
A De Novo PTEN Pathogenic Variant in a Young Girl with Sporadic Cowden Syndrome—A Case Report



Abstract
1. Introduction
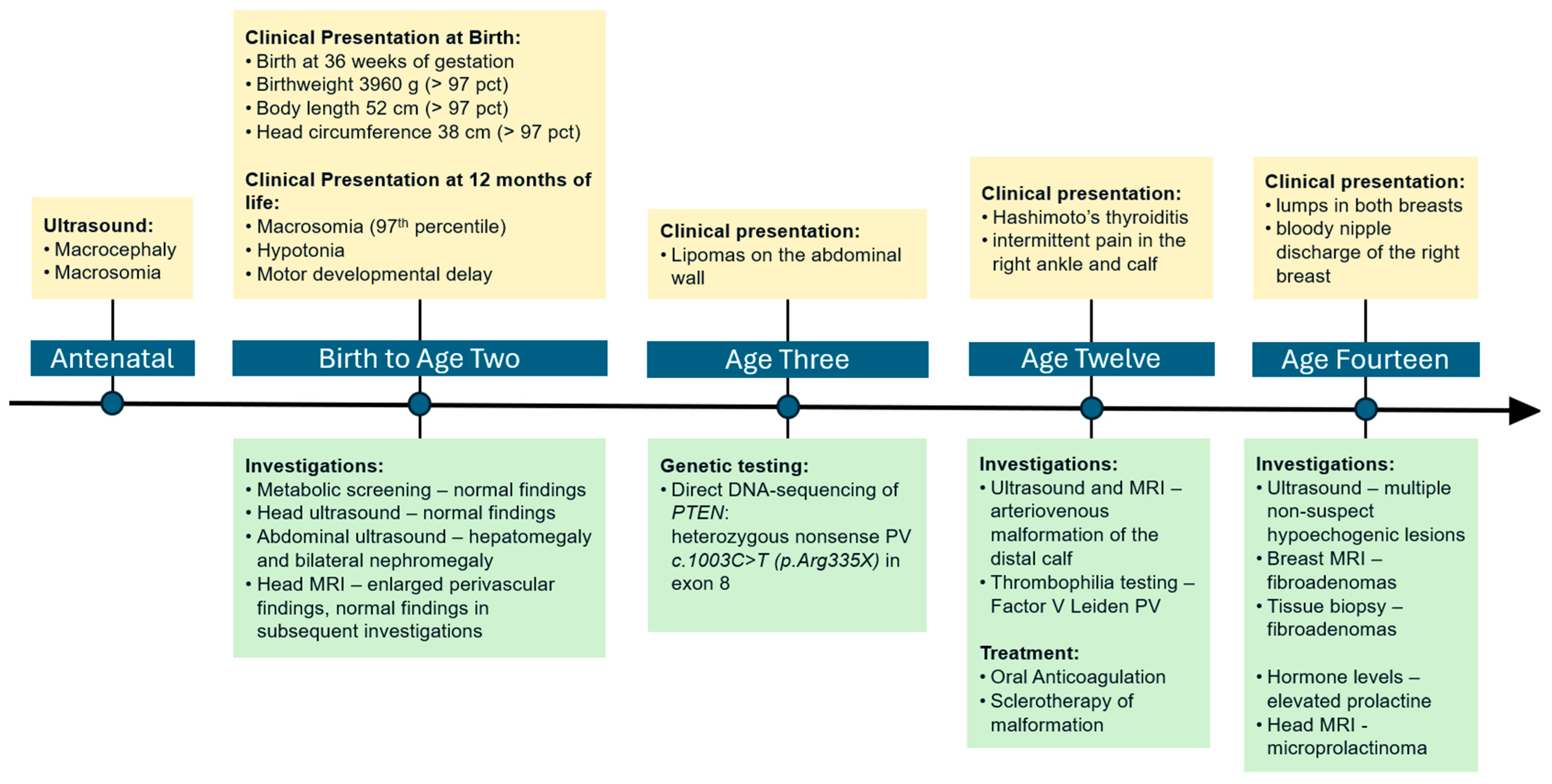
2. Case Presentation
2.1. Early Development
2.2. Genetic Testing
2.3. Clinical Course and Management
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yehia, L.; Ngeow, J.; Eng, C. PTEN-Opathies: From Biological Insights to Evidence-Based Precision Medicine. J. Clin. Investig. 2019, 129, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Keel, E.; Eng, C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 2020, 71, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, R. PTEN Hamartoma Tumor Syndrome: A Clinical Overview. Cancers 2019, 11, 844. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime Cancer Risks in Individuals with Germline PTEN Mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef]
- Hendricks, L.A.J.; Hoogerbrugge, N.; Schuurs-Hoeijmakers, J.H.M.; Vos, J.R. A Review on Age-related Cancer Risks in PTEN Hamartoma Tumor Syndrome. Clin. Genet. 2021, 99, 219–225. [Google Scholar] [CrossRef]
- Nelen, M.R.; Kremer, H.; Konings, I.B.M.; Schoute, F.; Van Essen, A.J.; Koch, R.; Woods, C.G.; Fryns, J.P.; Hamel, B.; Hoefsloot, L.H.; et al. Novel PTEN Mutations in Patients with Cowden Disease: Absence of Clear Genotype–Phenotype Correlations. Eur. J. Hum. Genet. 1999, 7, 267–273. [Google Scholar] [CrossRef]
- Eng, C. PTEN: One Gene, Many Syndromes. Hum. Mutat. 2003, 22, 183–198. [Google Scholar] [CrossRef]
- Tan, M.H.; Mester, J.; Peterson, C.; Yang, Y.; Chen, J.L.; Rybicki, L.A.; Milas, K.; Pederson, H.; Remzi, B.; Orloff, M.S.; et al. A Clinical Scoring System for Selection of Patients for PTEN Mutation Testing Is Proposed on the Basis of a Prospective Study of 3042 Probands. Am. J. Hum. Genet. 2011, 88, 42. [Google Scholar] [CrossRef]
- Mester, J.; Eng, C. Estimate of de Novo Mutation Frequency in Probands with PTEN Hamartoma Tumor Syndrome. Genet. Med. 2012, 14, 819–822. [Google Scholar] [CrossRef]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Sutera, S.; Giachino, D.F.; Pelle, A.; Zuntini, R.; Pentenero, M. Gingival Overgrowths Revealing PTEN Hamartoma Tumor Syndrome: Report of Novel PTEN Pathogenic Variants. Biomedicines 2022, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Bubien, V.; Bonnet, F.; Brouste, V.; Hoppe, S.; Barouk-Simonet, E.; David, A.; Edery, P.; Bottani, A.; Layet, V.; Caron, O.; et al. High Cumulative Risks of Cancer in Patients with PTEN Hamartoma Tumour Syndrome. J. Med. Genet. 2013, 50, 255–263. [Google Scholar] [CrossRef]
- Hendricks, L.A.J.; Hoogerbrugge, N.; Venselaar, H.; Aretz, S.; Spier, I.; Legius, E.; Brems, H.; de Putter, R.; Claes, K.B.M.; Evans, D.G.; et al. Genotype-Phenotype Associations in a Large PTEN Hamartoma Tumor Syndrome (PHTS) Patient Cohort. Eur. J. Med. Genet. 2022, 65, 104632. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, L.A.J.; Hoogerbrugge, N.; Mensenkamp, A.R.; Brunet, J.; Lleuger-Pujol, R.; Høberg-Vetti, H.; Tveit Haavind, M.; Innella, G.; Turchetti, D.; Aretz, S.; et al. Cancer Risks by Sex and Variant Type in PTEN Hamartoma Tumor Syndrome. J. Natl. Cancer Inst. 2023, 115, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Eng, C. PTEN Hamartoma Tumor Syndrome. 2001 Nov 29 [Updated 2021 Feb 11]. In GeneReviews®; Adam, M., Mirzaa, G., Pagon, R., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2021; pp. 87–100. ISBN 978-3-319-28103-2. [Google Scholar]
- Mester, J.L.; Ghosh, R.; Pesaran, T.; Huether, R.; Karam, R.; Hruska, K.S.; Costa, H.A.; Lachlan, K.; Ngeow, J.; Barnholtz-Sloan, J.; et al. Gene-Specific Criteria for PTEN Variant Curation: Recommendations from the ClinGen PTEN Expert Panel. Hum. Mutat. 2018, 39, 1581–1592. [Google Scholar] [CrossRef]
- Spinelli, L.; Black, F.M.; Berg, J.N.; Eickholt, B.J.; Leslie, N.R. Functionally Distinct Groups of Inherited PTEN Mutations in Autism and Tumour Syndromes. J. Med. Genet. 2015, 52, 128–134. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Colas, C.; Pouwels, S.; Hoogerbrugge, N.; Bisseling, T.; Bubien, V.; Caux, F.; Chabbert-Buffet, N.; Colas, C.; Da Mota Gomes, S.; et al. Cancer Surveillance Guideline for Individuals with PTEN Hamartoma Tumour Syndrome. Eur. J. Hum. Genet. 2020, 28, 1387. [Google Scholar] [CrossRef]
- Mester, J.; Eng, C. PTEN Hamartoma Tumor Syndrome. In Handb Clin Neurol; Elsevier: Amsterdam, The Netherlands, 2015; Volume 132, pp. 129–137. [Google Scholar]
- Plamper, M.; Gohlke, B.; Schreiner, F.; Woelfle, J. Phenotype-Driven Diagnostic of PTEN Hamartoma Tumor Syndrome: Macrocephaly, But Neither Height nor Weight Development, Is the Important Trait in Children. Cancers 2019, 11, 975. [Google Scholar] [CrossRef]
- Schultz, K.A.P.; MacFarland, S.P.; Perrino, M.R.; Mitchell, S.G.; Kamihara, J.; Nelson, A.T.; Mallinger, P.H.R.; Brzezinski, J.J.; Maxwell, K.N.; Woodward, E.R.; et al. Update on Pediatric Surveillance Recommendations for PTEN Hamartoma Tumor Syndrome, DICER1-Related Tumor Predisposition, and Tuberous Sclerosis Complex. Clin. Cancer Res. 2025, 31, 234–244. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Dwyer; Mary NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines ®) Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed on 11 May 2023).
- Pilarski, R.; Stephens, J.A.; Noss, R.; Fisher, J.L.; Prior, T.W. Predicting PTEN Mutations: An Evaluation of Cowden Syndrome and Bannayan–Riley–Ruvalcaba Syndrome Clinical Features. J. Med. Genet. 2011, 48, 505–512. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Sample Material | PTEN Status | |
|---|---|---|
| Proband | Blood | Nonsense Variant c.1003C>T(p.Arg335X) |
| Urine | Nonsense Variant c.1003C>T(p.Arg335X) | |
| Hair | n/a * | |
| Vaginal Swab | Nonsense Variant c.1003C>T(p.Arg335X) | |
| Mother | Blood | Wild-type |
| Urine | n/a * | |
| Hair | n/a * | |
| Vaginal Swab | Wild-type | |
| Father | Blood | Wild-type |
| Urine | Wild-type | |
| Hair | n/a * | |
| Sperm | Wild-type | |
| Sister | Blood | Wild-type |
| Urine | Wild-type | |
| Hair | Wild-type | |
| Vaginal Swab | Wild-type |
| Major Criterion (Required) | Secondary Criteria |
|---|---|
| Macrocephaly (≥2 standard deviations) | At least one of the following criteria:∙
|
| Pediatric-onset thyroid cancer and germ cell tumors are known to be associated with CS and should therefore also prompt consideration of PTEN genetic testing. | |
| Cancer Type | Risk Estimate | Surveillance | Starting Age (Years) | Frequency |
|---|---|---|---|---|
| Breast | 85% | Self-examination | 18 | |
| Clinical examination | 25 | Annual | ||
| MRI | 30 | Annual | ||
| Mammography | 40 | Every 2 years | ||
| Thyroid | 35% | Ultrasound | 12 | Every 3 years |
| Annual after age 18 | ||||
| Renal | 33% | Ultrasound | 40 | Every 2 years |
| Colorectal | 16% | Baseline colonoscopy | 35–40 | As required a |
| Melanoma | 5% | Skin examination | 18 b | Annual |
| Endometrial | 28% | Not recommended c |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gebhart, P.; Singer, C.; Muhr, D.; Stein, C.; Tan, Y.Y. A De Novo PTEN Pathogenic Variant in a Young Girl with Sporadic Cowden Syndrome—A Case Report. Pediatr. Rep. 2025, 17, 54. https://doi.org/10.3390/pediatric17030054
Gebhart P, Singer C, Muhr D, Stein C, Tan YY. A De Novo PTEN Pathogenic Variant in a Young Girl with Sporadic Cowden Syndrome—A Case Report. Pediatric Reports. 2025; 17(3):54. https://doi.org/10.3390/pediatric17030054
Chicago/Turabian StyleGebhart, Paulina, Christian Singer, Daniela Muhr, Christina Stein, and Yen Y. Tan. 2025. "A De Novo PTEN Pathogenic Variant in a Young Girl with Sporadic Cowden Syndrome—A Case Report" Pediatric Reports 17, no. 3: 54. https://doi.org/10.3390/pediatric17030054
APA StyleGebhart, P., Singer, C., Muhr, D., Stein, C., & Tan, Y. Y. (2025). A De Novo PTEN Pathogenic Variant in a Young Girl with Sporadic Cowden Syndrome—A Case Report. Pediatric Reports, 17(3), 54. https://doi.org/10.3390/pediatric17030054