrault Syndrome Diagnosis in a Patient Presenting to Her Primary Care Provider with Secondary Amenorrhea. Case Rep. Obstet. Gynecol. 2019, 2019, 9865281. [Google Scholar] [CrossRef] [PubMed]
- Oldak, M.; Ozieblo, D.; Pollak, A.; Stepniak, I.; Lazniewski, M.; Lechowicz, U.; Kochanek, K.; Furmanek, M.; Tacikowska, G.; Plewczynski, D.; et al. Novel neuro-audiological findings and further evidence for TWNK involvement in Perrault syndrome. J. Transl. Med. 2017, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Demain, L.A.M.; Gerkes, E.H.; Smith, R.J.H.; Molina-Ramirez, L.P.; O’Keefe, R.T.; Newman, W.G. A recurrent missense variant in HARS2 results in variable sensorineural hearing loss in three unrelated families. J. Hum. Genet. 2020, 65, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agha, R.A.; Franchi, T.; Sohrabi, C.; Mathew, G.; Kerwan, A.; Group, S. The SCARE 2020 Guideline: Updating Consensus Surgical CAse REport (SCARE) Guidelines. Int. J. Surg. 2020, 84, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Forli, F.; Lazzerini, F.; Canelli, R.; Lorenzoni, F.; Franciosi, B.; Berrettini, S.; Bruschini, L. Extended-hearing targeted screening for congenital cytomegalovirus infection. Minerva Pediatrics 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Berrettini, S.; Ghirri, P.; Lazzerini, F.; Lenzi, G.; Forli, F. Newborn hearing screening protocol in tuscany region. Ital. J. Pediatrics 2017, 43, 82. [Google Scholar] [CrossRef] [Green Version]
- Freni, F.; Gazia, F.; Slavutsky, V.; Scherdel, E.P.; Nicenboim, L.; Posada, R.; Portelli, D.; Galletti, B.; Galletti, F. Cochlear Implant Surgery: Endomeatal Approach versus Posterior Tympanotomy. Int. J. Environ. Res. Public Health 2020, 17. [Google Scholar] [CrossRef] [PubMed]
- Dursun, F.; Mohamoud, H.S.; Karim, N.; Naeem, M.; Jelani, M.; Kirmizibekmez, H. A Novel Missense Mutation in the CLPP Gene Causing Perrault Syndrome Type 3 in a Turkish Family. J. Clin. Res. Pediatric Endocrinol. 2016, 8, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Jelani, M.; Alrayes, N.; Mohamoud, H.S.; Almramhi, M.M.; Anshasi, W.; Ahmed, N.A.; Wang, J.; Nasir, J.; Al-Aama, J.Y. Exome analysis identified a novel missense mutation in the CLPP gene in a consanguineous Saudi family expanding the clinical spectrum of Perrault Syndrome type-3. J. Neurol. Sci. 2015, 353, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Brodie, E.J.; Zhan, H.; Saiyed, T.; Truscott, K.N.; Dougan, D.A. Perrault syndrome type 3 caused by diverse molecular defects in CLPP. Sci. Rep. 2018, 8, 12862. [Google Scholar] [CrossRef] [Green Version]
- Theunissen, T.E.J.; Szklarczyk, R.; Gerards, M.; Hellebrekers, D.M.E.I.; Mulder-Den Hartog, E.N.M.; Vanoevelen, J.; Kamps, R.; de Koning, B.; Rutledge, S.L.; Schmitt-Mechelke, T.; et al. Specific MRI Abnormalities Reveal Severe Perrault Syndrome due to CLPP Defects. Front. Neurol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Corydon, T.J.; Wilsbech, M.; Jespersgaard, C.; Andresen, B.S.; Borglum, A.D.; Pedersen, S.; Bolund, L.; Gregersen, N.; Bross, P. Human and mouse mitochondrial orthologs of bacterial ClpX. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2000, 11, 899–905. [Google Scholar] [CrossRef]
- Dominguez-Ruiz, M.; Garcia-Martinez, A.; Corral-Juan, M.; Perez-Alvarez, A.I.; Plasencia, A.M.; Villamar, M.; Moreno-Pelayo, M.A.; Matilla-Duenas, A.; Menendez-Gonzalez, M.; Del Castillo, I. Perrault syndrome with neurological features in a compound heterozygote for two TWNK mutations: Overlap of TWNK-related recessive disorders. J. Transl. Med. 2019, 17, 290. [Google Scholar] [CrossRef] [PubMed]
- Karstensen, H.G.; Rendtorff, N.D.; Hindbaek, L.S.; Colombo, R.; Stein, A.; Birkebaek, N.H.; Hartmann-Petersen, R.; Lindorff-Larsen, K.; Hojland, A.T.; Petersen, M.B.; et al. Novel HARS2 missense variants identified in individuals with sensorineural hearing impairment and Perrault syndrome. Eur. J. Med. Genet. 2020, 63, 103733. [Google Scholar] [CrossRef]
- Demain, L.A.; Urquhart, J.E.; O’Sullivan, J.; Williams, S.G.; Bhaskar, S.S.; Jenkinson, E.M.; Lourenco, C.M.; Heiberg, A.; Pearce, S.H.; Shalev, S.A.; et al. Expanding the genotypic spectrum of Perrault syndrome. Clin. Genet. 2017, 91, 302–312. [Google Scholar] [CrossRef]
- Lerat, J.; Jonard, L.; Loundon, N.; Christin-Maitre, S.; Lacombe, D.; Goizet, C.; Rouzier, C.; Van Maldergem, L.; Gherbi, S.; Garabedian, E.N.; et al. An Application of NGS for Molecular Investigations in Perrault Syndrome: Study of 14 Families and Review of the Literature. Hum. Mutat. 2016, 37, 1354–1362. [Google Scholar] [CrossRef]
- Carminho-Rodrigues, M.T.; Klee, P.; Laurent, S.; Guipponi, M.; Abramowicz, M.; Cao-van, H.; Guinand, N.; Paoloni-Giacobino, A. LARS2-Perrault syndrome: A new case report and literature review. BMC Med. Genet. 2020, 21, 109. [Google Scholar] [CrossRef] [PubMed]
- Gotta, F.; Lamp, M.; Geroldi, A.; Trevisan, L.; Origone, P.; Fugazza, G.; Fabbri, S.; Nesti, C.; Rubegni, A.; Morani, F.; et al. A novel mutation of Twinkle in Perrault syndrome: A not rare diagnosis? Ann. Hum. Genet. 2020, 84, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Faridi, R.; Rea, A.; Fenollar-Ferrer, C.; O’Keefe, R.T.; Gu, S.; Munir, Z.; Khan, A.A.; Riazuddin, S.; Hoa, M.; Naz, S.; et al. New insights into Perrault syndrome, a clinically and genetically heterogeneous disorder. Hum. Genet. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Kaga, K.; Nakamura, M.; Shinogami, M.; Tsuzuku, T.; Yamada, K.; Shindo, M. Auditory nerve disease of both ears revealed by auditory brainstem responses, electrocochleography and otoacoustic emissions. Scand. Audiol. 1996, 25, 233–238. [Google Scholar] [CrossRef]
- Dutt, S.N.; Kumar, A.; Mittal, A.A.; Vadlamani, S.; Gaur, S.K. Cochlear implantation in auditory neuropathy spectrum disorders: Role of transtympanic electrically evoked auditory brainstem responses and serial neural response telemetry. J. Laryngol. Otol. 2021, 135, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Breneman, A.I.; Gifford, R.H.; Dejong, M.D. Cochlear implantation in children with auditory neuropathy spectrum disorder: Long-term outcomes. J. Am. Acad. Audiol. 2012, 23, 5–17. [Google Scholar] [CrossRef]
- Trecca, E.M.C.; Riggs, W.J.; Mattingly, J.K.; Hiss, M.M.; Cassano, M.; Adunka, O.F. Electrocochleography and Cochlear Implantation: A Systematic Review. Otol. Neurotol. 2020, 41, 864–878. [Google Scholar] [CrossRef]
- Palmieri, M.; Berrettini, S.; Forli, F.; Trevisi, P.; Genovese, E.; Chilosi, A.M.; Arslan, E.; Martini, A. Evaluating benefits of cochlear implantation in deaf children with additional disabilities. Ear Hear. 2012, 33, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Berrettini, S.; Forli, F.; Genovese, E.; Santarelli, R.; Arslan, E.; Chilosi, A.M.; Cipriani, P. Cochlear implantation in deaf children with associated disabilities: Challenges and outcomes. Int. J. Audiol. 2008, 47, 199–208. [Google Scholar] [CrossRef]
- Ay, N.; Gehl, H.B.; Sudhoff, H.; Todt, I. Effect of head position on cochlear implant MRI artifact. Eur. Arch. Oto-Rhino-Laryngol. 2021, 278, 2763–2767. [Google Scholar] [CrossRef] [PubMed]
- Todt, I.; Rademacher, G.; Mittmann, P.; Wagner, J.; Mutze, S.; Ernst, A. MRI Artifacts and Cochlear Implant Positioning at 3 T In Vivo. Otol. Neurotol. 2015, 36, 972–976. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Article | Number of Cases | Grade of SNHL | Diagnosis of SNHL | Progression | Result of Newborn Hearing Screening | OEs or Cochlear Microphonic | ABR Track | ANSD | Hearing Restoration |
|---|---|---|---|---|---|---|---|---|---|
| Present article | #1 | Severe-to-profound | 24 months of age | Yes | Pass | Present | Absent | Yes | Cochlear Implant |
| Faridi 2021 [24] | #1 | Severe-to-profound | Childhood | Yes | Not reported | Not reported | Not reported | - | Not reported |
| Demain 2017 [20] | #1 | Severe-to-profound | 14 months of age | Yes | Not reported | Not reported | Not reported | - | Not reported |
| Dursun 2016 [13] | #2 | Not reported (2) | Not reported (2) | Not reported (2) | Not reported (2) | Not reported (2) | Not reported (2) | - | Not reported (2) |
| Lerat 2016 [21] | #2 | Severe (2) | 3 years of age (1) | Yes (2) | Not reported (2) | Not reported (2) | Not reported (2) | - | Not reported (2) |
| 6 years of age (1) | |||||||||
| Theunissen 2016 [16] | #5 | Profound (5) | Congenital (1) | Not reported (5) | Not reported (5) | Not reported (5) | Not reported (5) | - | Not reported (5) |
| 16 months of age (1) | |||||||||
| Not reported (3) | |||||||||
| Ahmed 2015 [14] | #2 | Severe-to-profound (2) | Not reported (2) | Not reported (2) | Not reported (2) | Not reported (2) | Not reported (2) | - | Cochlear implant (1) |
| Not reported (1) | |||||||||
| Jenkinson 2013 [2] | #7 | Profound (7) | Congenital (7) | Not reported (7) | Not reported (7) | Not reported (7) | Not reported (7) | - | Not reported (7) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forli, F.; Bruschini, L.; Franciosi, B.; Battini, R.; Marinella, G.; Berrettini, S.; Lazzerini, F. A Rare Case of Perrault Syndrome with Auditory Neuropathy Spectrum Disorder: Cochlear Implantation Treatment and Literature Review. Audiol. Res. 2021, 11, 609-617. https://doi.org/10.3390/audiolres11040055
Forli F, Bruschini L, Franciosi B, Battini R, Marinella G, Berrettini S, Lazzerini F. A Rare Case of Perrault Syndrome with Auditory Neuropathy Spectrum Disorder: Cochlear Implantation Treatment and Literature Review. Audiology Research. 2021; 11(4):609-617. https://doi.org/10.3390/audiolres11040055
Chicago/Turabian StyleForli, Francesca, Luca Bruschini, Beatrice Franciosi, Roberta Battini, Gemma Marinella, Stefano Berrettini, and Francesco Lazzerini. 2021. "A Rare Case of Perrault Syndrome with Auditory Neuropathy Spectrum Disorder: Cochlear Implantation Treatment and Literature Review" Audiology Research 11, no. 4: 609-617. https://doi.org/10.3390/audiolres11040055
APA StyleForli, F., Bruschini, L., Franciosi, B., Battini, R., Marinella, G., Berrettini, S., & Lazzerini, F. (2021). A Rare Case of Perrault Syndrome with Auditory Neuropathy Spectrum Disorder: Cochlear Implantation Treatment and Literature Review. Audiology Research, 11(4), 609-617. https://doi.org/10.3390/audiolres11040055