

A Case Report of a 5-Year-Old Girl with Self-Limited Epilepsy with Autonomic Seizures



Abstract
1. Introduction
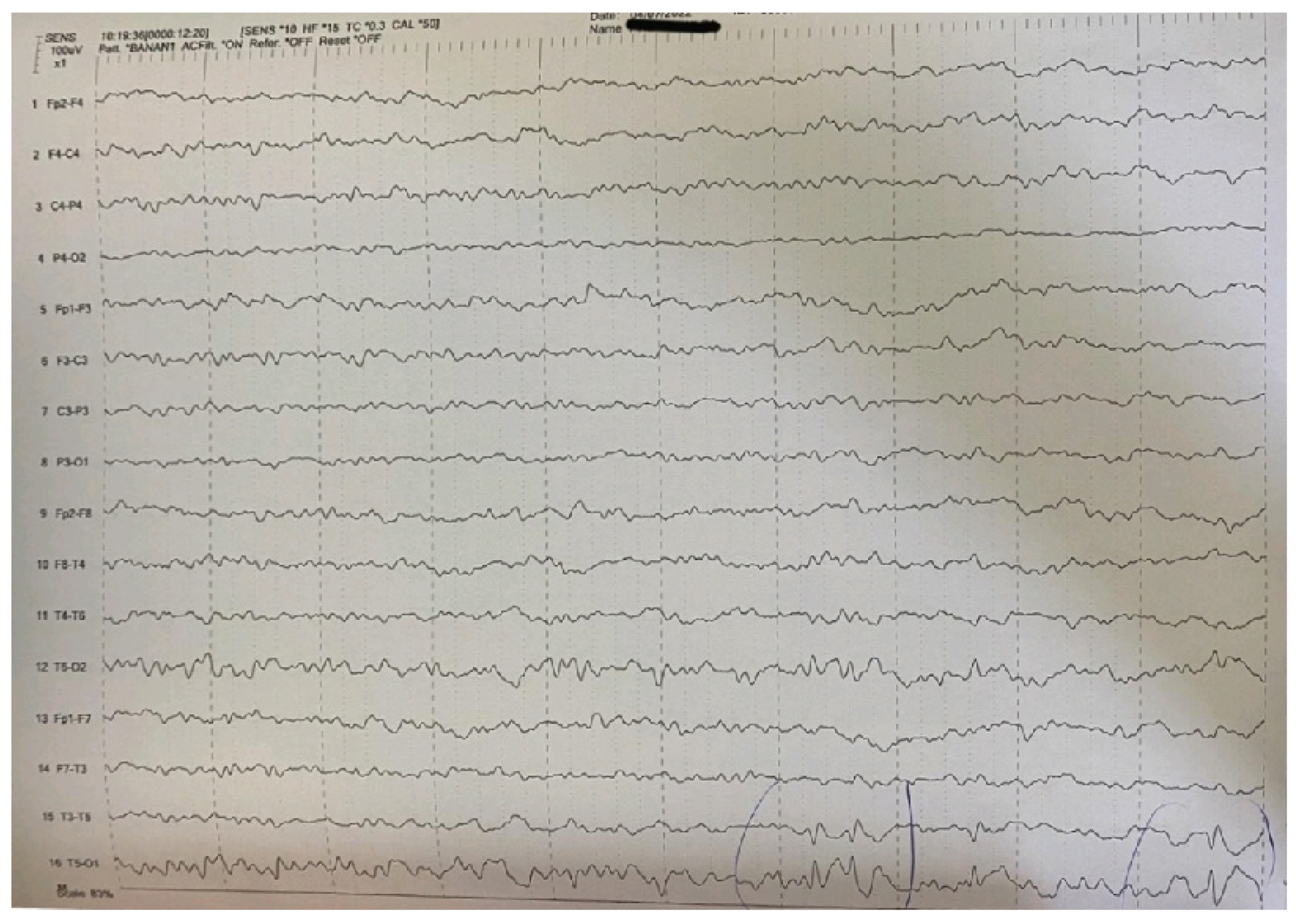
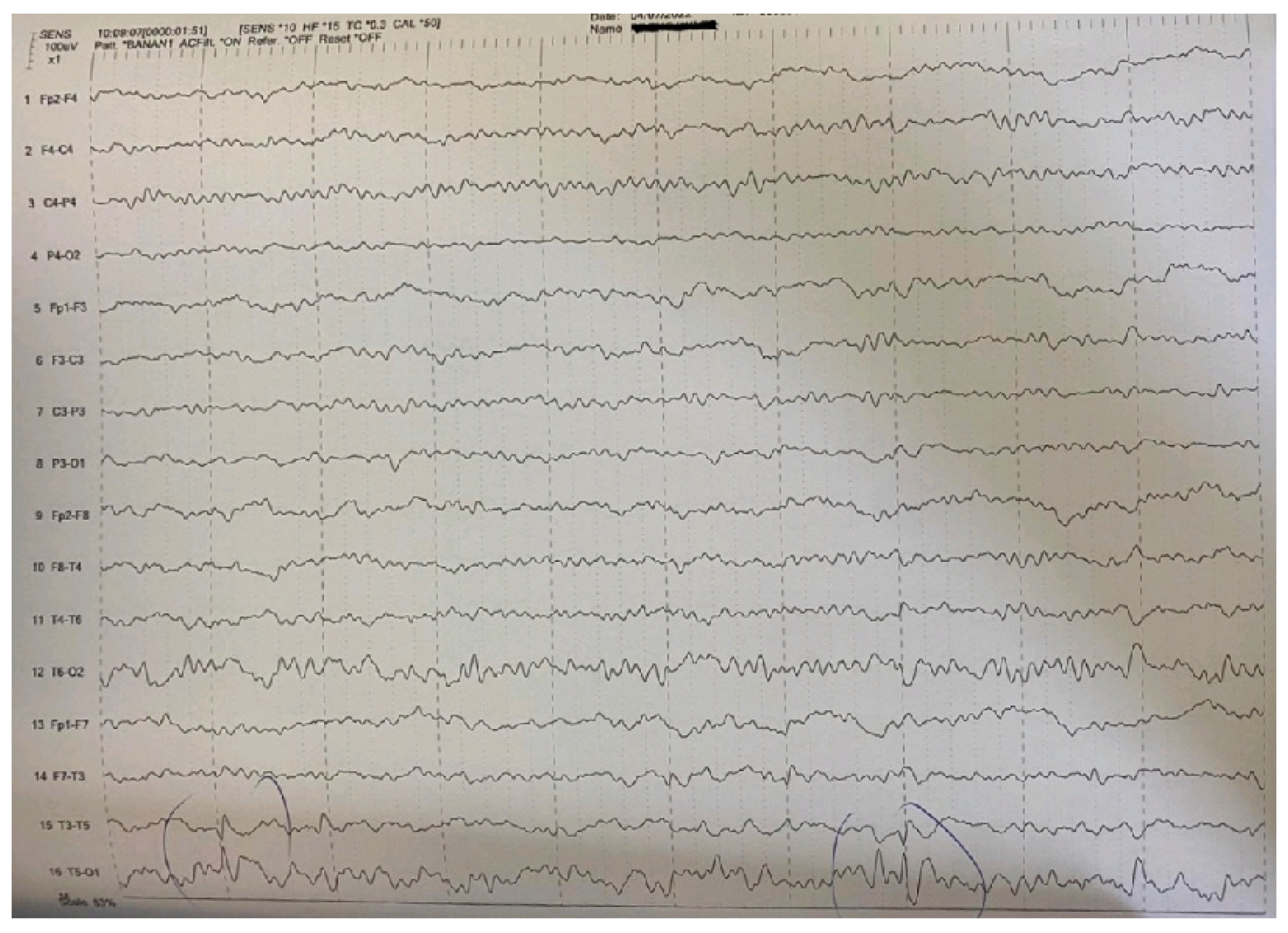
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Panayiotopoulos, C.P.; Aicardi, J. Benign Nocturnal Childhood Occipital Epilepsy: A New Syndrome with Nocturnal Seizures, Tonic Deviation of the Eyes, and Vomiting. J. Child Neurol. 1989, 4, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Covanis, A. Panayiotopoulos Syndrome: A Benign Childhood Autonomic Epilepsy Frequently Imitating Encephalitis, Syncope, Migraine, Sleep Disorder, or Gastroenteritis. Pediatrics 2006, 118, e1237-43. [Google Scholar] [CrossRef]
- Ferrie, C.D.; Caraballo, R.; Covanis, A.; Demirbilek, V.; Dervent, A.; Fejerman, N.; Fusco, L.; Grünewald, R.A.; Kanazawa, O.; Koutroumanidis, M.; et al. Autonomic Status Epilepticus in Panayiotopoulos Syndrome and Other Childhood and Adult Epilepsies: A Consensus View. Epilepsia 2007, 48, 1165–1172. [Google Scholar] [CrossRef]
- Caraballo, R.; Koutroumanidis, M.; Panayiotopoulos, C.P.; Fejerman, N. Idiopathic Childhood Occipital Epilepsy of Gastaut: A Review and Differentiation From Migraine and Other Epilepsies. J. Child Neurol. 2009, 24, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Specchio, N.; Wirrell, E.C.; Scheffer, I.E.; Nabbout, R.; Riney, K.; Samia, P.; Guerreiro, M.; Gwer, S.; Zuberi, S.M.; Wilmshurst, J.M.; et al. International League Against Epilepsy Classification and Definition of Epilepsy Syndromes with Onset in Childhood: Position Paper by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1398–1442. [Google Scholar] [CrossRef] [PubMed]
- Covanis, A.; Ferrie, C.D.; Koutroumanidis, M.; Oguni, H.; Panayiotopoulos, C.P. Panayiotopoulos Syndrome and Gastaut Type Idiopathic Childhood Occipital Epilepsy. In Epileptic Syndromes in Infancy, Childhood and Adolescence; Roger, J., Bureau, M., Dravet, C., Genton, P., Tassinari, C.A., Wolf, P., Eds.; John Libbey Eurotext: Montrouge, France, 2005; pp. 227–253. [Google Scholar]
- Panayiotopoulos, C.P. Panayiotopoulos Syndrome: A Common and Benign Childhood Epileptic Syndrome; John Libbey & Company: London, UK, 2002. [Google Scholar]
- Kawakami, S.; Kubota, M.; Terashima, H.; Nagata, C.; Ishiguro, A. Differentiating Early Clinical Features of Panayiotopoulos Syndrome from Acute Encephalopathy. Brain Dev. 2022, 44, 386–390. [Google Scholar] [CrossRef]
- Panayiotopoulos, C.P. Autonomic Seizures and Autonomic Status Epilepticus Peculiar to Childhood: Diagnosis and Management. Epilepsy Behav. 2004, 5, 286–295. [Google Scholar] [CrossRef]
- Panayiotopoulos, C.P. Vomiting as an Ictal Manifestation of Epileptic Seizures and Syndromes. J. Neurol. Neurosurg. Psychiatry 1988, 51, 1448–1451. [Google Scholar] [CrossRef] [PubMed]
- Panayiotopoulos, C.P. Extraoccipital Benign Childhood Partial Seizures with Ictal Vomiting and Excellent Prognosis. J. Neurol. Neurosurg. Psychiatry 1999, 66, 82–85. [Google Scholar] [CrossRef]
- Rubin, D.I.; Patterson, M.C.; Westmoreland, B.F.; Klass, D.W. Angelman’s Syndrome: Clinical and Electroencephalographic Findings. Electroencephalogr. Clin. Neurophysiol. 1997, 102, 299–302. [Google Scholar] [CrossRef]
- Viani, F.; Romeo, A.; Viri, M.; Mastrangelo, M.; Lalatta, F.; Briscioli, V.; Gobbi, G.; Lanzi, G.; Bettio, D. Seizure and EEG Patterns in Angelman’s Syndrome. J. Child Neurol. 1995, 10, 467–471. [Google Scholar] [CrossRef]
- Wilson, S.J. Rethinking Neurobehavioral Comorbidity in Panayiotopoulos Syndrome. Dev. Med. Child Neurol. 2020, 62, 893. [Google Scholar] [CrossRef] [PubMed]
- Kivity, S.; Ephraim, T.; Weitz, R.; Tamir, A. Childhood Epilepsy with Occipital Paroxysms: Clinical Variants in 134 Patients. Epilepsia 2000, 41, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.; Rowlinson, S.; Manidakis, I.; Ferrie, C.D.; Koutroumanidis, M. The Contribution of the EEG Technologists in the Diagnosis of Panayiotopoulos Syndrome (Susceptibility to Early Onset Benign Childhood Autonomic Seizures). Seizure 2004, 13, 565–573. [Google Scholar] [CrossRef][Green Version]
- Parisi, P.; Ferri, R.; Pagani, J.; Cecili, M.; Montemitro, E.; Villa, M.P. Ictal Video-Polysomnography and EEG Spectral Analysis in a Child with Severe Panayiotopoulos Syndrome. Epileptic Disord. 2005, 7, 333–339. [Google Scholar]
- Panayiotopoulos, C.P. Benign Childhood Focal Seizures and Related Epileptic Syndromes. In Clinical Guide to Epileptic Syndromes and Their Treatment; Springer: London, UK, 2007; pp. 285–318. [Google Scholar]
- Dalla Bernardina, B.; Sgro, V.; Fejerman, N. Epilepsy with Centrotemporal Spikes and Related Syndromes. In Epileptic Syndromes in Infancy, Childhood and Adolescence; John Libbey Eurotext: Paris, France, 2005; pp. 203–225. [Google Scholar]
- Marini, C.; Mei, D.; Temudo, T.; Ferrari, A.R.; Buti, D.; Dravet, C.; Dias, A.I.; Moreira, A.; Calado, E.; Seri, S.; et al. Idiopathic Epilepsies with Seizures Precipitated by Fever and SCN1A Abnormalities. Epilepsia 2007, 48, 1678–1685. [Google Scholar] [CrossRef]
- Mulley, J.C.; Scheffer, I.E.; Petrou, S.; Dibbens, L.M.; Berkovic, S.F.; Harkin, L.A. SCN1A Mutations and Epilepsy. Hum. Mutat. 2005, 25, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Livingston, J.H.; Cross, J.H.; Mclellan, A.; Birch, R.; Zuberi, S.M. A Novel Inherited Mutation in the Voltage Sensor Region of SCN1A Is Associated with Panayiotopoulos Syndrome in Siblings and Generalized Epilepsy with Febrile Seizures Plus. J. Child Neurol. 2009, 24, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.O.; Sotero de Menezes, M.A. SCN1A Seizure Disorders. University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Kivity, S.; Oliver, K.L.; Afawi, Z.; Damiano, J.A.; Arsov, T.; Bahlo, M.; Berkovic, S.F. SCN1A Clinical Spectrum Includes the Self-Limited Focal Epilepsies of Childhood. Epilepsy Res. 2017, 131, 9–14. [Google Scholar] [CrossRef]
- Grosso, S.; Orrico, A.; Galli, L.; Di Bartolo, R.; Sorrentino, V.; Balestri, P. SCN1A Mutation Associated with Atypical Panayiotopoulos Syndrome. Neurology 2007, 69, 609–611. [Google Scholar] [CrossRef]
- Panayiotopoulos, C.P.; Michael, M.; Sanders, S.; Valeta, T.; Koutroumanidis, M. Benign Childhood Focal Epilepsies: Assessment of Established and Newly Recognized Syndromes. Brain 2008, 131, 2264–2286. [Google Scholar] [CrossRef] [PubMed]
- Specchio, N.; Trivisano, M.; Di Ciommo, V.; Cappelletti, S.; Masciarelli, G.; Volkov, J.; Fusco, L.; Vigevano, F. Panayiotopoulos Syndrome: A Clinical, EEG, and Neuropsychological Study of 93 Consecutive Patients. Epilepsia 2010, 51, 2098–2107. [Google Scholar] [CrossRef] [PubMed]
- Valeta, T. Parental Attitude, Reaction and Education in Benign Childhood Focal Seizures. In The Epilepsies: Seizures, Syndromes and Management; Panayiotopoulos, C.P., Ed.; Bladon Medical Publishing: Oxford, UK, 2005; pp. 258–261. [Google Scholar]
- Ferrie, C.; Caraballo, R.; Covanis, A.; Demirbilek, V.; Dervent, A.; Kivity, S.; Koutroumanidis, M.; Martinovic, Z.; Oguni, H.; Verrotti, A.; et al. Panayiotopoulos Syndrome: A Consensus View. Dev. Med. Child Neurol. 2006, 48, 236–240. [Google Scholar] [CrossRef]
- Beardsley, S.J.; Dostal, I.; Cole, J.; Gutierrez, A.; Robson, J. Valproate Use in Women Aged 15–44 Years: An Observational Study in General Practice. BJGP Open 2021, 5, BJGPO.2020.0104. [Google Scholar] [CrossRef]
- Kikumoto, K.; Yoshinaga, H.; Oka, M.; Ito, M.; Endoh, F.; Akiyama, T.; Ohtsuka, Y. EEG and Seizure Exacerbation Induced by Carbamazepine in Panayiotopoulos Syndrome. Epileptic Disord. 2006, 8, 53–56. [Google Scholar]
- Mujawar, Q.M.; Sen, S.; Ali, M.D.; Balakrishnan, P.; Patil, S. Panayiotopoulos Syndrome Presenting with Status Epilepticus and Cardiorespiratory Arrest: A Case Report. Pediatr. Emerg. Care 2011, 27, 754–757. [Google Scholar] [CrossRef]
- García, C.; Rubio, G. Efficacy and Safety of Levetiracetam in the Treatment of Panayiotopoulos Syndrome. Epilepsy Res. 2009, 85, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, F.U. Febrile Seizures: Treatment and Prognosis. Epilepsia 2000, 41, 2–9. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Symptoms and Manifestations of SeLEAS | Characteristics and Frequency of Symptoms and Manifestations of SeLEAS | Symptoms and Manifestations of Our Patient 1 |
|---|---|---|
| Age of onset (years) | 1–14 (Mean: 4–5) | 5 years old |
| Duration of seizures | ||
| Less than 4 min | Rare | <5 min duration of seizures |
| ≥5 min | As a rule | |
| Seizures during sleep | >2/3 of cases | Seizures during sleep |
| Features of seizures | ||
| Ictal vomiting | Frequent | Ictal vomiting |
| Deviation of the eyes | Frequent | Deviation of the eyes |
| Impairment of consciousness | Frequent | Drowsiness |
| Visual hallucinations | Rare | |
| Loss of vision | Rare | |
| Autonomic semiology | As a rule | Urinary and faecal incontinence |
| Postictal migraine-like headache | Rare | |
| Seizures evolving into autonomic status epilepticus | Frequent | |
| Main interictal EEG | Multifocal spikes | High-amplitude sharp waves and spike-wave complexes in temporal-occipital areas of the left hemisphere in the EEG. Enhancement of focal abnormalities in temporal-occipital areas of the left hemisphere in the EEG during sleep. |
| Clinical Epilepsy Syndromes | Less Common Presentations |
|---|---|
| Febrile seizures (simple or complex) | Epilepsy with focal seizures |
| Febrile seizures plus (FS+) | Myoclonic–astatic epilepsy (MAE, Doose syndrome) |
| Generalized epilepsy | Lennox–Gastaut syndrome |
| Generalized epilepsy with febrile seizures plus (GEFS+) | Infantile spasms |
| Dravet syndrome | Vaccine-related encephalopathy and seizures |
| Severe myoclonic epilepsy, borderline (SMEB) | |
| Intractable childhood epilepsy with generalized tonic–clonic seizures (ICE-GTC) | |
| Infantile partial seizures with variable foci |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsaras, G.; Samartzi, P.; Tsitsani, P. A Case Report of a 5-Year-Old Girl with Self-Limited Epilepsy with Autonomic Seizures. Pediatr. Rep. 2023, 15, 494-501. https://doi.org/10.3390/pediatric15030045
Katsaras G, Samartzi P, Tsitsani P. A Case Report of a 5-Year-Old Girl with Self-Limited Epilepsy with Autonomic Seizures. Pediatric Reports. 2023; 15(3):494-501. https://doi.org/10.3390/pediatric15030045
Chicago/Turabian StyleKatsaras, Georgios, Petrina Samartzi, and Pelagia Tsitsani. 2023. "A Case Report of a 5-Year-Old Girl with Self-Limited Epilepsy with Autonomic Seizures" Pediatric Reports 15, no. 3: 494-501. https://doi.org/10.3390/pediatric15030045
APA StyleKatsaras, G., Samartzi, P., & Tsitsani, P. (2023). A Case Report of a 5-Year-Old Girl with Self-Limited Epilepsy with Autonomic Seizures. Pediatric Reports, 15(3), 494-501. https://doi.org/10.3390/pediatric15030045