
Genetic Characteristics and Enzymatic Activities of Bacillus velezensis KS04AU as a Stable Biocontrol Agent against Phytopathogens

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Sequencing, Assembly, Genome Annotation and Gene Prediction
2.2. Genome Annotation, Gene Prediction and Comparative Genomic Analysis
2.3. Antagonistic and Hydrolytic Activities
2.3.1. Bacterial Suspension Preparation
2.3.2. Hydrolytic Activities
2.3.3. Antagonistic Activity
2.4. Biocontrol Ability of B. velezensis KS04AU to Suppress Tomato Foot and Root Rot
3. Result
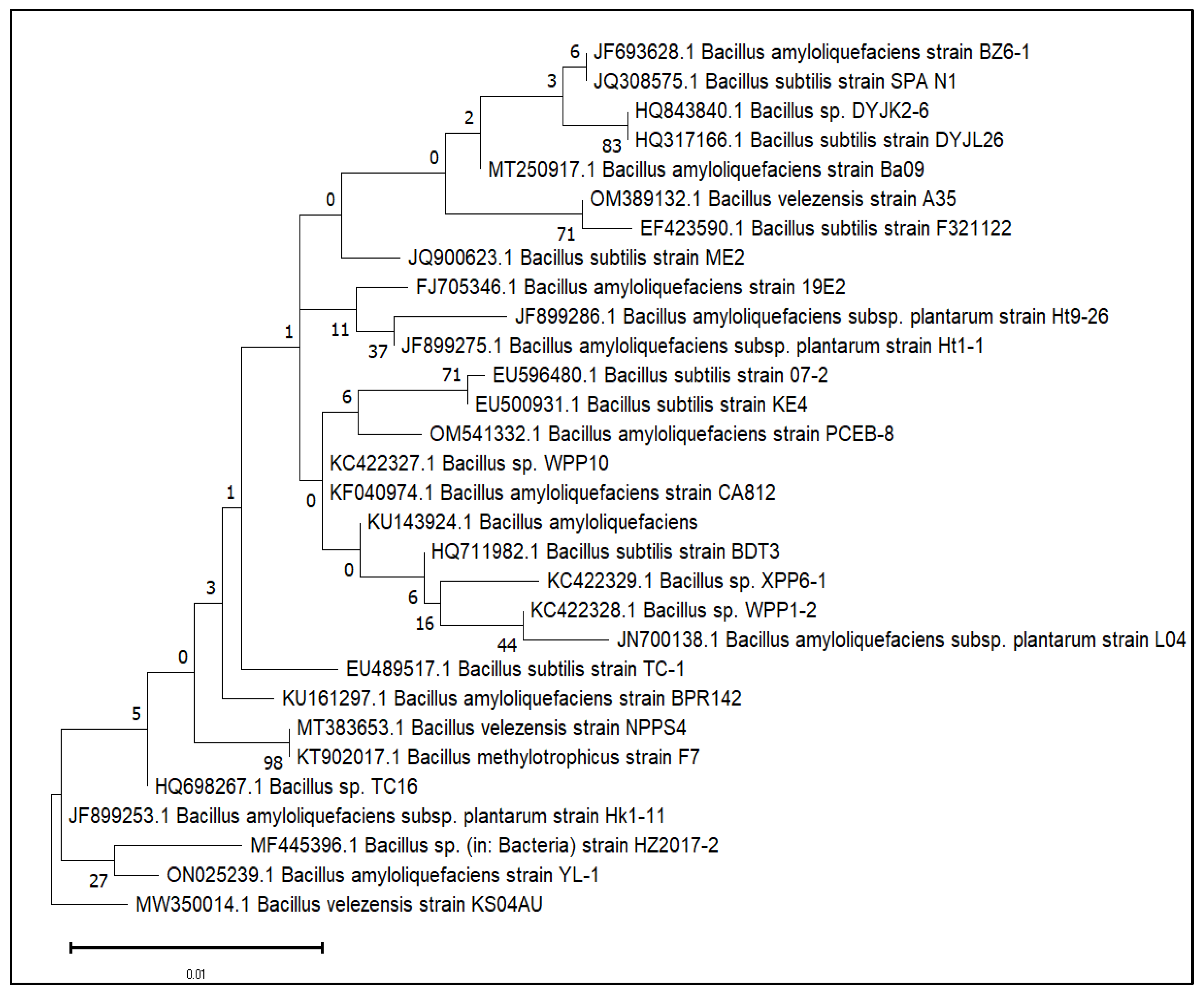
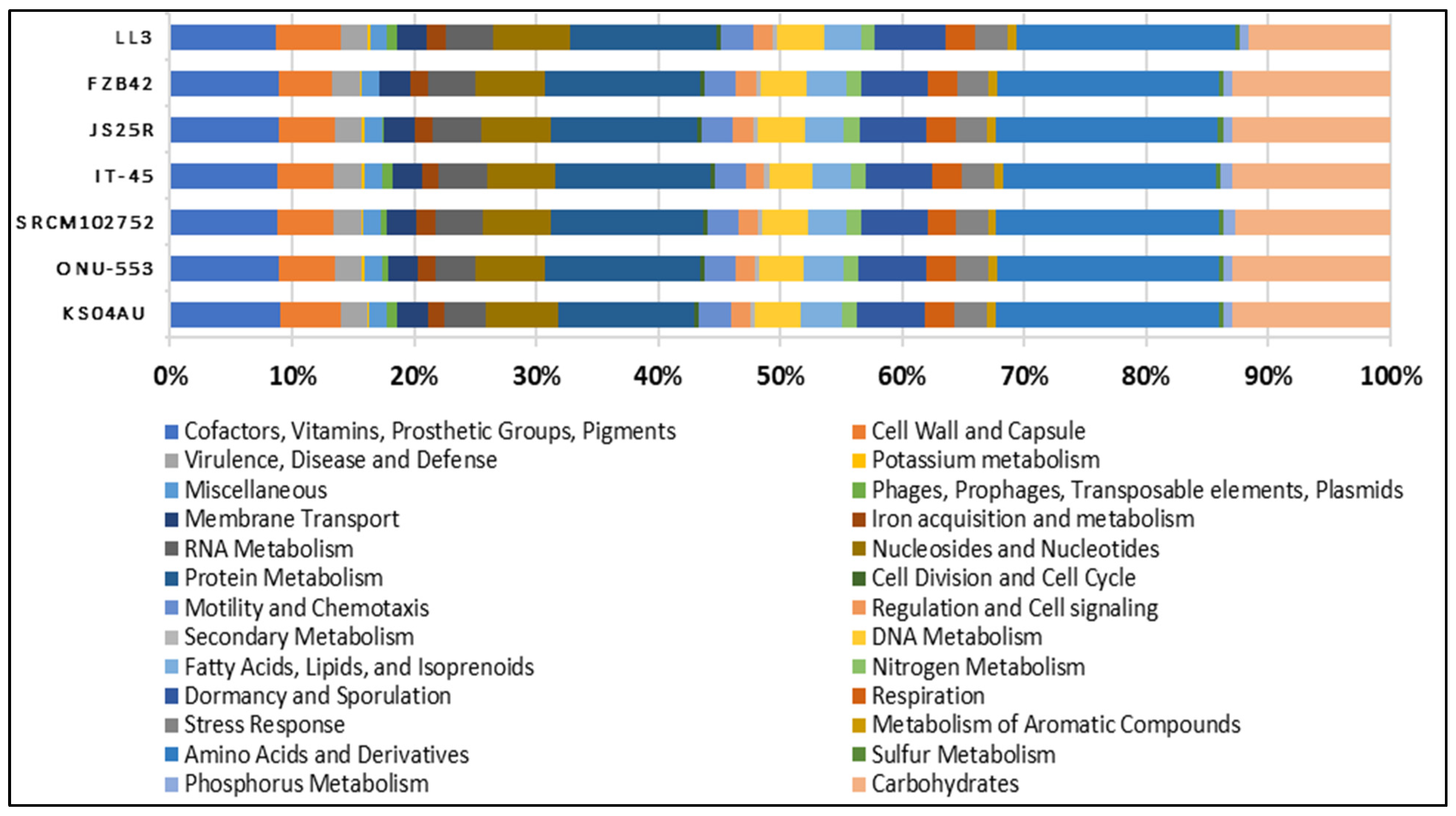
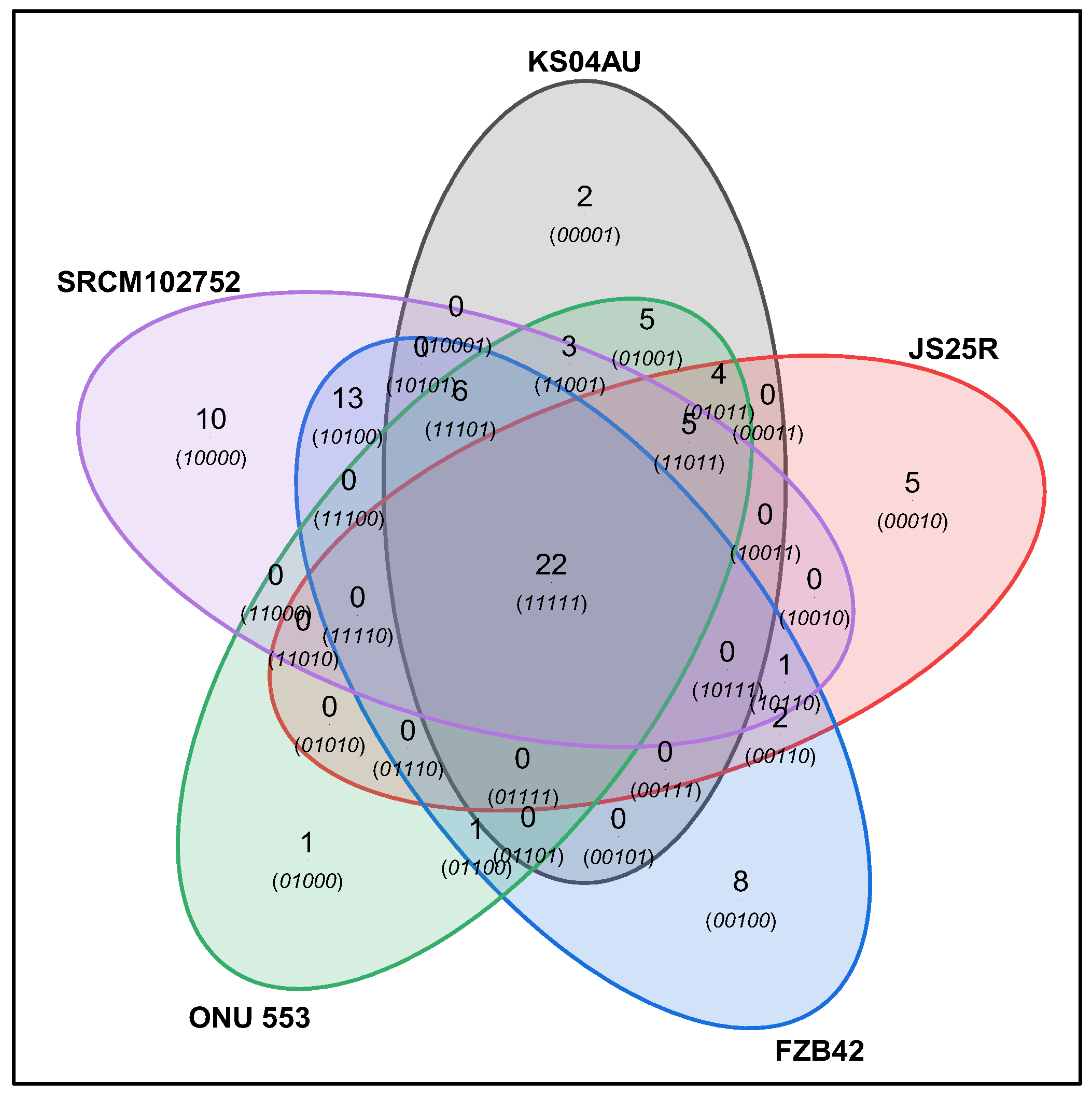
3.1. Genome Sequencing, Assembly and Comparison
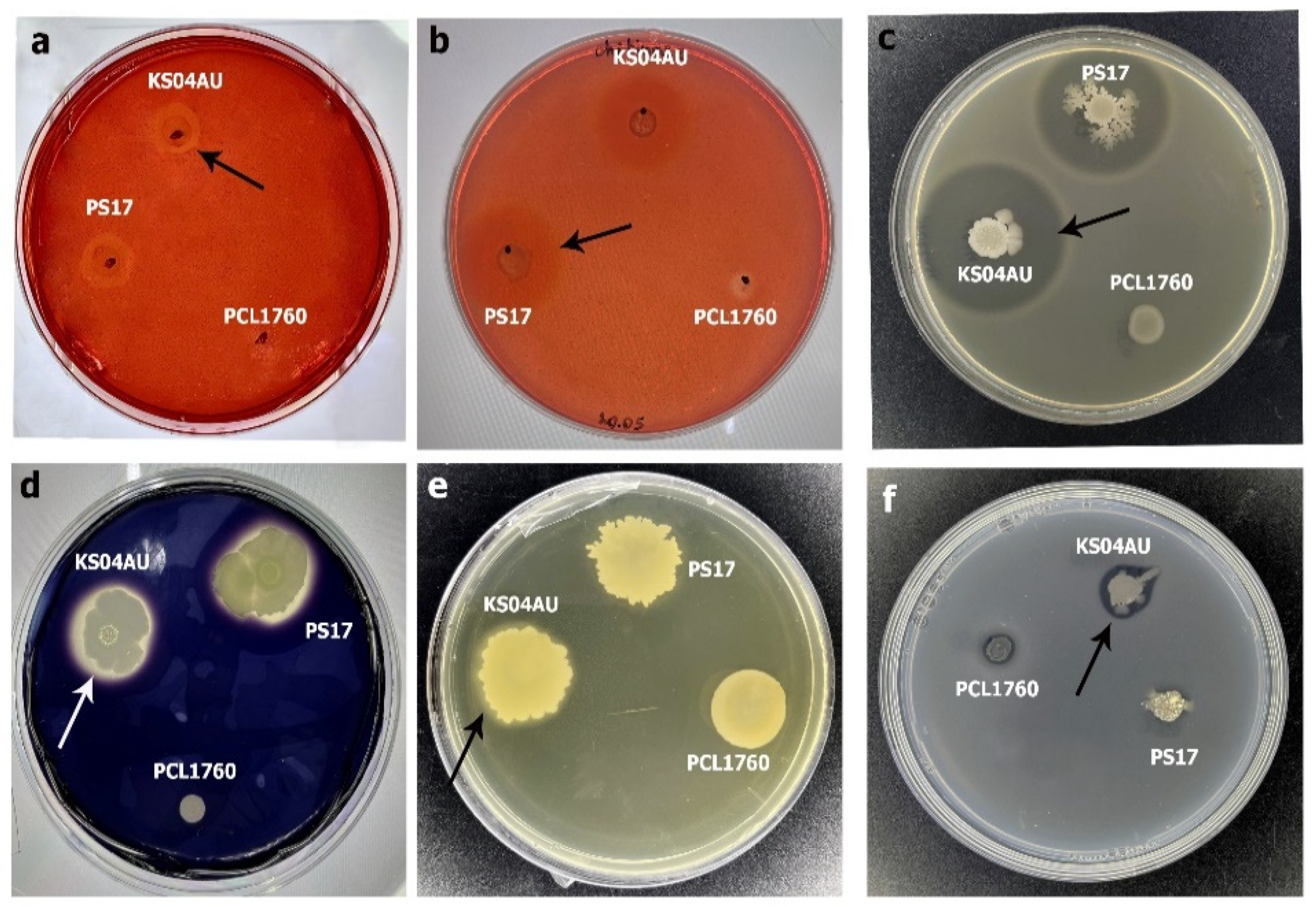
3.2. Antagonistic and Hydrolytic Activities
3.3. Biocontrol Ability of B. velezensis KS04AU to Suppress Tomato Foot and Root Rot
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fira, D.; Dimkić, I.; Berić, T.; Lozo, J.; Stanković, S. Biological control of plant pathogens by Bacillus species. J. Biotechnol. 2018, 285, 44–55. [Google Scholar] [CrossRef]
- Ongena, M.; Jacques, P. Bacillus lipopeptides: Versatile weapons for plant disease biocontrol. Trends Microbiol. 2008, 16, 115–125. [Google Scholar] [CrossRef]
- Ruiz-Garcia, C.; Bejar, V.; Martinez-Checa, F.; Llamas, I.; Quesada, E. Bacillus velezensis sp. nov., a surfactant-producing bacterium isolated from the river Velez in Malaga, southern Spain. Int. J. Syst. Evol. Microbiol. 2005, 55, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Pi, H.; Chandrangsu, P.; Li, Y.; Wang, Y.; Zhou, H.; Xiong, H.; Helmann, J.D.; Cai, Y. Antagonism of two plant-growth promoting Bacillus velezensis isolates against Ralstonia solanacearum and Fusarium oxysporum. Sci. Rep. 2018, 8, 4360. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, X.; Ma, Q.; Bian, L.; Liu, X.; Xu, Y.; Zhang, H.; Shao, J.; Liu, Y. Bacillus velezensis CLA178-induced systemic resistance of Rosa multiflora against crown gall disease. Front. Microbiol. 2020, 11, 11:587667. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zhang, Z.; Zhao, F.; Liu, H.; Yu, L.; Zha, J.; Wang, G. Probiotic potential of Bacillus velezensis JW: Antimicrobial activity against fish pathogenic bacteria and immune enhancement effects on Carassius auratus. Fish Shellfish Immunol. 2018, 78, 322–330. [Google Scholar] [CrossRef]
- Chen, L.; Heng, J.; Qin, S.; Bian, K. A comprehensive understanding of the biocontrol potential of Bacillus velezensis LM2303 against Fusarium head blight. PLoS ONE 2018, 13, e0198560. [Google Scholar] [CrossRef]
- Dunlap, C.A.; Kim, S.J.; Kwon, S.W.; Rooney, A.P. Phylogenomic analysis shows that Bacillus amyloliquefaciens subsp. plantarum is a later heterotypic synonym of Bacillus methylotrophicus. Int. J. Syst. Evol. 2015, 65 Pt 7, 2104–2109. [Google Scholar] [CrossRef]
- Dunlap, C.; Kim, S.J.; Kwon, S.W.; Rooney, A. Bacillus velezensis is not a later heterotypic synonym of Bacillus amyloliquefaciens, Bacillus methylotrophicus, Bacillus amyloliquefaciens subsp. plantarum and ‘Bacillus oryzicola’ are later heterotypic synonyms of Bacillus velezensis based on phylogenomics. Int. J. Syst. Evol. Microbiol. 2016, 66, 1212–1217. [Google Scholar] [CrossRef]
- Kaltz, O.; Escobar-Páramo, P.; Hochberg, M.E.; Cohen, J.E. Bacterial microcosms obey Taylor’s law: Effects of abiotic and biotic stress and genetics on mean and variance of population density. Ecol. Process. 2012, 1, 5. [Google Scholar] [CrossRef][Green Version]
- Adeniji, A.A.; Loots, D.T.; Babalola, O.O. Bacillus velezensis: Phylogeny, useful applications, and avenues for exploitation. Appl. Microbiol. Biotechnol. 2019, 103, 3669–3682. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e100559. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Li, X.; Wu, Y. GAPPadder: A sensitive approach for closing gaps on draft genomes with short sequence reads. BMC Genom. 2019, 20, 426. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Wright, G.D. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39 (Suppl. S2), W347–W352. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Thomson, N.; Bleasby, A.; Berriman, M.; Parkhill, J. DNAPlotter: Circular and linear interactive genome visualization. Bioinformatics 2009, 25, 119–120. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Wang, Y. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N.S. Using repeat masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 5, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Zagnitko, O. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Teather, R.M.; Wood, P.J. Use of Congo red-polysaccharide interactions in enumeration and characterization of cellulolytic bacteria from the bovine rumen. Appl. Environ. Microbiol. 1982, 43, 777–780. [Google Scholar] [CrossRef]
- Howson, S.J.; Davis, R.P. Production of phytate-hydrolysing enzyme by some fungi. Enzym. Microb. Technol. 1983, 5, 377–382. [Google Scholar] [CrossRef]
- Huang, L.; Li, Q.C.; Hou, Y.; Li, G.Q.; Yang, J.Y.; Li, D.W.; Ye, J.R. Bacillus velezensis strain HYEB5-6 as a potential biocontrol agent against anthracnose on Euonymus japonicus. Biocontrol. Sci. Technol. 2017, 27, 636–653. [Google Scholar] [CrossRef]
- Myo, E.M.; Liu, B.; Ma, J.; Shi, L.; Jiang, M.; Zhang, K.; Ge, B. Evaluation of Bacillus velezensis NKG-2 for bio-control activities against fungal diseases and potential plant growth promotion. Biol. Control 2019, 134, 23–31. [Google Scholar] [CrossRef]
- Oda, Y.; Larimer, F.W.; Chain, P.S.; Malfatti, S.; Shin, M.V.; Vergez, L.M.; Hauser, L.; Land, M.L.; Braatsch, S.; Beatty, J.T.; et al. Multiple genome sequences reveal adaptations of a phototrophic bacterium to sediment microenvironments. Proc. Natl. Acad. Sci. USA 2008, 105, 18543–18548. [Google Scholar] [CrossRef]
- Rooney, A.P.; Price, N.P.; Ehrhardt, C.; Swezey, J.L.; Bannan, J.D. Phylogeny and molecular taxonomy of the Bacillus subtilis species complex and description of Bacillus subtilis subsp. inaquosorum subsp. nov. Int. J. Syst. Evol. Microbiol. 2009, 59, 2429–2436. [Google Scholar] [CrossRef]
- Borriss, R.; Chen, X.H.; Rueckert, C.; Blom, J.; Becker, A.; Baumgarth, B.; Fan, B.; Pukall, R.; Schumann, P.; Spröer, C.; et al. Relationship of Bacillus amyloliquefaciens clades associated with strains DSM 7T and FZB42T: A proposal for Bacillus amyloliquefaciens subsp. amyloliquefaciens subsp. nov. and Bacillus amyloliquefaciens subsp. plantarum subsp. nov. based on complete genome sequence comparisons. Int. J. Syst. Evol. Microbiol. 2011, 61, 786–1801. [Google Scholar] [CrossRef]
- Pattenden, T.; Eagles, C.; Wahl, L.M. Host life-history traits influence the distribution of prophages and the genes they carry. Philos. Trans. R. Soc. B 2022, 377, 20200465. [Google Scholar] [CrossRef]
- Morozova, V.; Bokovaya, O.; Kozlova, Y.; Kurilshikov, A.; Babkin, I.; Tupikin, A.; Bondar, A.; Ryabchikova, E.; Brayanskaya, A.; Peltek, S.; et al. A novel thermophilic Aeribacillus bacteriophage AP45 isolated from the Valley of Geysers, Kamchatka: Genome analysis suggests the existence of a new genus within the Siphoviridae family. Extremophiles 2019, 23, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Bousquet, A.C.; Bruff, E.; Carson, N.; Clark, A.; Connell, A.; Davis, Z.; Dums, J.; Everington, C.; Groth, A.; et al. Paenibacillus larvae phage Tripp genome has 378-base-pair terminal repeats. Genome Announc. 2016, 4, e01498-15. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A. CRISPR-Cas immunity against phages: Its effects on the evolution and survival of bacterial pathogens. PLoS Pathog. 2013, 9, e1003765. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Liu, Y.; Xu, Y.; Zhang, G.; Shen, Q.; Zhang, R. Exploring elicitors of the beneficial rhizobacterium Bacillus amyloliquefaciens SQR9 to induce plant systemic resistance and their interactions with plant signaling pathways. Mol. Plant-Microbe Interact. 2018, 31, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Camele, I.; Elshafie, H.S.; Caputo, L.; Sakr, S.H.; De Feo, V. Bacillus mojavensis: Biofilm formation and biochemical investigation of its bioactive metabolites. J. Biol. Res. 2019, 92, 39–45. [Google Scholar] [CrossRef]
- Elshafie, H.S.; Sakr, S.; Bufo, S.A.; Camele, I. An attempt of biocontrol the tomato-wilt disease caused by Verticillium dahliae using Burkholderia gladioli pv. agaricicola and its bioactive secondary metabolites. Int. J. Plant Biol. 2017, 8, 57–60. [Google Scholar] [CrossRef]
- Kruszewska, D.; Sahl, H.G.; Bierbaum, G.; Pag, U.; Hynes, S.O.; Ljungh, A. Mersacidin eradicates methicillin-resistant Staphylococcus aureus (MRSA) in a mouse rhinitis model. J. Antimicrob. Chemother. 2004, 54, 648–653. [Google Scholar] [CrossRef]
- Borriss, R.; Wu, H.; Gao, X. Secondary Metabolites of the Plant Growth Promoting Model Rhizobacterium Bacillus velezensis FZB42 Are Involved in Direct Suppression of Plant Pathogens and in Stimulation of Plant-Induced Systemic Resistance; Springer: Singapore, 2019; pp. 147–168. [Google Scholar] [CrossRef]
- Herzner, A.M.; Dischinger, J.; Szekat, C.; Josten, M.; Schmitz, S.; Yakeleba, A.; Reinartz, R.; Jansen, A.; Sahl, H.G.; Piel, J.; et al. Expression of the lantibiotic mersacidin in Bacillus amyloliquefaciens FZB42. PLoS ONE 2011, 6, e22389. [Google Scholar] [CrossRef]
- Touchon, M.; Rocha, E.P.C. Causes of insertion sequences abundance in prokaryotic genomes. Mol. Biol. Evol. 2007, 24, 969–981. [Google Scholar] [CrossRef]
- Siguier, P.; Gourbeyre, E.; Chandler, M. Bacterial insertion sequences: Their genomic impact and diversity. FEMS Microbiol. Rev. 2014, 38, 865–891. [Google Scholar] [CrossRef] [PubMed]
- Medhioub, I.; Cheffi, M.; Tounsi, S.; Triki, M.A. Study of Bacillus velezensis OEE1 potentialities in the biocontrol against Erwinia amylovora, causal agent of fire blight disease of rosaceous plants. Biol. Control 2022, 167, 104842. [Google Scholar] [CrossRef]
- Ye, L.; Hildebrand, F.; Dingemans, J.; Ballet, S.; Laus, G.; Matthijs, S.; Berendsen, R.; Cornelis, P. Draft genome sequence analysis of a Pseudomonas putida W15Oct28 strain with antagonistic activity to Gram-positive and Pseudomonas sp. pathogens. PLoS ONE 2014, 9, e110038. [Google Scholar] [CrossRef] [PubMed]
- Validov, S.Z.; Kamilova, F.; Lugtenberg, B.J. Pseudomonas putida strain PCL1760 controls tomato foot and root rot in stonewool under industrial conditions in a certified greenhouse. Biol. Control 2009, 48, 6–11. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microbial Strain | NCBI Reference Sequence |
|---|---|
| B. velezensis SRCM102752 | NZ_CP028961.1 |
| B. velezensis FZB42 | NC_009725.2 |
| B. velezensis JS25R | NZ_CP009679.1 |
| B. velezensis KS04AU | NZ_CP092750.1 |
| B. velezensis ONU-553 | NZ_CP043416.1 |
| B. amyloliquefaciens IT-45 | NC_020272.1 |
| B. amyloliquefaciens LL3 | NC_017190.1 |
| Features | KS04AU | SRCM102752 | ONU-553 | FZB42 | JS25R | LL3 | IT-45 |
|---|---|---|---|---|---|---|---|
| Genome (bp) | 4,063,541 | 3,971,509 | 3,934,563 | 3,918,596 | 4,006,002 | 3,995,227 | 3,928,857 |
| G + C (%) | 46.5 | 46.40 | 46.70 | 46.50 | 46.39 | 45.69 | 46.59 |
| Genes (total) | 4028 | 3950 | 3889 | 3870 | 3933 | 4151 | 3927 |
| Total CDS | 3941 | 3832 | 3771 | 3749 | 3826 | 4052 | 3797 |
| CDS coding | 3860 | 3761 | 3706 | 3676 | 3768 | 3943 | 3733 |
| Genes (RNA) | 87 | 118 | 118 | 121 | 107 | 99 | 130 |
| tRNA | 79 | 86 | 86 | 88 | 81 | 72 | 95 |
| ncRNA | 5 | 5 | 5 | 4 | 5 | 5 | 5 |
| Pseudo Genes | 81 | 71 | 65 | 73 | 58 | 109 | 64 |
| B. velezensis KS04AU | B. velezensis JS25R | B. velezensis FZB42 | B. velezensis ONU-553 | B. velezensis SRCM102752 | B. amyloliquefaciens LL3 | B. amyloliquefaciens IT-45 | |
|---|---|---|---|---|---|---|---|
| B. velezensis KS04AU | –– | 98.19 (91.60) | 98.66 (91.09) | 99.53 (95.32) | 98.31 (90.54) | 93.29 (86.11) | 97.38 (90.99) |
| B. velezensis JS25R | 98.20 (92.47) | –– | 98.19 (92.03) | 98.20 (92.58) | 97.88 (91.14) | 93.34 (86.41) | 97.47 (92.00) |
| B. velezensis FZB42 | 98.76 (93.84) | 98.26 (94.17) | –– | 98.77 (94.59) | 98.62 (93.57) | 93.30 (87.70) | 97.51 (93.32) |
| B. velezensis ONU-553 | 99.65 (97.83) | 98.31 (94.21) | 98.78 (94.25) | –– | 98.44 (93.53) | 93.40 (88.43) | 97.53 (94.09) |
| B. velezensis SRCM102752 | 98.34 (92.08) | 97.80 (92.01) | 98.56 (92.41) | 98.39 (92.62) | –– | 93.32 (87.67) | 97.18 (91.83) |
| B. amyloliquefaciens LL3 | 93.78 (86.01) | 93.77 (85.59) | 93.74 (84.70) | 93.82 (86.01) | 93.80 (86.04) | –– | 93.65 (85.92) |
| B. amyloliquefaciens IT-45. | 97.59 (93.22) | 97.67 (93.34) | 97.60 (92.86) | 97.62 (93.94) | 97.40 (92.58) | 93.31 (88.40) | –– |
| Region | Region Length | Completeness | Phage Hit Protein | Hypothetical Protein | Specific Keyword | Region Position | Possible Phage | G + C Percentage |
|---|---|---|---|---|---|---|---|---|
| 1 | 18.1 Kb | Incomplete (10) | 13 | 5 | NA | 3336–21,513 | PHAGE_Bacill_SPP1_NC_004166 | 44.55% |
| 2 | 49.1 Kb | intact (120) | 41 | 31 | integrase, terminase, tail | 1,107,820–1,157,010 | PHAGE_Aeriba_AP45_NC_048651 | 41.77% |
| 3 | 31.3 Kb | questionable | 29 | 16 | tail, plate, capsid | 1,203,112–1,234,419 | PHAGE_Brevib_Jimmer2_NC_041976 | 46.98% |
| 4 | 97.5 Kb | intact | 61 | 41 | integrase, tail, terminase, capsid | 3,892,492–3,990,010 | PHAGE_Paenib_Tripp_NC_028930 | 47.43% |
| Phage | Presence (+) or Absence (−) in Related Strains | ||||||
|---|---|---|---|---|---|---|---|
| KS04AU | SRCM102752 | ONU 553 | FZB42 | JS25R | LL3 | IT-45 | |
| PHAGE_Aeriba_AP45_NC_048651 | + | − | − | − | − | − | − |
| PHAGE_Brevib_Jimmer2_NC_041976 | + | + | + | − | − | − | − |
| PHAGE_Paenib_Tripp_NC_028930 | + | − | − | − | − | − | − |
| PHAGE_Bacill_SPP1_NC_004166 | + | − | + | − | − | − | − |
| PHAGE_Thermu_OH2_NC_021784 | − | − | − | − | − | + | − |
| PHAGE_Thermu_TMA_NC_015937 | − | − | − | − | + | − | − |
| PHAGE_Brevib_Osiris_NC_028969 | − | − | − | − | + | + | − |
| PHAGE_Bacill_phi105_NC_004167 | − | − | − | − | − | + | − |
| Genomic Region | Type | From | To | Most Similar Known Cluster | Similarity | |
|---|---|---|---|---|---|---|
| Region 1 | NRPS | 297,001 | 359,149 | surfactin | NRP: Lipopeptide | 95% |
| Region 2 | PKS-like | 881,875 | 923,119 | butirosin A/butirosin B | Saccharide | 7% |
| Region 3 | terpene | 1,009,298 | 1,026,466 | |||
| Region 4 | transAT-PKS | 1,379,829 | 1,467,645 | macrolactin H | Polyketide | 100% |
| Region 5 | transAT-PKS, T3PKS, NRPS | 1,689,828 | 1,790,022 | bacillaene | Polyketide + NRP | 100% |
| Region 6 | NRPS, transAT-PKS, betalactone | 1,856,677 | 1,988,381 | fengycin | NRP | 100% |
| Region 7 | terpene | 2,011,406 | 2,033,289 | |||
| Region 8 | T3PKS | 2,083,724 | 2,124,824 | |||
| Region 9 | transAT-PKS | 2,252,798 | 2,344,192 | difficidin | Polyketide + NRP | 100% |
| Region 10 | NRPS, RiPP-like | 2,955,287 | 3,005,799 | bacillibactin | NRP | 100% |
| Region 11 | NRPS | 3,284,182 | 3,330,146 | |||
| Region 12 | other | 3,550,785 | 3,592,203 | bacilysin | Other | 100% |
| Region 13 | lanthipeptide-class-ii | 3,740,316 | 3,763,504 | mersacidin | RiPP: Lanthipeptide | 100% |
| Presence (+) or Absence (−) of Secondary Metabolite Clusters in Related Strains | ||||||||
|---|---|---|---|---|---|---|---|---|
| Synthetase | Metabolites | KS04AU | SRCM102752 | ONU-553 | FZB42 | JS25R | LL3 | IT-45 |
| PKS-like | surfactin | + | + | + | + | + | + | + |
| terpene | − | + | + | + | + | + | + | + |
| transAT-PKS | butirosin A/butirosin B | + | + | + | + | + | + | + |
| transAT-PKS, T3PKS, NRPS | macrolactin H | + | − | + | + | + | − | + |
| NRPS, transAT-PKS, betalactone | bacillaene | + | + | + | + | + | + | + |
| terpene | − | + | + | + | + | + | + | + |
| T3PKS | − | + | + | + | + | + | ||
| transAT-PKS | fengycin | + | + | + | + | + | ||
| NRPS, RiPP-like | − | + | + | + | + | + | + | + |
| NRPS | difficidin | + | + | + | + | + | ||
| NRPS, RiPP-like | bacillibactin | + | + | + | + | + | + | + |
| NRPS | + | + | + | + | + | |||
| other | bacilysin | + | + | + | + | + | + | + |
| lanthipeptide-class-ii | mersacidin | + | − | − | − | − | − | − |
| cyclic-lactone-autoinducer, lanthipeptide-class-II | kijanimicin | − | + | − | − | − | − | − |
| NRPS, transAT-PKS | rhizocticin A | − | + | − | − | − | − | − |
| RRE-containing, LAP | plantazolicin | − | − | − | + | − | − | − |
| ARO Term ARM | Gene Family | Drug Class | Resistance Mechanism | Presence (+) or Absence (−) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| KS04AU | SRCM102752 | ONU 553 | FZB42 | JS25R | LL3 | IT-45 | ||||
| clbA | Cfr 23S ribosomal RNA methyltransferase | Incosamide antibiotic, streptogramin antibiotic, streptogramin A antibiotic, oxazolidinone antibiotic, phenicol antibiotic, pleuromutilin antibiotic | Antibiotic target alteration | + | + | + | + | + | + | + |
| tet (45) | Major facilitator superfamily (MFS) antibiotic efflux pump | Tetracycline antibiotic | Antibiotic efflux | + | + | + | + | + | + | + |
| qacJ | small multidrug resistance (SMR) antibiotic efflux pump | Disinfecting agents and antiseptics | Antibiotic efflux | + | + | + | + | + | + | + |
| qacG | small multidrug resistance (SMR) antibiotic efflux pump | Disinfecting agents and antiseptics | Antibiotic efflux | + | + | + | + | + | + | + |
| qacJ | small multidrug resistance (SMR) antibiotic efflux pump | Disinfecting agents and antiseptics | Antibiotic efflux | + | + | + | + | + | + | + |
| qacJ | small multidrug resistance (SMR) antibiotic efflux pump | Disinfecting agents and antiseptics | Antibiotic efflux | − | − | − | − | − | − | + |
| BcI | class A Bacillus cereus Bc beta-lactamase | cephalosporin, penem | antibiotic inactivation | + | + | + | + | + | + | + |
| Strain | Number of CRISPR/CAS | Element | Start | End | Spacer/Gene | Repeat Consensus/Cas Genes | Direction |
|---|---|---|---|---|---|---|---|
| KS04AU | 2 | Cas | 66,995 | 3,697,966 | 12 | Cas3_TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI | (–)—8 Cas genes (+)—4 Cas genes |
| CRISPR | 665,256 | 665,363 | 1 | CGGAGGATATCCGGGATACGGTTT | ND | ||
| CRISPR | 712,560 | 712,654 | 1 | TTCACCGGGGCAACGGGGCTGAC | ND | ||
| SRCM102752 | 1 | CAS | 61,088 | 3,747,587 | 12 | Cas3_TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI | (–)—8 Cas genes (+)—4 Cas genes |
| CRISPR | 780,220 | 780,314 | 1 | TTCACCGGGGCAACGGGGCTGAC | ND | ||
| ONU 553 | 1 | CAS | 61,088 | 3,747,587 | 12 | Cas3_TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI | (–)—8 Cas genes (+)—4 Cas genes |
| CRISPR | TTCACCGGGGCAACGGGGCTGAC | ND | |||||
| FZB42 | 0 | ||||||
| JS25R | 1 | CAS | 61,500 | 3,812,920 | 13 | Cas3_TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI | (–)—7 Cas genes (+)—6 Cas genes |
| CRISPR | 447,873 | 447,955 | 1 | AAGAAATCGGCCAAAAAGGCGGA | ND | ||
| CAS-TypeID | 2,171,815 | 2,174,061 | 1 | cas3_TypeID | - | ||
| LL3 | 0 | ||||||
| IT-45 | 2 | CAS | 13 | Cas3_TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ TypeI, Cas3_ Type | (–)—6 Cas genes (+)—7 Cas genes | ||
| CRISPR | 2,680,276 | 2,680,402 | 1 | TGCTCGCAATCTCGTCCGCTTTTCCCATGAATGAGGTCGTGAACTT | ND | ||
| CRISPR | 3,044,191 | 3,044,320 | 1 | AACAGGCTTTCAGCGGGGAATCCGGCGGACAGCAGCA | ND | ||
| CAS-TypeID | 1,779,683 | 1,781,929 | 1 | cas3_TypeID |
| Species | Gene Clusters | Singletons |
|---|---|---|
| KS04AU | 3727 | 158 |
| ONU-553 | 3685 | 33 |
| FZB42 | 3614 | 82 |
| JS25R | 3635 | 137 |
| SRCM102752 | 3666 | 126 |
| IT-45 | 3662 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diabankana, R.G.C.; Shulga, E.U.; Validov, S.Z.; Afordoanyi, D.M. Genetic Characteristics and Enzymatic Activities of Bacillus velezensis KS04AU as a Stable Biocontrol Agent against Phytopathogens. Int. J. Plant Biol. 2022, 13, 201-222. https://doi.org/10.3390/ijpb13030018
Diabankana RGC, Shulga EU, Validov SZ, Afordoanyi DM. Genetic Characteristics and Enzymatic Activities of Bacillus velezensis KS04AU as a Stable Biocontrol Agent against Phytopathogens. International Journal of Plant Biology. 2022; 13(3):201-222. https://doi.org/10.3390/ijpb13030018
Chicago/Turabian StyleDiabankana, Roderic Gilles Claret, Elena Urievna Shulga, Shamil Zavdatovich Validov, and Daniel Mawuena Afordoanyi. 2022. "Genetic Characteristics and Enzymatic Activities of Bacillus velezensis KS04AU as a Stable Biocontrol Agent against Phytopathogens" International Journal of Plant Biology 13, no. 3: 201-222. https://doi.org/10.3390/ijpb13030018
APA StyleDiabankana, R. G. C., Shulga, E. U., Validov, S. Z., & Afordoanyi, D. M. (2022). Genetic Characteristics and Enzymatic Activities of Bacillus velezensis KS04AU as a Stable Biocontrol Agent against Phytopathogens. International Journal of Plant Biology, 13(3), 201-222. https://doi.org/10.3390/ijpb13030018